Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications


 , ,
, ,  ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Poly(alkylene succinate) Polyesters
2.3. Characterization of Poly(alkylene succinate) Polyesters
2.3.1. Intrinsic Viscosity
2.3.2. Size Exclusion Chromatography (SEC)
2.3.3. Nuclear Magnetic Resonance Spectroscopy (1H-NMR)
2.3.4. DSC Measurements
2.4. Preparation of PVA-Polyesters Melt Mixtures
2.5. Miscibility Evaluation
2.5.1. Theoretical Evaluation
2.5.2. Experimental Evaluation
2.6. Thermal Properties Evaluation
2.7. Physical State Evaluation
2.8. Molecular Interactions Evaluation
2.8.1. Experimental Evaluation
2.8.2. Theoretical Evaluation
2.9. Molecular Dynamics (MD) Simulations
2.9.1. Preparation of Initial Structures
2.9.2. Contraction of Amorphous Cells
2.9.3. MD Simulation Runs
2.10. Melt Flow Index (MFI)
3. Results and Discussion
3.1. Characterization of Poly(alkylene succinate) Polyesters
3.1.1. H NMR Results
3.1.2. Intrinsic Viscosity—Molecular Weight
3.1.3. Thermal Properties
3.2. PVA- Poly(alkylene succinate) Polyesters Characterization
3.2.1. Miscibility Evaluation
Theoretical Evaluation
Experimental Evaluation
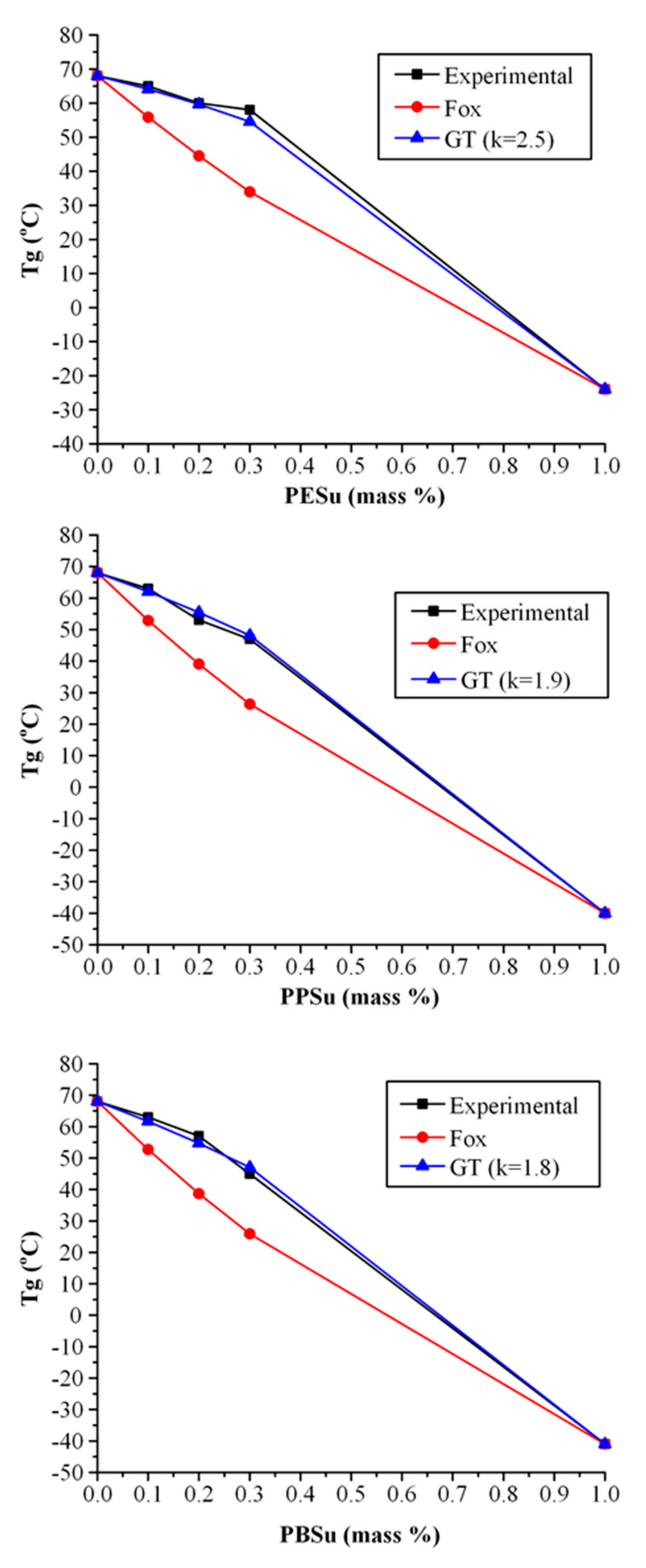
3.2.2. Evaluation of Thermal Properties
TGA Results
DSC Results
3.2.3. Physical State Evaluation
3.2.4. Molecular Interactions
DSC Analysis
ATR-FTIR Analysis
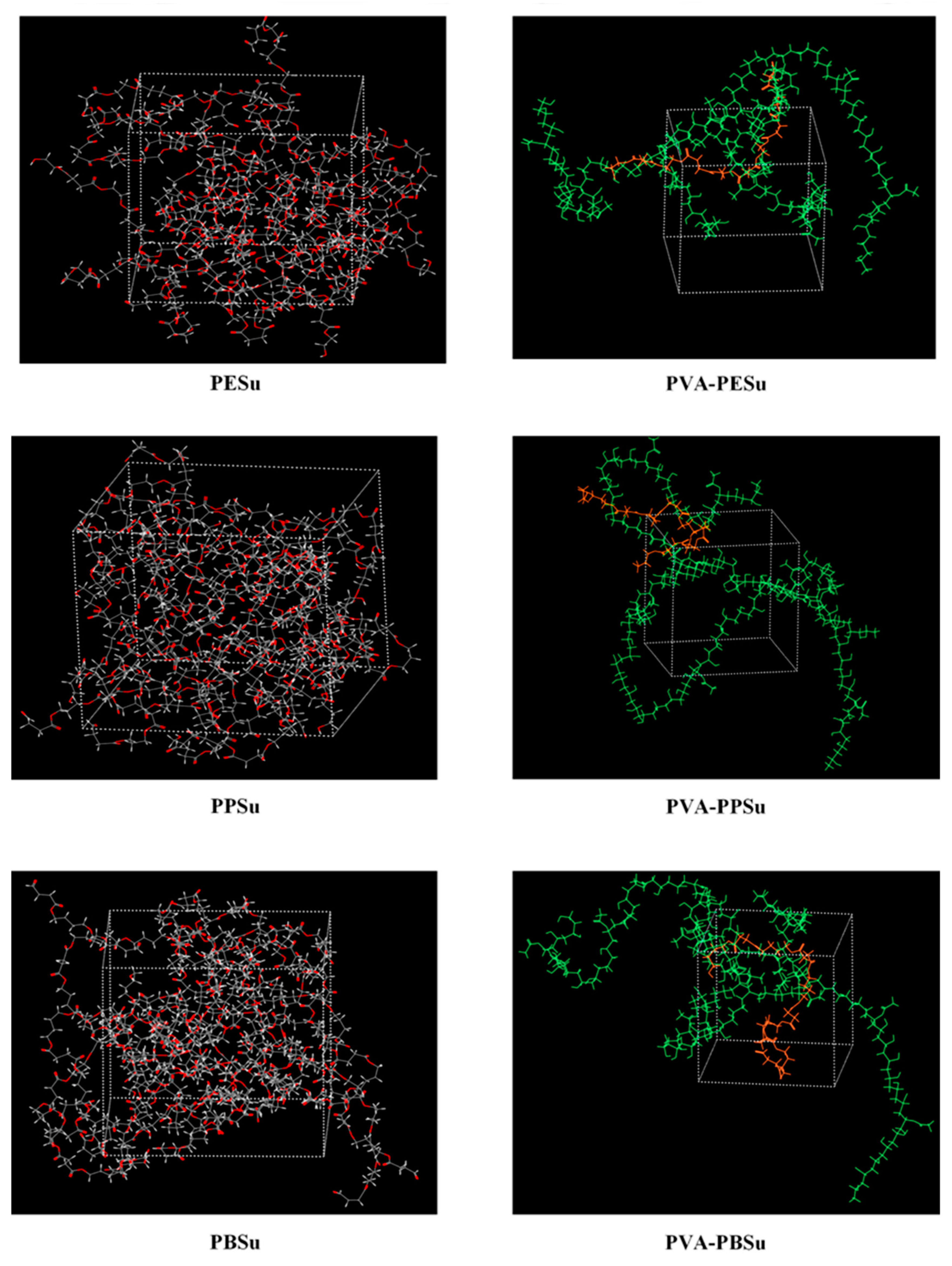
MD Simulations
3.2.5. Melt Flow Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Swain, S.; Rizwan, M.; Irfanuddin, M.; Malini, D.S. Bioavailability Enhancement Strategies: Basics, Formulation Approaches and Regulatory Considerations. Curr. Drug Deliv. 2011, 8, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Siahi-Shadbad, M.; Adibkia, K.; Barzegar-Jalali, M. Recent advances in improving oral drug bioavailability by cocrystals. BioImpacts. 2018, 8, 305–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release. 2007, 123, 78–99. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today. 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics. 2019, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Williams, R.O. Effects of the Preparation Process on the Properties of Amorphous Solid Dispersions. AAPS PharmSciTech 2018, 19, 1971–1984. [Google Scholar] [CrossRef]
- Mahmah, O.; Tabbakh, R.; Kelly, A.; Paradkar, A. A comparative study of the effect of spray drying and hot-melt extrusion on the properties of amorphous solid dispersions containing felodipine. J. Pharm. Pharmacol. 2014, 66, 275–284. [Google Scholar] [CrossRef]
