1. Introduction
Chitosan (CS) is a natural biopolymer composed of glucosamine and
N-acetylglucosamine units, that gained the interest of scientists over the last four decades for its promises in pharmaceutics and medicine [
1]. This polymer is produced on a commercial scale by the deacetylation of chitin, which is widely present in crustacea’s shells and fungi. The abundance of seafood waste represents a large reservoir of raw material for the production of CS, at low cost and in large quantities [
2]. CS exhibits high biocompatibility as well as favorable adsorption properties and biodegradability [
3,
4], which make it a versatile biomaterial for the development of biomedical and pharmaceutical products [
5]. For example, biomaterials based on CS display promising anti-bacterial and anti-oxidant properties that led to the development of dressing for wound healing [
6]. Other applications use CS gels for drug-delivery systems and scaffold for bone treatment and biomedical imaging [
3,
7,
8].
In the field of gene therapy, CS attracted much interest as a potential non-viral DNA vector. Most current strategies make use of viral vectors that can generate changes in the genome of the host cell with serious consequences [
9]. Non-viral DNA, on the other hand, does not present the risk of uncontrolled genome insertion and is therefore considered safer than its viral counterpart. While naked DNA is unable to penetrate cells due to its high negative charge density, CS–DNA complexes assembled by electrostatic interactions were reported for cell transfection whilst protecting the nucleic acid from degradation [
10].
The applications of CS are severely restricted by its poor solubility in the physiological environment and the lack of standardized characterizations. CS is only soluble in diluted acidic aqueous media and displays a complex behavior in solution such as macromolecular association (H-bond) [
11] and polyelectrolyte effect [
12], which is a challenge for characterization and its possible application as a functional material. The quality of CS should be described at least by the molecular weight (MW), polydispersity (PD) and degree of deacetylation (DD). Moreover, the purity level has a great influence on the biological characteristics of CS and CS derivatives, such as immunogenicity or biodegradability, but also has a profound effect on its solubility and stability [
13]. Various commercial sources of CS are available, but specification data provided by the suppliers is often incomplete, which leads to misleading and inconsistent results [
14,
15,
16,
17]. Studies suggest that the properties of CS and its derivatives, in particular their antimicrobial properties, depend on the natural sources used for CS production (shellfish, fungi or bacteria) [
18]. A refined conformational analysis reported by Weinhold et al. [
19] on different CS preparations showed the absence of correlation between their conformation in solution with several parameters such as the number of acetyl groups, the pattern of acetylation and aggregation processes. This can be partially explained by the difficulties of fitting theoretical models to the non-standard behavior of CS [
15]. The establishment of clear predictions is impeded by the varying characteristics of commercial CS preparations (such as solubility, DD, MW and PD), leading to results that might deviate from pharmacopeial standards. Despite the high promise of CS for biomedical applications, the current variability in the characteristics of commercial and further purified polymer sources represents a serious limitation towards its translation into the clinics.
The MW is considered to be one of the main factors determining the solubility of the polymer. Other parameters such as the DD or the pattern of acetylation are also expected to have an effect, although less pronounced [
20]. Their impact on the solubility of CS preparations in aqueous solutions is currently strongly debated in the field [
21,
22,
23]. The reduction of the MW of commercial CS is a common way to improve the polymer solubility and biological properties. Degradation methods include chemical depolymerization under oxidative, basic or acidic conditions, enzymatic treatment, or irradiation. Physical irradiation is less common, as it requires specific and expensive materials, extended reaction times and leads to an irregular degradation rate [
24,
25]. Similarly, the high cost of degrading enzymes is a considerable drawback of enzymatic treatment. In addition, their substrate specificity limits the range of the low MW which can be achieved [
26]. Chemical degradation has been more widely studied and presents the most promising results [
12,
27,
28]. Basic conditions are considered to reduce the DD faster than the MW, compared to acid hydrolysis of the 1,4-glycosidic bonds [
29]. While lowering the MW of CS samples, oxidative degradations were reported to concomitantly lead to the loss of amino groups and the formation of carboxyls [
30]. More recently, oxidative degradation (using oxygen peroxide) coupled with microwave treatment attracted considerable interest. This dual method has been shown to shorten reaction times, reduce the amount of oxidative agent needed and provide higher yields [
31,
32,
33]. The factors influencing the depolymerization include the reaction time, the concentration of oxidative or acidic reagent, the quality and MW of the starting material and the liquid–solid ratio. However, uncontrolled chemical transformations (such as ring opening) as observed during oxidative degradation add to the list of undesirable unknown parameters of CS preparations and should be avoided.
