The Importance of the Mixing Energy in Ionized Superabsorbent Polymer Swelling Models


Abstract
:
1. Introduction
2. Methods
2.1. Swelling Model
2.1.1. Equilibrium Conditions
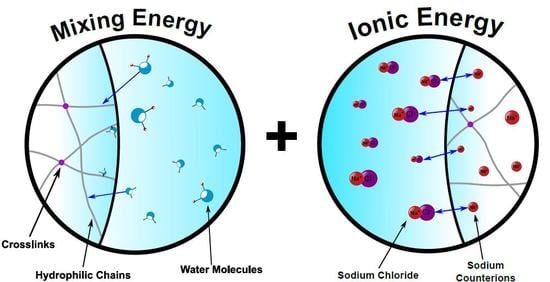
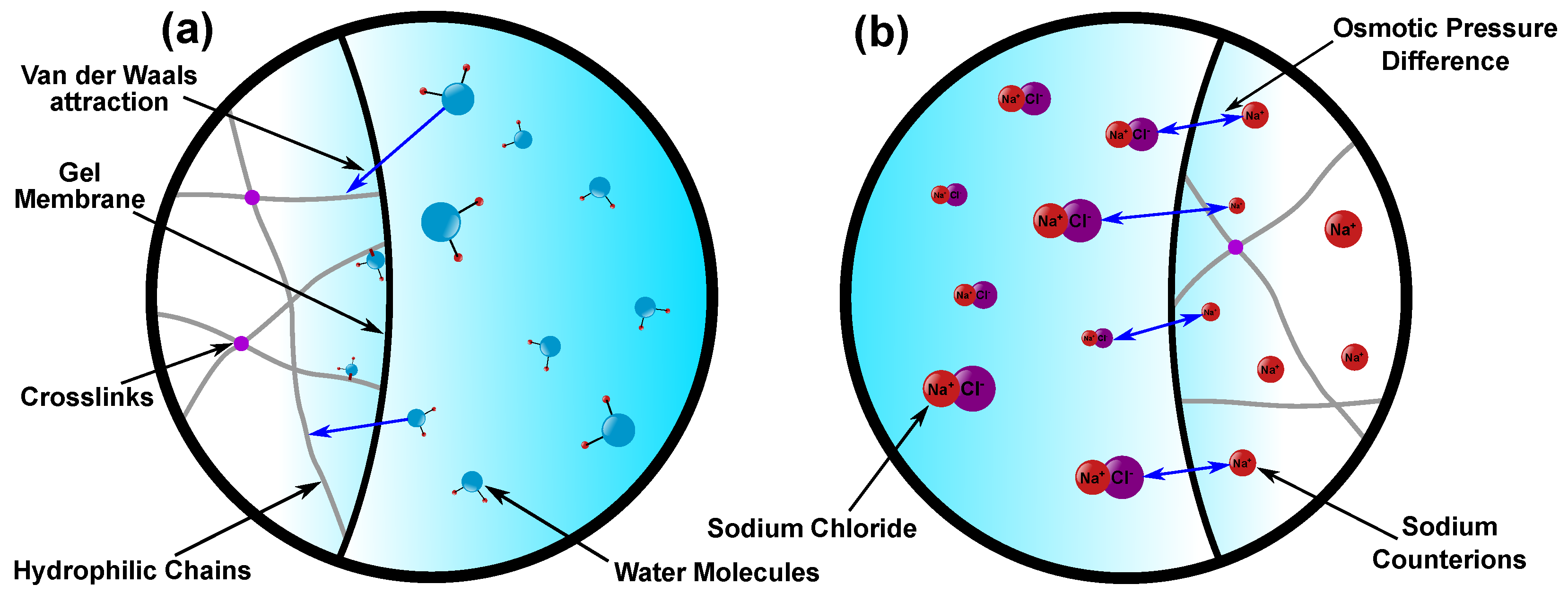
2.1.2. Mixing Energy
2.1.3. Ionic Energy
2.1.4. Elastic Energy
2.1.5. Kinematics
2.2. Finite Element Model & Model Parameters
2.3. Solutions
2.4. Experimental Validation
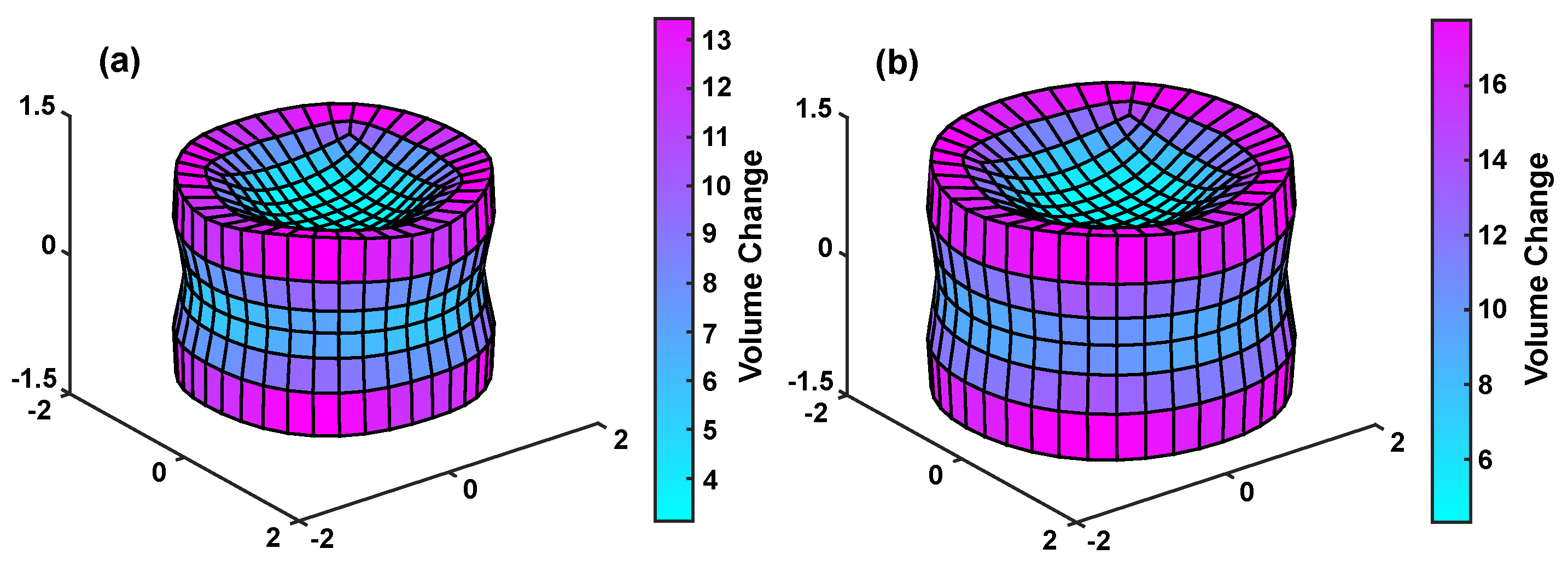
3. Simulation Results
3.1. Univalent Solution
3.2. Divalent vs. Univalent Solutions
3.3. Transient Surface Instabilities
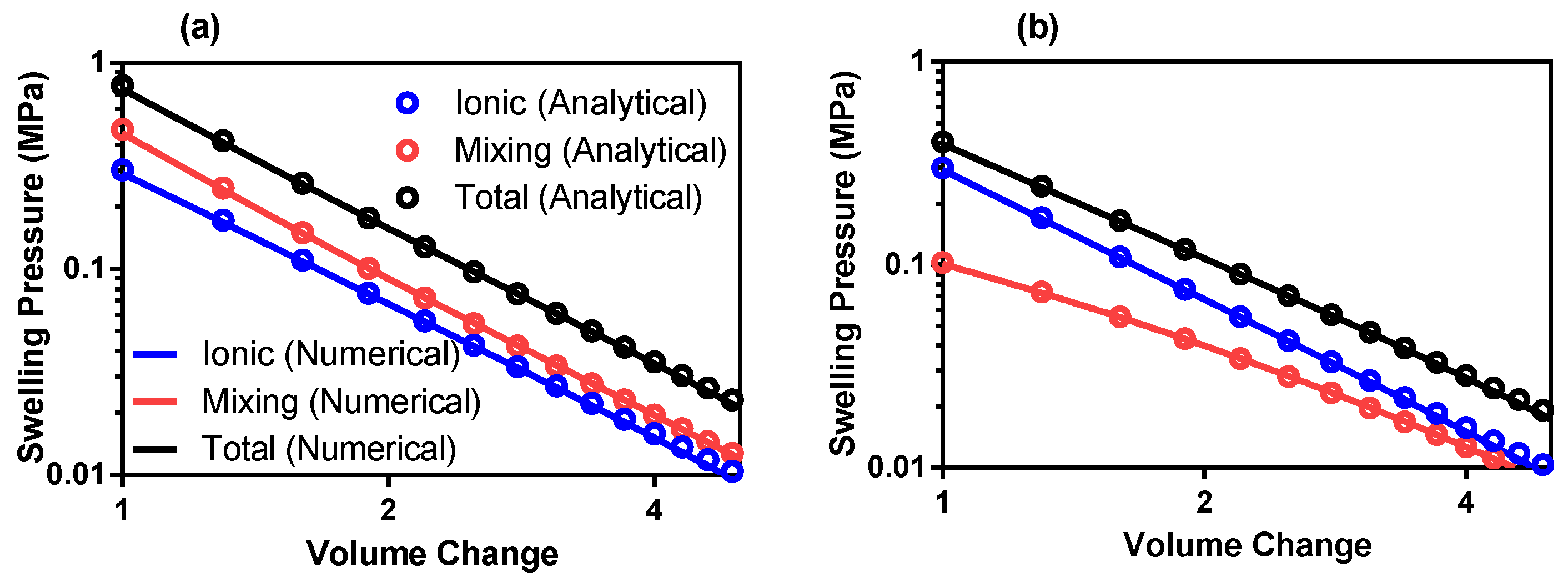
4. Comparison to Theory
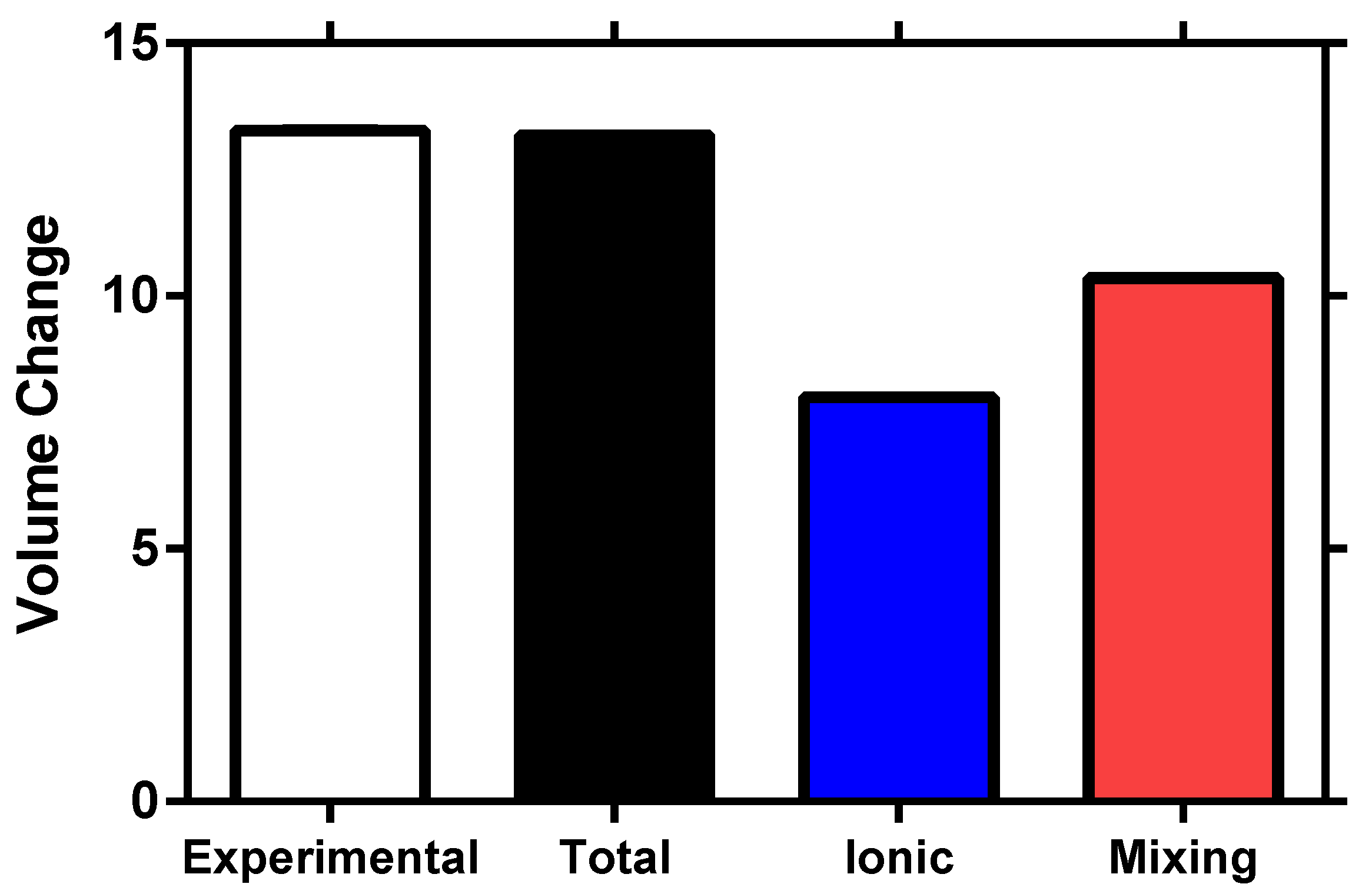
5. Experimental Validation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Calcium Chloride | |
| MHFEM | Mixed Hybrid Finite Element Method |
| NaCl | Sodium Chloride |
| pHEMA | Polyhydroxyethylmethacrylate |
References
- Buchholz, F.L.; Graham, A.T. Modern Superabsorbent Polymer Technology; John Wiley & Sons, Inc.: New York, NY, USA, 1998; p. 279. [Google Scholar]
- Rimmer, S. Biomedical Hydrogels: Biochemistry, Manufacture and Medical Applications; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Narjary, B.; Aggarwal, P.; Singh, A.; Chakraborty, D.; Singh, R. Water availability in different soils in relation to hydrogel application. Geoderma 2012, 187, 94–101. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T. Gels. Sci. Am. 1981, 244, 124. [Google Scholar] [CrossRef]
- Drozdov, A.D. Mechanical behavior of temperature-sensitive gels under equilibrium and transient swelling. Int. J. Eng. Sci. 2018, 128, 79–100. [Google Scholar] [CrossRef]