- Keen, J.M.; McGinity, J.W.; Williams, R.O., III. Enhancing bioavailability through thermal processing. Int. J. Pharm. 2013, 450, 185–196. [Google Scholar] [CrossRef]
- Tiwari, R.V.; Patil, H.; Repka, M.A. Contribution of hot-melt extrusion technology to advance drug delivery in the 21st century. Expert Opin. Drug Deliv. 2016, 13, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical Stability of Amorphous Solid Dispersions: A Physicochemical Perspective with Thermodynamic, Kinetic and Environmental Aspects. Pharm Res. 2018, 35, 125. [Google Scholar] [CrossRef] [PubMed]
- Edueng, K.; Mahlin, D.; Bergström, C.A.S. The Need for Restructuring the Disordered Science of Amorphous Drug Formulations. Pharm. Res. 2017, 34, 1754–1772. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xie, X.; Zhao, Y.; Gao, Y.; Cai, C.; Zhang, Q.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; et al. Effect of plasticizers on manufacturing ritonavir/copovidone solid dispersions via hot-melt extrusion: Preformulation, physicochemical characterization, and pharmacokinetics in rats. Eur. J. Pharm. Sci. 2019, 127, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Kapourani, A.; Vardaka, E.; Katopodis, K.; Kachrimanis, K.; Barmpalexis, P. Crystallization tendency of APIs possessing different thermal and glass related properties in amorphous solid dispersions. Int. J. Pharm. 2020, 579, 119149. [Google Scholar] [CrossRef] [PubMed]
- Gaaz, T.S.; Sulong, A.B.; Akhtar, M.N.; Kadhum, A.A.H.; Mohamad, A.B.; Al-Amiery, A.A. Properties and Applications of Polyvinyl Alcohol, Halloysite Nanotubes and Their Nanocomposites. Molecules 2015, 20, 22833–22847. [Google Scholar] [CrossRef] [Green Version]
- DeMerlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- LaFountaine, J.S.; Jermain, S.V.; Prasad, L.K.; Brough, C.; Miller, D.A.; Lubda, D.; McGinity, J.W.; Williams, R.O. Enabling thermal processing of ritonavir–polyvinyl alcohol amorphous solid dispersions by KinetiSol® Dispersing. Eur. J. Pharm. Biopharm. 2016, 101, 72–81. [Google Scholar] [CrossRef]
- Brough, C.; Miller, D.A.; Keen, J.M.; Kucera, S.A.; Lubda, D.; Williams, R.O. Use of Polyvinyl Alcohol as a Solubility-Enhancing Polymer for Poorly Water Soluble Drug Delivery (Part 1). AAPS PharmSciTech 2016, 17, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Thellen, C.; Cheney, S.; Ratto, J.A. Melt processing and characterization of polyvinyl alcohol and polyhydroxyalkanoate multilayer films. J. Appl. Polym. Sci. 2013, 127, 2314–2324. [Google Scholar] [CrossRef]
- Alexy, P.; Káchová, D.; Kršiak, M.; Bakoš, D.; Šimková, B. Poly(vinyl alcohol) stabilisation in thermoplastic processing. Polym. Degrad. Stab. 2002, 78, 413–421. [Google Scholar] [CrossRef]