For biological applications, the properties of CS are often further improved by chemical functionalization on the amino or hydroxyl groups. Despite an effort by the scientific community to improve the characterization for a given CS and CS derivative, chemical analyses are still lacking which complicates the evaluation of such chemical transformation. The extremely low solubility of CS in organic solvents and in pure water prevents the straightforward characterization by nuclear magnetic resonance (NMR) of the new grafted polymers, which is the most commonly used analytical technique to probe chemical transformations. Our work not only aims at investigating the production of depolymerized chitosan (dCS), but also the production of CS and CS derivatives soluble in water, a solvent suitable for NMR analysis and compatible with biological applications.
Acidic conditions are considered to favor chain depolymerization over the removal of acetyl groups and reduce side reactions which are often associated with oxidative degradation [
34]. We therefore studied the degradation of CS samples from two different suppliers in acidic conditions (HCl) and investigated the effect of microwave-assisted heating on the yield and characteristics of the resulting polymers. The characterization of the dCS preparations included the systematic determination of the MW (Mp, Mn and Mw), PD, DD and solubility in water, properties which are often partially described in previous publications. Other acidic reagents, such as nitrous acid, were reported for CS chain degradation and fractionation into neutral-soluble and neutral-insoluble dCS materials [
35]. However, this method resulted in the formation of end-carbonyl groups, requiring an additional reductive step to prevent side-transformations on the highly reactive aldehydes. In addition, we herein report on the formation of dCS aggregates in solution, resulting from the removal of ions and sample freezing, and discuss the consequences of this change of crystallinity with regards to the discrepancies between the results reported in literature.
2. Materials and Methods
2.1. General Conditions
CS-A refers to the commercial CS from Tokyo Chemical International (TCI) with the following characteristics: η = 5–20 mPa·s (0.5% w/v in 0.5% acetic acid, 20 °C), DD = 84.5% (guaranteed ≥ 80%). CS-B refers to the commercial CS “for biochemistry” from Roth AG with the following characteristics: DD ≥ 90%. Depolymerized chitosan produced from CS-A and CS-B are referred to as dCS-A and dCS-B, respectively.
The commercial sources for the following chemicals and consumables were: fuming HCl (37%), deuterium chloride (DCl): Sigma Aldrich; NaOH: Alfa Aesar; glacial acetic acid: Merck KGaA; sodium acetate trihydrate 99%: ABCR GmbH; deuterated water (D2O): Cambridge Isotope Laboratories, Inc. Dialysis membranes of regenerated cellulose (RC) of 3.5 and 7 kDa MWCO (Spectra/Por® 3 and 1 respectively): Fisher scientific. Dialysis membranes of 14 kDa MWCO (Membra-Cel™, RC, packaged dry): Roth AG.
Aqueous solutions were produced with ultrapure water obtained from Milli-Q Integral System (Merck/Millipore, Schaffhausen, Switzerland). Centrifugations were performed with an Allegra X-30R centrifuge (Beckman Coulter, Nyon, Switzerland). pH monitoring during neutralization operations was done with a pH/Ion bench meter SC S220-K (Mettler-Toledo AG, Greifensee, Switzerland).
2.2. Chitosan Depolymerization
The depolymerization of CS by conventional heating was conducted as follows. The polymer was dissolved over a 1 h period in 1M HCl to a final concentration of 1% w/v. The suspension was heated to 80 °C and then further stirred under reflux for 48 h. The solution was cooled to 30 °C before being neutralized to pH 7 using NaOH solution. The resulting light yellow suspension was centrifuged (10 min, 4700 ω, 20 °C) to isolate the liquid phase. Water was added to the precipitate up to the initial volume, shaken to obtain a homogeneous suspension and centrifuged a second time under the same conditions as above. The liquids were combined and transferred to a dialysis membrane (3.5 kDa MWCO), then dialyzed against water for a minimum of 3 days, changing the water three times per day and lyophilized. In case the solution was colored (dark yellow), it was filtered through a sterile 0.22 mm polyethersulfone (PES) membrane before lyophilization. In case of analysis of the precipitate, the precipitate isolated by centrifugation was suspended in water, dialyzed and lyophilized in the same way as the soluble fraction of dCS.
