Biodegradable Chitosan-graft-Poly(l-lactide) Copolymers For Bone Tissue Engineering

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
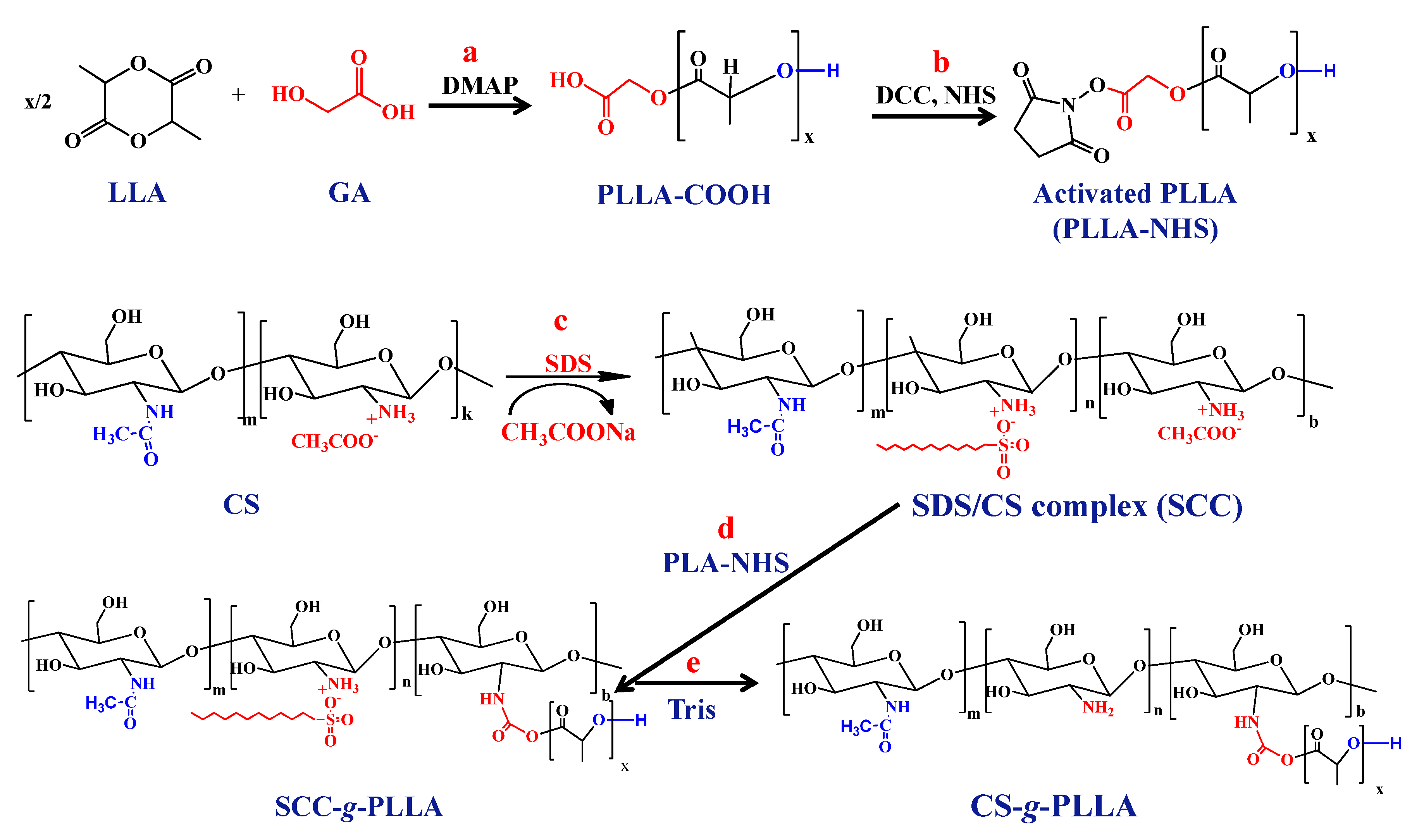
2.2. Synthesis of Carboxyl Terminated PLLA
2.3. Synthesis of CS-g-PLLA Copolymers
2.4. Characterization Techniques
2.5. Preparation and Characterization of the CS-g-PLLA Copolymer Films and Discs
2.6. Nanoindentation Testing in the Dry and Wet State
2.7. Degradation Study
2.8. Cell Culture of MC3T3-E1 pre-Osteoblastic Cells on the CS-g-PLLA Films
2.9. Adhesion and Morphology of the MC3T3-E1 Cells on the CS-g-PLLA Films
2.10. Viability and Proliferation of the Pre-Osteoblastic Cells on the CS-g-PLLA Films
2.11. Statistical Analysis
3. Results
3.1. Synthesis of Carboxyl Terminated Poly(L-Lactide)
3.2. Synthesis of the Chitosan-g-poly(l-lactide) Copolymers
3.3. Characterization of the CS-g-PLLA Graft Copolymers
3.4. Thermal Properties of the CS-g-PLLA Copolymers
3.5. Characterization of the Copolymer Films
3.6. Nanoindentation on the CS-g-PLLA Discs in the Dry and Wet State
3.7. Degradation Profile of the CS-g-PLLA Discs
3.8. Viability and Proliferation of Pre-osteoblastic Cells on the CS-g-PLLA Films
3.9. Morphology of ΜC3T3-E1 Cells on the CS-g-PLLA Film
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Logeart-Avramoglou, D.; Anagnostou, F.; Bizios, R.; Petite, H. Engineering bone: challenges and obstacles. J. Cell. Mol. Med. 2005, 9, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.M. Biomaterials for bone tissue engineering. Mater. Today 2008, 11, 18–25. [Google Scholar] [CrossRef]
- Farokhi, M.; Mottaghitalab, F.; Shokrgozar, M.A.; Ou, K.-L.; Mao, C.; Hosseinkhani, H. Importance of dual delivery systems for bone tissue engineering. J. Controlled Release 2016, 225, 152–169. [Google Scholar] [CrossRef]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Lee, E.J.; Kasper, F.K.; Mikos, A.G. Biomaterials for Tissue Engineering. Ann. Biomed. Eng. 2014, 42, 323–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LogithKumar, R.; KeshavNarayan, A.; Dhivya, S.; Chawla, A.; Saravanan, S.; Selvamurugan, N. A review of chitosan and its derivatives in bone tissue engineering. Carbohydr. Polym. 2016, 151, 172–188. [Google Scholar] [CrossRef]
- Levengood, S.K.L.; Zhang, M. Chitosan-based scaffolds for bone tissue engineering. J. Mat. Chem. B 2014, 2, 3161–3184. [Google Scholar] [CrossRef]
- Di Martino, A.; Sittinger, M.; Risbud, M.V. Chitosan: A versatile biopolymer for orthopaedic tissue-engineering. Biomaterials 2005, 26, 5983–5990. [Google Scholar] [CrossRef]
- Pillai, C.K.S.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Saravanan, S.; Leena, R.S.; Selvamurugan, N. Chitosan based biocomposite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1354–1365. [Google Scholar] [CrossRef]