- Wu, W.; Tian, H.; Xiang, A. Influence of Polyol Plasticizers on the Properties of Polyvinyl Alcohol Films Fabricated by Melt Processing. J. Polym. Environ. 2012, 20, 63–69. [Google Scholar] [CrossRef]
- Jang, J.; Lee, D.K. Plasticizer effect on the melting and crystallization behavior of polyvinyl alcohol. Polymer 2003, 44, 8139–8146. [Google Scholar] [CrossRef]
- Katopodis, K.; Kapourani, A.; Vardaka, E.; Karagianni, A.; Chorianopoulou, C.; Kontogiannopoulos, K.N.; Bikiaris, D.N.; Kachrimanis, K.; Barmpalexis, P. Partially hydrolyzed polyvinyl alcohol for fusion-based pharmaceutical formulation processes: Evaluation of suitable plasticizers. Int. J. Pharm. 2020, 578, 119121. [Google Scholar] [CrossRef] [PubMed]
- Yasuniwa, M.; Satou, T. Multiple melting behavior of poly(butylene succinate). I. Thermal analysis of melt-crystallized samples. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2411–2420. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and melting behavior of three biodegradable poly(alkylene succinates). A comparative study. Polymer 2005, 46, 12081–12092. [Google Scholar] [CrossRef]
- Karayannidis, G.P.; Roupakias, C.P.; Bikiaris, D.N.; Achilias, D.S. Study of various catalysts in the synthesis of poly(propylene terephthalate) and mathematical modeling of the esterification reaction. Polymer 2003, 44, 931–942. [Google Scholar] [CrossRef]
- Bikiaris, D.; Karayannidis, G. Synthesis and characterisation of branched and partially crosslinked poly(ethylene terephthalate). Polym. Int. 2003, 52, 1230–1239. [Google Scholar] [CrossRef]
- Solomon, O.F.; Ciutǎ, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Breitkreutz, J. Prediction of Intestinal Drug Absorption Properties by Three-Dimensional Solubility Parameters. Pharm. Res. 1998, 15, 1370–1375. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA–An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Mumby, S.J.; Maple, J.R.; Hagler, A.T. An ab Initio CFF93 All-Atom Force Field for Polycarbonates. J. Am. Chem. Soc. 1994, 116, 2978–2987. [Google Scholar] [CrossRef]
- Shenogin, S.; Ozisik, R. XenoView: Visualization for Atomistic Simulations. Available online: http://www.vemmer.org/xenoview/xenoview.html (accessed on 15 September 2019).
- Li, D.; Panchal, K.; Mafi, R.; Xi, L. An Atomistic Evaluation of the Compatibility and Plasticization Efficacy of Phthalates in Poly(vinyl chloride). Macromolecules 2018, 51, 6997–7012. [Google Scholar] [CrossRef]