The optimized conditions for the depolymerization of CS via microwave-assisted heating are described as follows. The CS was suspended in 1 M HCl to a final concentration of 1% w/v, typically 200 mg in 20 mL of solution. Commercial CS sources were dissolved in 1 M HCl solution, but required long hydration times (a few hours) or heating for approximately 20 min. In order to minimize acid-driven side reactions, CS (as a suspension) was transferred to a microwave-safe 30 mL vial and placed in the cavity of the microwave reactor (Monowave 400, Anton Paar). Video monitoring of the solution showed that the sample was entirely solubilized one minute after the start of the depolymerization program, at which point the CS solution had reached 60–70 °C. The optimal cycle of microwave depolymerization (under constant stirring of 300 rpm) followed the program below:
Heat as fast as possible to 100 °C;
Hold at 100 °C for 19 min;
Cool down to 35 °C as fast as possible.
Once the resulting dCS solution reached a temperature of 30 °C, the solution was neutralized in two steps: 10 M NaOH solution was added until the solution reached pH 6.7. The suspension was left to equilibrate at room temperature for 40 min before being centrifuged (10 min, 4700 ω, 20 °C) to remove the precipitate. The pH of the liquid fraction was then further increased to 7.0 by dropwise addition of a 0.05 M NaOH solution. The white suspension was centrifuged a second time under the same conditions to remove the second precipitate, if any. The soluble dCS fraction was transferred to a dialysis membrane (3.5 kDa MWCO unless otherwise stated) and dialyzed against water for a minimum of 3 days, changing the water three times per day. In case of analysis of the precipitate, the precipitate isolated by centrifugation was suspended in water and dialyzed in the same way as the soluble fraction of dCS.
2.3. Evaluation of the Solubility of dCS in Acidic Aqueous Solutions and in DMSO
The solubility of dCS-A was first evaluated in aqueous acidic solution using 1M HCl. The solubility of the starting CS-A and CS-B were also evaluated for comparison. For each polymer, 30 mg were dissolved in 3 mL of pure water and left to dissolve for 15 min at room temperature under gentle shaking. The pH of the resulting suspensions was measured and indicated as starting pH. Then, 4, 8, 16 or 32 μL of 1M HCl aqueous solution were added and the resulting suspensions were again gently shaken over 10 min. The addition of 1M HCl and subsequent solution homogenization were repeated until a clear solution was obtained in each case. The pH was measured between each addition-shaking cycle and the last one corresponding to a clear solution was indicated as the final pH.
To test the solubility of the chitosan in DMSO, 20 mg of CS-A, CS-B or dCS-A were poured in 3 mL of DMSO, leading to a concentration of 6.7 mg/mL. The suspensions were gently shaken for 10 min and then left on the side for 10 additional minutes. A picture of the resulting solutions was taken (see
Supporting information S-10, Figure S3).