- Kim, I.-Y.; Seo, S.-J.; Moon, H.-S.; Yoo, M.-K.; Park, I.-Y.; Kim, B.-C.; Cho, C.-S. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv. 2008, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Van der Schueren, L.; Steyaert, I.; De Schoenmaker, B.; De Clerck, K. Polycaprolactone/chitosan blend nanofibres electrospun from an acetic acid/formic acid solvent system. Carbohydr. Polym. 2012, 88, 1221–1226. [Google Scholar] [CrossRef]
- Li, A.D.; Sun, Z.Z.; Zhou, M.; Xu, X.X.; Ma, J.Y.; Zheng, W.; Zhou, H.M.; Li, L.; Zheng, Y.F. Electrospun Chitosan-graft-PLGA nanofibres with significantly enhanced hydrophilicity and improved mechanical property. Colloids Surf. B 2013, 102, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, C.; Luo, B.; Wen, W.; Wang, X.; Ding, S.; Zhou, C. Enhancement of growth and osteogenic differentiation of MC3T3-E1 cells via facile surface functionalization of polylactide membrane with chitooligosaccharide based on polydopamine adhesive coating. Appl. Surf. Sci. 2016, 360, 858–865. [Google Scholar] [CrossRef]
- Niu, X.; Li, X.; Liu, H.; Zhou, G.; Feng, Q.; Cui, F.; Fan, Y. Homogeneous Chitosan/Poly(L-Lactide) Composite Scaffolds Prepared by Emulsion Freeze-Drying. J. Biomater. Sci. Polym. Ed. 2012, 23, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, H.; Yu, A.; Wen, D. Biodegradable Polylactide/Chitosan Blend Membranes. Biomacromolecules 2006, 7, 1362–1372. [Google Scholar] [CrossRef]
- Shan, X.; Li, F.; Liu, C.; Gao, Q. Electrospinning of chitosan/poly(lactic acid) nanofibers: The favorable effect of nonionic surfactant. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Chen, S.; Hao, Y.; Cui, W.; Chang, J.; Zhou, Y. Biodegradable electrospun PLLA/chitosan membrane as guided tissue regeneration membrane for treating periodontitis. J. Mater. Sci. 2013, 48, 6567–6577. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.L.; Qi, A.D.; Liu, W.G.; Wang, X.H.; Wang, H.; Ma, D.M.; Yao, K.D. Biomimetic surface modification of poly(l-lactic acid) with chitosan and its effects on articular chondrocytes in vitro. Biomaterials 2003, 24, 3859–3868. [Google Scholar] [CrossRef]
- Rogina, A.; Pribolšan, L.; Hanžek, A.; Gόmez-Estrada, L.; Gallego Ferrer, G.; Marijanović, I.; Ivanković, M.; Ivanković, H. Macroporous poly(lactic acid) construct supporting the osteoinductive porous chitosan-based hydrogel for bone tissue engineering. Polymer 2016, 98, 172–181. [Google Scholar] [CrossRef]
- Lin, C.-C.; Fu, S.-J.; Lin, Y.-C.; Yang, I.K.; Gu, Y. Chitosan-coated electrospun PLA fibers for rapid mineralization of calcium phosphate. Int. J. Biol. Macromol. 2014, 68, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Maharana, T.; Pattanaik, S.; Routaray, A.; Nath, N.; Sutar, A.K. Synthesis and characterization of poly(lactic acid) based graft copolymers. React. Funct. Polym. 2015, 93, 47–67. [Google Scholar] [CrossRef]
- Li, G.; Zhuang, Y.; Mu, Q.; Wang, M. Preparation, characterization and aggregation behavior of amphiphilic chitosan derivative having poly (l-lactic acid) side chains. Carbohydr. Polym. 2008, 72, 60–66. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S. Synthesis and characterization of a novel amphiphilic chitosan–polylactide graft copolymer. Carbohydr. Polym. 2005, 59, 165–171. [Google Scholar] [CrossRef]