- Gunsteren, W.F.v.; Mark, A.E. Validation of molecular dynamics simulation. J. Chem. Phys. 1998, 108, 6109–6116. [Google Scholar] [CrossRef] [Green Version]
- Macháčková, M.; Tokarský, J.; Čapková, P. A simple molecular modeling method for the characterization of polymeric drug carriers. Eur. J. Pharm. Sci. 2013, 48, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, G.Z.; Bikiaris, D.N. Biodegradable poly(alkylene succinate) blends: Thermal behavior and miscibility study. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 584–597. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Papageorgiou, G.Z.; Papadimitriou, S.A.; Karavas, E.; Avgoustakis, K. Novel biodegradable polyester poly(propylene succinate): Synthesis and application in the preparation of solid dispersions and nanoparticles of a water-soluble drug. AAPS PharmSciTech 2009, 10, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Bikiaris, D.N.; Papageorgiou, G.Z.; Achilias, D.S. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym. Degrad. Stab. 2006, 91, 31–43. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Nanoscale Infrared, Thermal, and Mechanical Characterization of Telaprevir–Polymer Miscibility in Amorphous Solid Dispersions Prepared by Solvent Evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef]
- Anderson, B.D. Predicting Solubility/Miscibility in Amorphous Dispersions: It Is Time to Move Beyond Regular Solution Theories. J. Pharm. Sci. 2018, 107, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Huang, J.; Zhu, Q.; Haddadin, R.; Gawel, J.; Garmise, R.; Hussain, M. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int. J. Pharm. 2010, 395, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci 1999, 88, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, S.; Tsakiridou, G.; Ditzinger, F.; Koehl, N.J.; Price, D.J.; Ilie, A.-R.; Kalantzi, L.; Kimpe, K.; Holm, R.; Nair, A.; et al. Application of the solubility parameter concept to assist with oral delivery of poorly water-soluble drugs—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 441–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapourani, A.; Chatzitheodoridou, M.; Kontogiannopoulos, K.N.; Barmpalexis, P. Experimental, Thermodynamic, and Molecular Modeling Evaluation of Amorphous Simvastatin-Poly(vinylpyrrolidone) Solid Dispersions. Mol. Pharm. 2020, 17, 2703–2720. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating Miscibility and Molecular Mobility of Nifedipine-PVP Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kapourani, A.; Vardaka, E.; Katopodis, K.; Kachrimanis, K.; Barmpalexis, P. Rivaroxaban polymeric amorphous solid dispersions: Moisture-induced thermodynamic phase behavior and intermolecular interactions. Eur. J. Pharm. Biopharm. 2019, 145, 98–112. [Google Scholar] [CrossRef]