2.4. Protocol for Complete Study of Post-Dialysis dCS Aggregation
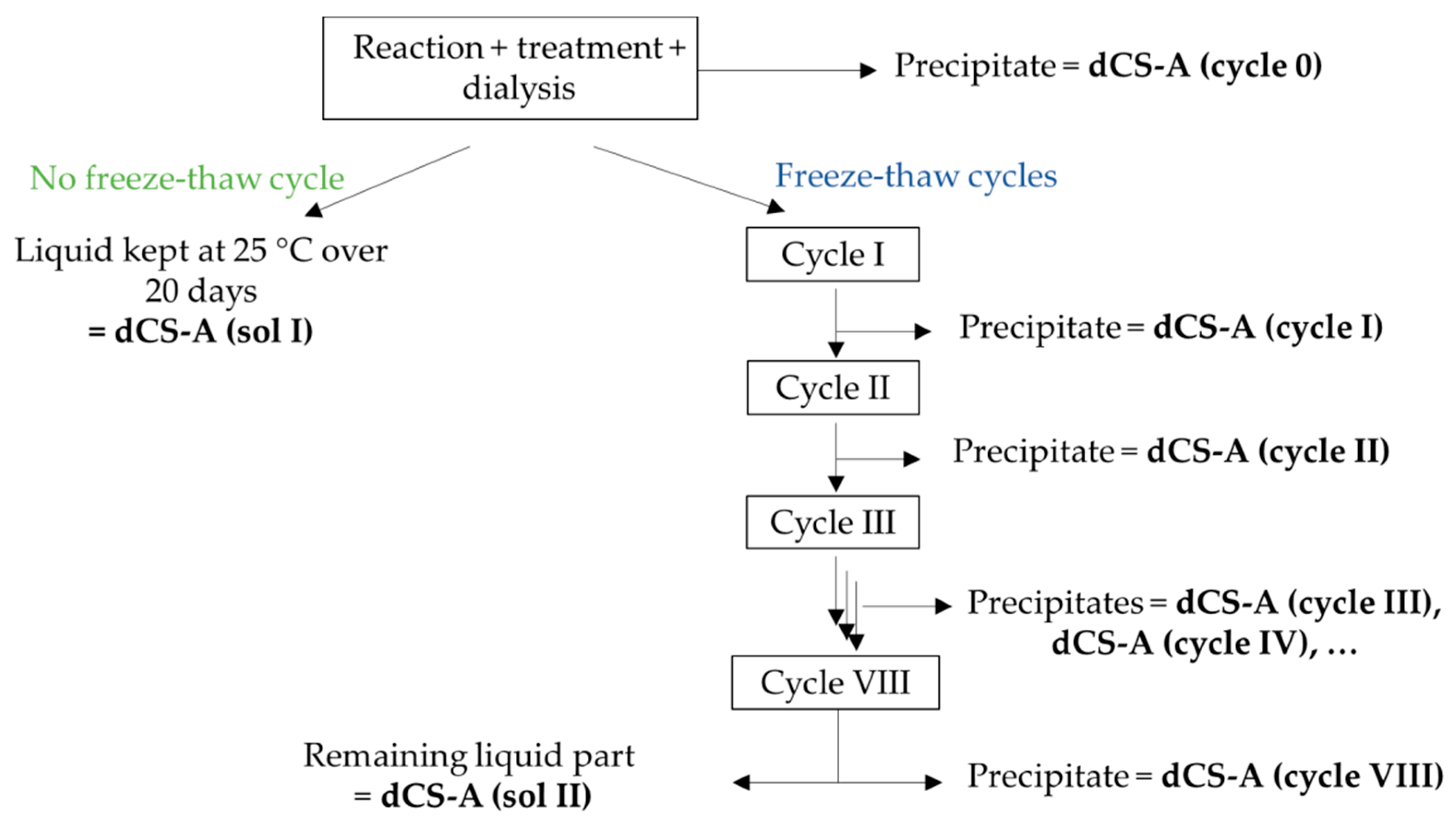
CS-A was depolymerized in the microwave reactor at 100 °C for 19 min as described above. Once the solution reached 30 °C, 10M NaOH was added until the pH 6.7 was reached. The turbid solution was stirred for 45 min before being centrifuged (10 min, 4700 ω, 20 °C), and the aqueous phase was gently poured out to isolate the precipitate. The liquid phase was neutralized up to the pH 7 with a 0.05M NaOH solution and stirred for another 45 min. The solution was centrifuged a second time and the precipitate was discarded. The soluble dCS fraction was collected and dialyzed, at the end of which a new precipitate appeared in the dialysis membrane. After the isolation of this precipitate by centrifugation (named dCS (cycle 0)), the aqueous solution was divided in two equal volumes: one half of the volume was frozen whilst the second half remained at 25 °C overnight. The frozen solutions were then allowed to reach room temperature and centrifuged to collect the precipitate that had formed overnight before freezing the solution again. The numbering of the freeze–thaw cycle (cycle I, II, III, etc.) indicates how many times the dCS solution was subjected to freezing. All the samples went through a total of eight successive overnight freeze–thaw cycles and were centrifuged each time. No more precipitation was observed in the samples that remained at 25 °C over 20 days, or after four freeze–thaw cycles. Each precipitate was lyophilized and weighted individually. The concentration of dCS after each cycle was calculated by subtracting the mass of precipitate collected (after cycle I, II, etc.) to the mass of dCS (sol I). As a control, the concentration was also calculated by adding the amount of precipitate removed at each freeze–thaw cycle to the mass of dCS (sol II). Both calculations provided the same concentration of dCS in water, expressed in mg/mL. The protocol was performed four times and the presented values are the averages of four experiments.