- Fennell, E.; Huyghe, J.M. Chemically responsive hydrogel deformation mechanics: A review. Molecules 2019, 24, 3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Flory, P.J.; Rehner, J., Jr. Statistical mechanics of cross-linked polymer networks I. Rubberlike elasticity. J. Chem. Phys. 1943, 11, 512–520. [Google Scholar] [CrossRef]
- Yu, C.; Malakpoor, K.; Huyghe, J.M. A three-dimensional transient mixed hybrid finite element model for superabsorbent polymers with strain-dependent permeability. Soft Matter 2018, 14, 3834–3848. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, T.; Peixinho, J.; Mukhopadhyay, S.; MacMinn, C.W. Dynamics of swelling and drying in a spherical gel. Phys. Rev. Appl. 2016, 6, 064010. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Malakpoor, K.; Huyghe, J.M. A mixed hybrid finite element framework for the simulation of swelling ionized hydrogels. Comput. Mech. 2019, 63, 835–852. [Google Scholar] [CrossRef] [Green Version]
- Levenston, M.; Frank, E.; Grodzinsky, A. Variationally derived 3-field finite element formulations for quasistatic poroelastic analysis of hydrated biological tissues. Comput. Methods Appl. Mech. Eng. 1998, 156, 231–246. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, X.; Suo, Z.; Jiang, H. A finite element method for transient analysis of concurrent large deformation and mass transport in gels. J. Appl. Phys. 2009, 105, 093522. [Google Scholar] [CrossRef]
- Capriles-González, D.; Sierra-Martín, B.; Fernández-Nieves, A.; Fernández-Barbero, A. Coupled deswelling of multiresponse microgels. J. Phys. Chem. B 2008, 112, 12195–12200. [Google Scholar] [CrossRef] [PubMed]