- Skotak, M.; Leonov, A.P.; Larsen, G.; Noriega, S.; Subramanian, A. Biocompatible and Biodegradable Ultrafine Fibrillar Scaffold Materials for Tissue Engineering by Facile Grafting of l-Lactide onto Chitosan. Biomacromolecules 2008, 9, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, A.; Guo, S.; Chen, S.; Li, J. Preparation of chitosan-g-polylactide graft copolymers via self-catalysis of phthaloylchitosan and their complexation with DNA. React. Funct. Polym. 2010, 70, 301–305. [Google Scholar] [CrossRef]
- Su, F.; Wang, J.; Zhu, S.; Liu, S.; Yu, X.; Li, S. Synthesis and characterization of novel carboxymethyl chitosan grafted polylactide hydrogels for controlled drug delivery. Polym. Adv. Technol. 2015, 26, 924–931. [Google Scholar] [CrossRef]
- Ge, W.; Li, D.; Chen, M.; Wang, X.; Liu, S.; Sun, R. Characterization and antioxidant activity of β-carotene loaded chitosan-graft-poly(lactide) nanomicelles. Carbohydr. Polym. 2015, 117, 169–176. [Google Scholar] [CrossRef]
- Di Martino, A.; Sedlarik, V. Amphiphilic chitosan-grafted-functionalized polylactic acid based nanoparticles as a delivery system for doxorubicin and temozolomide co-therapy. Int. J. Pharm. 2014, 474, 134–145. [Google Scholar] [CrossRef]
- Zhang, Z.; Cui, H. Biodegradability and Biocompatibility Study of Poly(Chitosan-g-lactic Acid) Scaffolds. Molecules 2012, 17, 3243–3258. [Google Scholar] [CrossRef]
- Wan, Y.; Cao, X.; Zhang, S.; Wang, S.; Wu, Q. Fibrous poly(chitosan-g-dl-lactic acid) scaffolds prepared via electro-wet-spinning. Acta Biomater. 2008, 4, 876–886. [Google Scholar] [CrossRef]
- Qi, W.; Pei, L.; Peifeng, L.; Tao, G.; Suming, L.; Yourong, D.; Zhirong, Z. Preparation, blood coagulation and cell compatibility evaluation of chitosan-graft-polylactide copolymers. Biomed. Mater. 2014, 9, 015007. [Google Scholar]
- Kadota, J.; Pavlovic, D.; Hirano, H.; Okada, A.; Agari, Y.; Bibal, B.; Deffieux, A.; Peruch, F. Controlled bulk polymerization of l-lactide and lactones by dual activation with organo-catalytic systems. RSC Adv. 2014, 4, 14725–14732. [Google Scholar] [CrossRef]
- Xie, D.; Huang, H.; Blackwood, K.; MacNeil, S. A novel route for the production of chitosan/poly(lactide-co-glycolide) graft copolymers for electrospinning. Biomed. Mater. 2010, 5, 065016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzinikolaidou, M.; Kaliva, M.; Batsali, A.; Pontikoglou, C.; Vamvakaki, M. Wharton’s Jelly Mesenchymal Stem Cell Response on Chitosan-graft-poly (ε-caprolactone) Copolymer for Myocardium Tissue Engineering. Curr. Pharm. Des. 2014, 20, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Dragatogiannis, D.A.; Koumoulos, E.; Ellinas, K.; Tserepi, A.; Gogolides, E.; Charitidis, C.A. Nanoscale Mechanical and Tribological Properties of Plasma Nanotextured COP Surfaces with Hydrophobic Coatings. Plasma Processes Polym. 2015, 12, 1271–1283. [Google Scholar] [CrossRef]