2.5. NMR Analysis
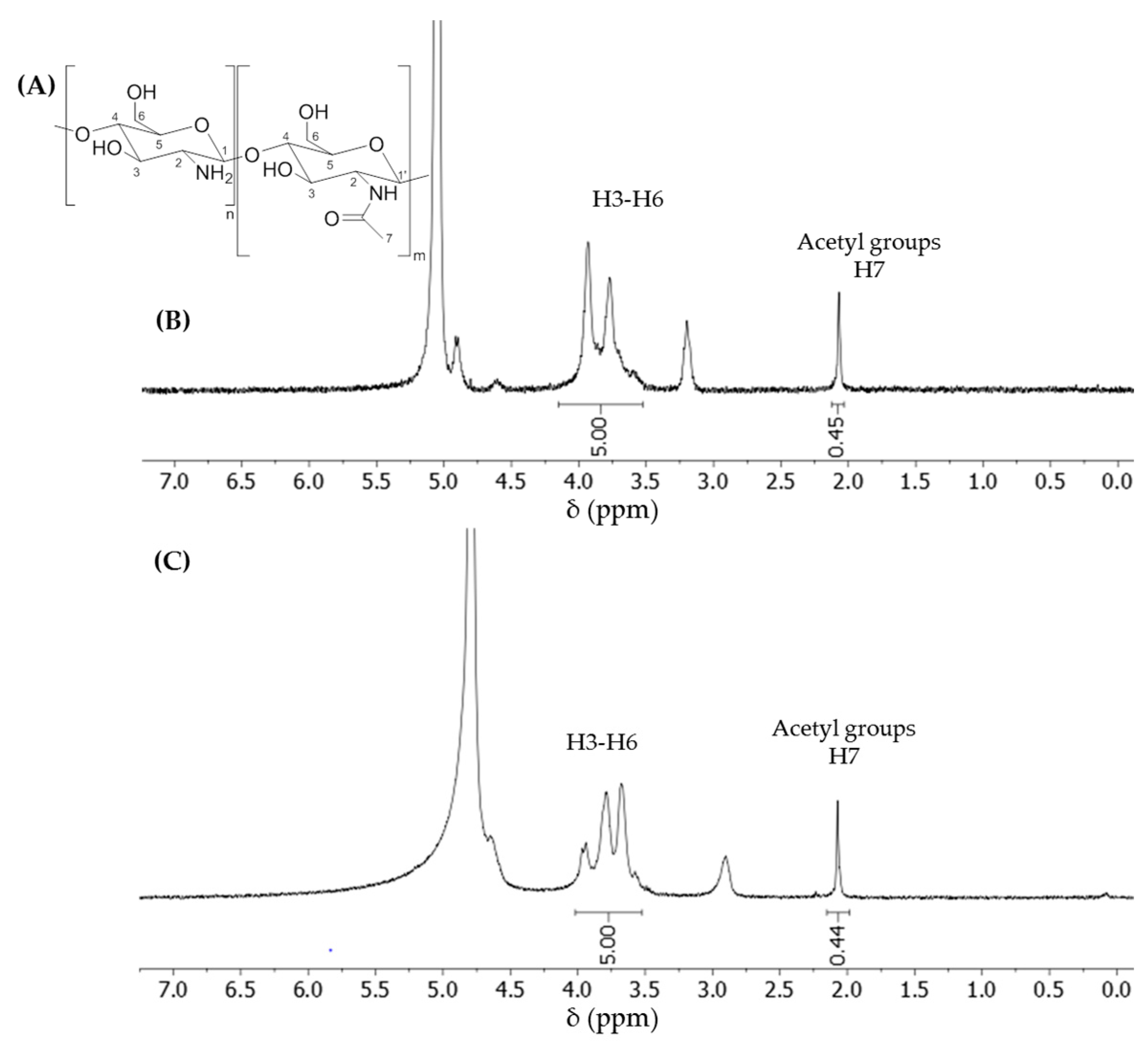
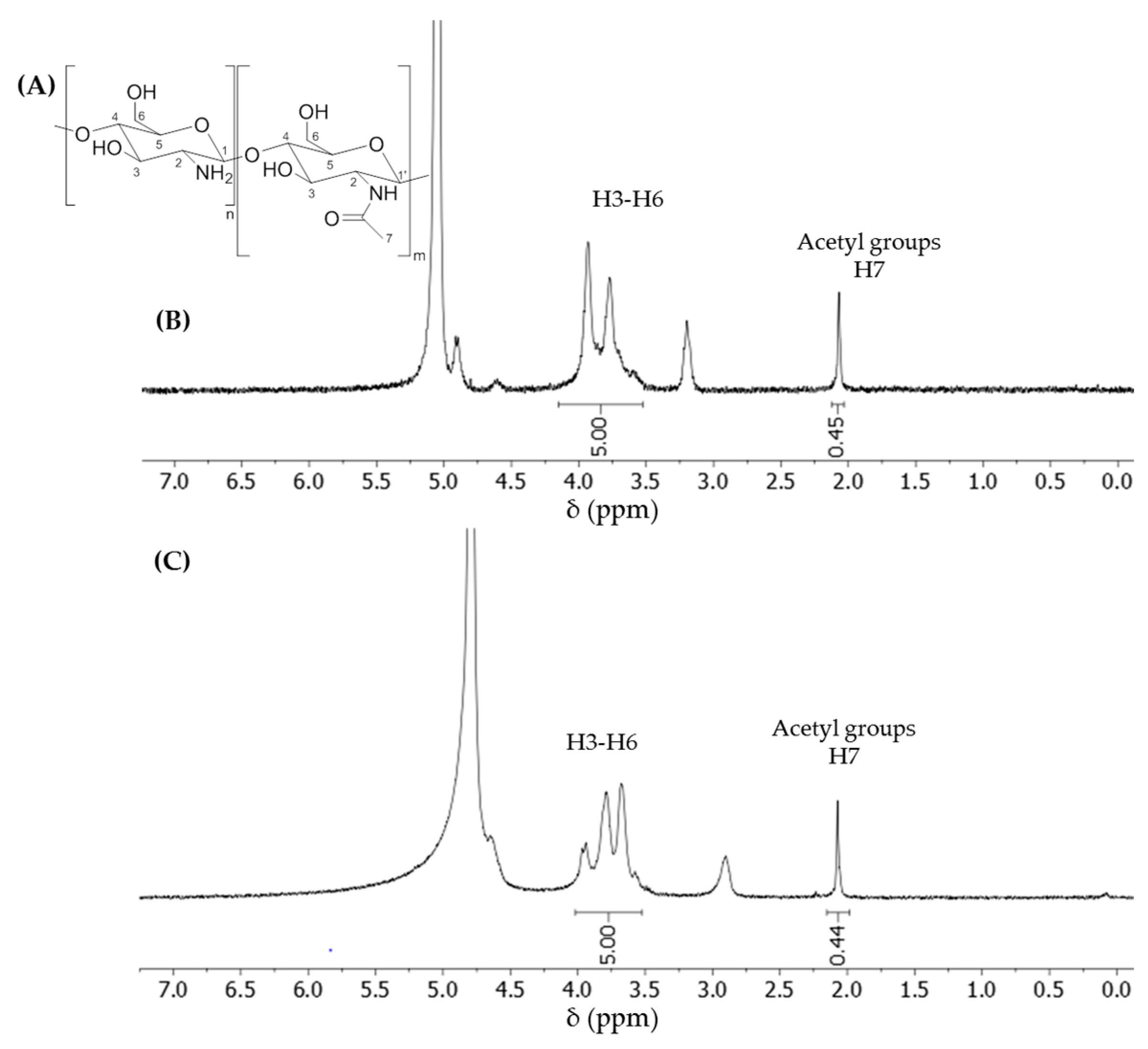
1H-NMR spectra were recorded on a Bruker Avance III-HD spectrometer (Bruker, Billerica, MA, USA) at room temperature unless stated otherwise. The 1H frequency was at 400.13 MHz. The chemical shifts were reported downfield from tetramethylsilane. 1H signals were reported in ppm in pure D2O using the water’s residual signal at 4.79 ppm as the internal reference, or in a mixture of D2O + 1% DCl with the acetyl signal as a reference at 2.07 ppm. The shift of acetyl at 2.07 ppm corresponds to its position in pure deuterated water; we reported the same value in the spectrum recorded in D2O + 1% DCl. All the dCS samples were recorded in both solvents to compare the calculated DD values. The DDs calculated in D2O or D2O + 1% DCl were similar.