- Neuburger, N.A.; Eichinger, B.E. Critical experimental test of the Flory–Rehner theory of swelling. Macromolecules 1988, 21, 3060–3070. [Google Scholar] [CrossRef]
- Horkay, F.; Hecht, A.M.; Geissler, E. Effect of cross-links on the swelling equation of state: polyacrylamide hydrogels. Macromolecules 1989, 22, 2007–2009. [Google Scholar] [CrossRef]
- Roos, R.W.; Petterson, R.; Huyghe, J.M. Confined compression and torsion experiments on a pHEMA gel in various bath concentrations. Biomech. Model. Mechanobiol. 2013, 12, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.A.; Holm, C.; Kremer, K. Swelling of polyelectrolyte networks. J. Chem. Phys. 2005, 122, 154903. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F.; Tasaki, I.; Basser, P.J. Osmotic swelling of polyacrylate hydrogels in physiological salt solutions. Biomacromolecules 2000, 1, 84–90. [Google Scholar] [CrossRef]
- Horkay, F.; Tasaki, I.; Basser, P.J. Effect of monovalent- divalent cation exchange on the swelling of polyacrylate hydrogels in physiological salt solutions. Biomacromolecules 2001, 2, 195–199. [Google Scholar] [CrossRef]
- Horkay, F.; Basser, P.J. Osmotic observations on chemically cross-linked DNA gels in physiological salt solutions. Biomacromolecules 2004, 5, 232–237. [Google Scholar] [CrossRef]
- Yin, D.W.; Horkay, F.; Douglas, J.F.; de Pablo, J.J. Molecular simulation of the swelling of polyelectrolyte gels by monovalent and divalent counterions. J. Chem. Phys. 2008, 129, 154902. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.N.; Wang, W.B.; Wang, A.Q. Effect of surfactant on porosity and swelling behaviors of guar gum-g-poly (sodium acrylate-co-styrene)/attapulgite superabsorbent hydrogels. Colloids Surf. B Biointerfaces 2011, 88, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.; Bordado, J.; Casquilho, M. Dynamic and equilibrium swelling of a sulfonic acid superabsorbent copolymer in salt solutions. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 505–514. [Google Scholar] [CrossRef]
- Wilson, W.; Donkelaar, C.C.V.; Rietbergen, B.V.; Huiskes, R. A fibril-reinforced poroviscoelastic swelling model for articular cartilage. J. Biomech. 2005, 38, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Cegoñino, J.; Moramarco, V.; Calvo-Echenique, A.; Pappalettere, C.; Palomar, A.P.D. A constitutive model for the annulus of human intervertebral disc: Implications for developing a degeneration model and its influence on lumbar spine functioning. J. Appl. Math. 2014, 2014, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cortes, D.H.; Jacobs, N.T.; DeLucca, J.F.; Elliott, D.M. Elastic, permeability and swelling properties of human intervertebral disc tissues: A benchmark for tissue engineering. J. Biomech. 2014, 47, 2088–2094. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Huggins, M.L. Some properties of solutions of long-chain compounds. J. Phys. Chem. 1942, 46, 151–158. [Google Scholar] [CrossRef]
- Coussy, O. Poromechanics; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Loon, R.V.; Huyghe, J.M.; Wijlaars, M.W.; Baaijens, F.P.T. 3D FE implementation of an incompressible quadriphasic mixture model. Int. J. Numer. Methods Eng. 2003, 57, 1243–1258. [Google Scholar] [CrossRef]
- Wilson, W.; Huyghe, J.M.; Donkelaar, C.C.V. Depth-dependent compressive equilibrium properties of articular cartilage explained by its composition. Biomech. Model. Mechanobiol. 2007, 6, 43–53. [Google Scholar] [CrossRef]