- King, R.B. Elastic analysis of some punch problems for a layered medium. Int. J. Solids Struct. 1987, 23, 1657–1664. [Google Scholar] [CrossRef]
- Oliver, W.C.; Pharr, G.M. An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 1992, 7, 1564–1583. [Google Scholar] [CrossRef]
- Sneddon, I.N. Boussinesq’s problem for a rigid cone. Math. Proc. Cambridge Phil. Soc. 2008, 44, 492–507. [Google Scholar] [CrossRef]
- Li, J.; Kong, M.; Cheng, X.J.; Li, J.J.; Liu, W.F.; Chen, X.G. A facile method for preparing biodegradable chitosan derivatives with low grafting degree of poly(lactic acid). Int. J. Biol. Macromol. 2011, 49, 1016–1021. [Google Scholar] [CrossRef]
- Feng, H.; Dong, C.-M. Preparation, Characterization, and Self-Assembled Properties of Biodegradable Chitosan−Poly(lL-lactide) Hybrid Amphiphiles. Biomacromolecules 2006, 7, 3069–3075. [Google Scholar] [CrossRef]
- Shao, J.; Chen, C.; Wang, Y.; Chen, X.; Du, C. Structure and surface nanomechanics of poly(L-lactide) from thermally induced phase separation process. Appl. Surf. Sci. 2012, 258, 6665–6671. [Google Scholar] [CrossRef]
- Huda, M.S.; Yasui, M.; Mohri, N.; Fujimura, T.; Kimura, Y. Dynamic mechanical properties of solution-cast poly(L-lactide) films. Mater. Sci. Eng. A 2002, 333, 98–105. [Google Scholar] [CrossRef]
- Wang, S.-F.; Shen, L.; Zhang, W.-D.; Tong, Y.-J. Preparation and Mechanical Properties of Chitosan/Carbon Nanotubes Composites. Biomacromolecules 2005, 6, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chesnutt, B.M.; Haggard, W.O.; Bumgardner, J.D. Deacetylation of Chitosan: Material Characterization and in vitro Evaluation via Albumin Adsorption and Pre-Osteoblastic Cell Cultures. Materials 2011, 4, 1399–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xue, C.; Xue, Y.; Gao, R.; Zhang, X. Determination of the degree of deacetylation of chitin and chitosan by X-ray powder diffraction. Carbohydr. Res. 2005, 340, 1914–1917. [Google Scholar] [CrossRef]
- De Britto, D.; Campana-Filho, S.P. Kinetics of the thermal degradation of chitosan. Thermochim. Acta 2007, 465, 73–82. [Google Scholar] [CrossRef]
- Cai, G.; Jiang, H.; Chen, Z.; Tu, K.; Wang, L.; Zhu, K. Synthesis, characterization and self-assemble behavior of chitosan-O-poly(ε-caprolactone). Eur. Polym. J. 2009, 45, 1674–1680. [Google Scholar] [CrossRef]
- Wan, Y.; Cao, X.; Wu, Q.; Zhang, S.; Wang, S. Preparation and mechanical properties of poly(chitosan-g-DL-lactic acid) fibrous mesh scaffolds. Polym. Adv. Technol. 2008, 19, 114–123. [Google Scholar] [CrossRef]
- Dee, K.C.; Puleo, D.A.; Bizios, R. An Introduction To Tissue-Biomaterial Interactions; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp. 1–13. [Google Scholar]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.-L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Robert, L.; Mikos, A.G. In vitro and in vivo degradation of porous poly(dl-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Charitidis, C.A.; Dragatogiannis, D.A.; Milioni, E.; Kaliva, M.; Vamvakaki, M.; Chatzinikolaidou, M. Synthesis, Nanomechanical Characterization and Biocompatibility of a Chitosan-Graft-Poly(ε-caprolactone) Copolymer for Soft Tissue Regeneration. Materials 2019, 12, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckachan, G.E.; Pillai, C.K.S. Chitosan/oligo L-lactide graft copolymers: Effect of hydrophobic side chains on the physico-chemical properties and biodegradability. Carbohydr. Polym. 2006, 64, 254–266. [Google Scholar] [CrossRef]