DD was calculated by comparing the integration of the protons from H3–H6 (4.00–3.49 ppm in D
2O; 4.15–3.53 in D
2O + 1% DCl,) set at a value of 5, and the relative integration of the acetyl signal (2.07 ppm, 3 protons). The percentage of the acetyl group in a given sample of dCS corresponded to the value of the integrated acetyl signal (
) divided by 3. The DD can then be expressed as the following equation:
2.6. Gel Permeation Chromatography (GPC) Analysis
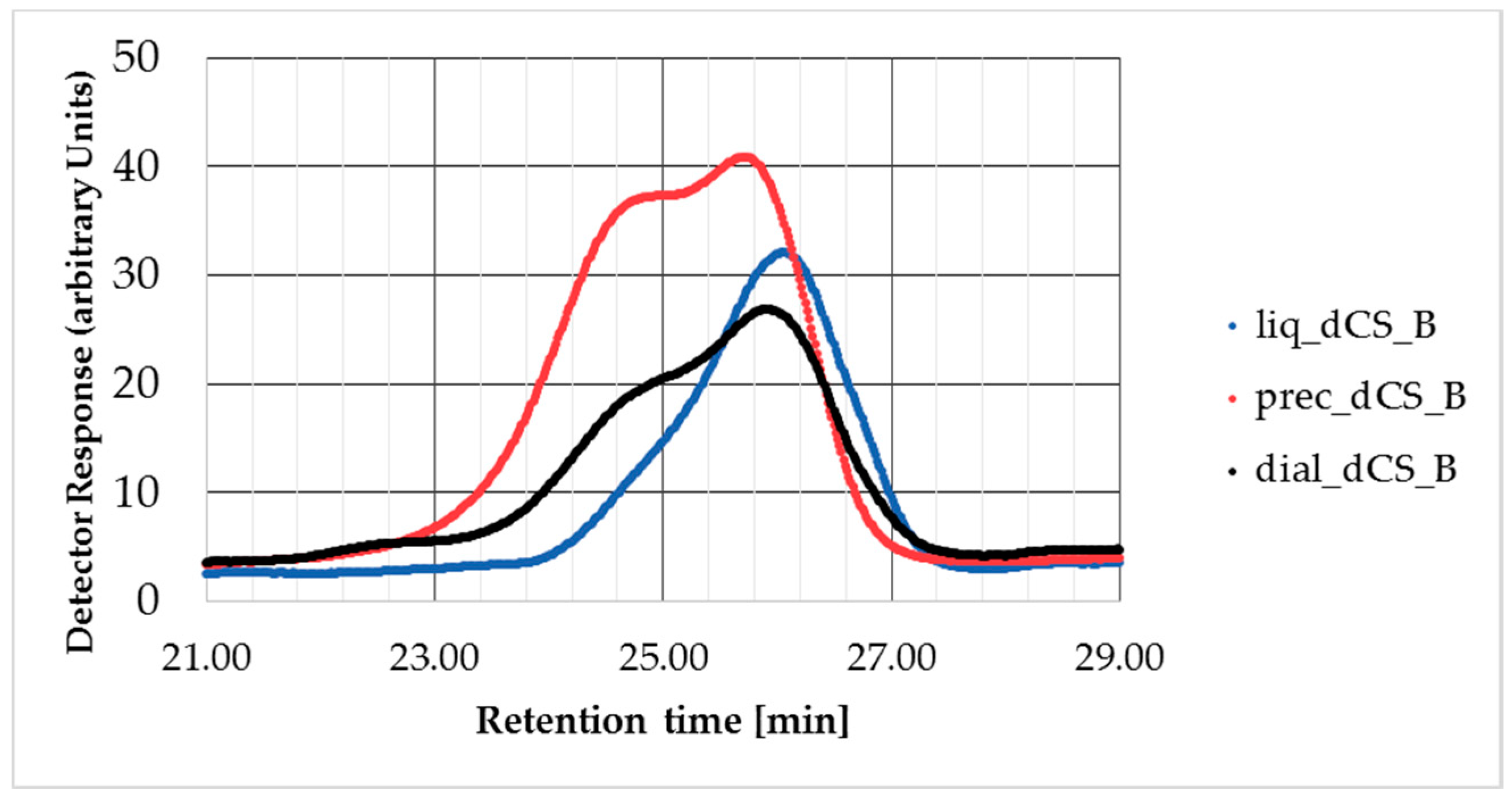
GPC analysis was performed on a PL-GPC 50 integrated GPC/SEC system (Agilent, Basel, Switzerland) equipped with a refractive index detector, a PSS NOVEMA MAX columns set (1× guard column, 10 μm; 2× analytical columns 1000 Å, 10 μm; 1× analytical column 30 Å, 10 μm), using 0.3 M aqueous AcOH and the 0.2 M aqueous NaOAc as eluents. The system was calibrated with pullulan from 180 to 1,220,000 Da. The samples of CS and dCS (2 mg each) were dissolved overnight in 1.5 mL of mobile phase. The solutions were filtered through a sterile 0.22 µm PTFE filter followed by injection (100 µL). The flow rate of analysis was 1 mL/min at 40 °C. The results of the GPC analysis were expressed as the number of the average molecular weight (Mn), the weight average of the molecular weight (Mw), and the molecular weight of the highest peak (Mp) and polydispersity index (PD).
5. Conclusions
CS is known for being a highly valuable biopolymer candidate for medical and pharmaceutical applications. However, we believe that the scientific community needs to increase awareness concerning the lack of characterization and standardization of this polymer. The most obvious challenge of CS relies in its very low solubility in organic solvents and neutral aqueous solutions. Its solubility can be increased by degrading the CS chains into smaller, depolymerized polymer chains (dCS). Depolymerization in acidic conditions (1M HCl) was studied either using conventional heating or microwave-assisted heating. The use of the microwave proved to be superior to conventional heating as it reduced the reaction time from 48 h to 19 min whilst preserving the