- Tokita, M.; Tanaka, T. Friction coefficient of polymer networks of gels. J. Chem. Phys. 1991, 95, 4613–4619. [Google Scholar] [CrossRef]
- Grattoni, C.A.; Al-Sharji, H.H.; Yang, C.; Muggeridge, A.H.; Zimmerman, R.W. Rheology and permeability of crosslinked polyacrylamide gel. J. Colloid Interface Sci. 2001, 240, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Engelsberg, M.; Barros, W., Jr. Free-evolution kinetics in a high-swelling polymeric hydrogel. Phys. Rev. E 2013, 88, 062602. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.; Chatterjee, A.; Dziezok, P.; Ehrnsperger, B.J.; Kamphus, J.; Lutsche, M. Absorbent Cores Having Material Free Areas. U.S. Patent 10,130,527, 20 November 2018. [Google Scholar]
- Zohourian, M.M.; Kabiri, K. Superabsorbent polymer materials: A review. Iran. Polym. J. 2008, 17, 447–451. [Google Scholar]
- Sweijen, T.; Hassanizadeh, S.M.; Aslannejad, H.; Leszczynski, S. Grain-scale modelling of swelling granular materials using the discrete element method and the multi-sphere approximation. Poromechanics VI 2017, 329–336. [Google Scholar]
- Lopez, C.G.; Lohmeier, T.; Wong, J.E.; Richtering, W. Electrostatic expansion of polyelectrolyte microgels: Effect of solvent quality and added salt. J. Colloid Interface Sci. 2019, 558, 200–210. [Google Scholar] [CrossRef]
- Ullner, M.; Qamhieh, K.; Cabane, B. Osmotic pressure in polyelectrolyte solutions: Cell-model and bulk simulations. Soft Matter 2018, 14, 5832–5846. [Google Scholar] [CrossRef] [Green Version]
- Feher, J.J. Quantitative Human Physiology: An Introduction; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NaCl | NaCl + CaCl |