- Duarte, A.R.C.; Mano, J.F.; Reis, R.L. Novel 3D scaffolds of chitosan–PLLA blends for tissue engineering applications: Preparation and characterization. J. Supercrit. Fluids 2010, 54, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Yang, H.; Yang, D.; Yu, Z.-Z. Polylactic Acid Nanofiber Scaffold Decorated with Chitosan Islandlike Topography for Bone Tissue Engineering. ACS Appl. Mater. Interfaces 2017, 9, 21094–21104. [Google Scholar] [CrossRef]
- Georgopoulou, A.; Kaliva, M.; Vamvakaki, M.; Chatzinikolaidou, M. Osteogenic Potential of Pre-Osteoblastic Cells on a Chitosan-graft-Polycaprolactone Copolymer. Materials (Basel) 2018, 11, 490. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, L.; Kaliva, M.; Vamvakaki, M.; Chatzinikolaidou, M. Immunomodulatory Potential of Chitosan-graft-poly(ε-caprolactone) Copolymers toward the Polarization of Bone-Marrow-Derived Macrophages. ACS Biomater Sci. Eng. 2017, 3, 1341–1349. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tm (°C) | ΔHf (J/g) a | Χc |
|---|---|---|---|
| PLLA-COOH | 154 | 62.1 | 66% |
| CS-g-PLLA(80/20) | 155 | 44.2 | 47% |
| CS-g-PLLA(50/50) | 155 | 53.8 | 57% |
| Sample | Er (GPa) | E (GPa) | H (GPa) |
|---|---|---|---|
| CS-g-PLLA(80/20) | 0.53 ± 0.11 | 0.48 ± 0.01 | 0.06 ± 0.02 |
| CS-g-PLLA(50/50) | 0.14 ± 0.01 | 0.13 ± 0.02 | 0.03 ± 0.01 |
| Time (h) | Er (MPa) | E (MPa) | H (MPa) | |||
|---|---|---|---|---|---|---|
| CS-g-PLLA (80/20) | CS-g-PLLA (50/50) | CS-g-PLLA (80/20) | CS-g-PLLA (50/50) | CS-g-PLLA (80/20) | CS-g-PLLA (50/50) | |
| 0 | 785 ± 20 | 250 ± 50 | 715 ± 70 | 226 ± 50 | 112 ± 10 | 63 ± 0 |
| 24 | 3.7 ± 2 | 4.5 ± 1.5 | 3.3 ± 1.5 | 4.1 ± 1.5 | 0.04 ± 0.02 | 0.04 ± 0.02 |
| 648 | 1.1 ± 0.4 | 4.2 ± 2 | 1.0 ± 0.2 | 3.8 ± 2 | 0.01 ± 0.01 | 0.04 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaliva, M.; Georgopoulou, A.; Dragatogiannis, D.A.; Charitidis, C.A.; Chatzinikolaidou, M.; Vamvakaki, M. Biodegradable Chitosan-graft-Poly(l-lactide) Copolymers For Bone Tissue Engineering. Polymers 2020, 12, 316. https://doi.org/10.3390/polym12020316
Kaliva M, Georgopoulou A, Dragatogiannis DA, Charitidis CA, Chatzinikolaidou M, Vamvakaki M. Biodegradable Chitosan-graft-Poly(l-lactide) Copolymers For Bone Tissue Engineering. Polymers. 2020; 12(2):316. https://doi.org/10.3390/polym12020316
Chicago/Turabian StyleKaliva, Maria, Anthie Georgopoulou, Dimitrios A. Dragatogiannis, Costas A. Charitidis, Maria Chatzinikolaidou, and Maria Vamvakaki. 2020. "Biodegradable Chitosan-graft-Poly(l-lactide) Copolymers For Bone Tissue Engineering" Polymers 12, no. 2: 316. https://doi.org/10.3390/polym12020316