N-acetyl groups on the polymer backbone and preventing degradation. Our results suggest that the depolymerization in acidic conditions needs to be adapted to the CS of choice, as the behavior of this polymer changed with the source (and batch) of the commercial source. Although we cannot comment on the kinetics of the 1,4-glycosidic bond breakage, we postulate that the very low amount of acetyl groups in CS-B is the determining factor for its apparent resistance to chain breakage. A high DD was linked to a higher crystallinity of CS [
36,
43], which reduces the surface of the polymer available for nucleophilic attack and the likelihood of complete 1,4-glycosidic bond breakage [
37].
Beyond the demonstration that microwave-assisted depolymerization provides a fast access to dCS preparations which are fully soluble in pure water, we also highlighted that the average MW of dCS can be selected by isolating soluble fractions at pH between 6.6 and 7.0. The higher the pH at which dCS is collected, the smaller the MW will be. In the neutralization process, precipitation was observed from pH 6.7, increased up to pH 6.9, and then reduced linearly up to pH 7.4. From pH 7.0, almost no precipitate was formed anymore. The selection of the soluble fraction at each step of the precipitation process allowed the highly reproducible isolation of dCS with a MW of 12.6 ± 0.6 kDa, and a PD of 1.41 ± 0.05. To our knowledge, this is the first time that CS samples of meaningful quantities—enough for further study and experiment—were reported to be isolated based on their MW.
Furthermore, it was determined that the selection of soluble dCS at a neutral pH is not enough to guarantee a fully soluble dCS in pure water. Both the removal of ions and phase change (from liquid to solid) of the dCS solutions were responsible for the formation of irreversible aggregated dCS particles in water. The addition post dialysis and lyophilization of sodium chloride salt in water was not efficient to reverse the crystallized dCS.
The solid form of a product is the most convenient and often most stable form for packaging and shipping [
44,
45] for industrial applications. Regarding potential CS-derived drug applications, the results presented herein would suggest that great attention will have to be given to the change in crystallinity of CS-based formulations. In order to ensure the selection of dCS samples which remain fully soluble in pure water independently of phase changes, successive freezing and thawing cycles might offer a valuable technique.






{kind=link}
{kind=link}
{kind=link}