|---|---|---|
| Flory Huggins Parameter 1 () | 0.45 | 0.453 |
| Flory Huggins Parameter 2 () | 0.21 | 0.53 |
| Molar Concentration | 0.154 M | 0.154 M + 8 × 10 M |
| Parameter | Value |
|---|---|
| Shear Modulus | 18 kPa |
| Degree of Neutralisation | 0.85 |
| Initial Counterion Concentration | 0.3 M |
| Permeability Parameter () | 2 |
| Temperature | 298 K |
| Universal Gas Constant | 8.3145 J/K.mol |
| Solvent Molar Volume | 18 mol/m |
| Initial Hydraulic Permeability | 10 mm/Ns |
| Model | RMSE (MPa) |
|---|---|
| Univalent—Total Swelling Pressure | 0.085 |
| Univalent—Ionic Pressure | 0.037 |
| Univalent—Mixing Pressure | 0.047 |
| Divalent—Total Swelling Pressure | 0.069 |
| Divalent—Ionic Pressure | 0.054 |
| Divalent—Mixing Pressure | 0.016 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fennell, E.; Kamphus, J.; Huyghe, J.M. The Importance of the Mixing Energy in Ionized Superabsorbent Polymer Swelling Models. Polymers 2020, 12, 609. https://doi.org/10.3390/polym12030609
Fennell E, Kamphus J, Huyghe JM. The Importance of the Mixing Energy in Ionized Superabsorbent Polymer Swelling Models. Polymers. 2020; 12(3):609. https://doi.org/10.3390/polym12030609
Chicago/Turabian StyleFennell, Eanna, Juliane Kamphus, and Jacques M. Huyghe. 2020. "The Importance of the Mixing Energy in Ionized Superabsorbent Polymer Swelling Models" Polymers 12, no. 3: 609. https://doi.org/10.3390/polym12030609
APA StyleFennell, E., Kamphus, J., & Huyghe, J. M. (2020). The Importance of the Mixing Energy in Ionized Superabsorbent Polymer Swelling Models. Polymers, 12(3), 609. https://doi.org/10.3390/polym12030609