- Ihn, K.J.; Yoo, E.S.; Im, S.S. Structure and Morphology of Poly(tetramethylene succinate) Crystals. Macromolecules. 1995, 28, 2460–2464. [Google Scholar] [CrossRef]
- Fox, T.G. Influence of Diluent and of Copolymer Composition on the Glass Temperature of a Poly-mer System. Bull. Am. Phys. Soc. 1956, 1, 123. [Google Scholar]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. i. non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Eguiazabal, J.I.; Iruin, J.J.; Cortazar, M.; Guzmán, G.M. Glass transition temperatures in blends of poly(N-vinyl-2-pyrrolidone) with a copolymer of bisphenol A and epichlorohydrin or with poly(vinyl butyral). Die Makromol. Chem. 1984, 185, 1761–1766. [Google Scholar] [CrossRef]
- Ferg, E.E.; Bolo, L.L. A correlation between the variable melt flow index and the molecular mass distribution of virgin and recycled polypropylene used in the manufacturing of battery cases. Polym. Test. 2013, 32, 1452–1459. [Google Scholar] [CrossRef]
- Dutta, A. On viscosity—melt flow index relationship. Rheol. Acta. 1984, 23, 565–569. [Google Scholar] [CrossRef]
- Aho, J.; Boetker, J.P.; Baldursdottir, S.; Rantanen, J. Rheology as a tool for evaluation of melt processability of innovative dosage forms. Int. J. Pharm. 2015, 494, 623–642. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyester | [η] (dL/g) | Mn (g/mol) | Mw (g/mol) | Mw/Mn | Tm (oC) | Tg (oC) | Tc (oC) | ΔH (J/g) | CFc (%) |
|---|---|---|---|---|---|---|---|---|---|
| PESu | 0.13 | 1196 | 3056 | 2.56 | 93 | −24 | 27 | 68 | 40 |
| PPSu | 0.07 | 2823 | 6551 | 2.36 | 45 | −40 | - | - | - |
| PBSu | 0.16 | 4238 | 8833 | 2.08 | 111 | −41 | 77 | 85 | 5 |
| Substances | ||||
|---|---|---|---|---|
| PVA | PESu | PPSu | PBSu | |
| HVK group contribution method | ||||
| HSPs (MPa1/2) | 34.0 | 21.6 | 21.9 | 22.1 |
| Δδt (MP1/2) | - | 12.4 | 12.1 | 11.9 |
| MD simulations | ||||
| δMD (MPa1/2) | 26.1 | 26.8 | 25.5 | 24.4 |
| Δδt (MP1/2) | - | 0.7 | 0.6 | 1.7 |
| PVA-Plasticizer | Einter (kcal/mol) |
|---|---|
| PVA-PESu | −2201 |
| PVA-PPSu | −2308 |
| PVA-PBSu | −2274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palamidi, A.; Kapourani, A.; Christodoulou, E.; Klonos, P.A.; Kontogiannopoulos, K.N.; Kyritsis, A.; Bikiaris, D.N.; Barmpalexis, P. Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications. Polymers 2021, 13, 146. https://doi.org/10.3390/polym13010146
Palamidi A, Kapourani A, Christodoulou E, Klonos PA, Kontogiannopoulos KN, Kyritsis A, Bikiaris DN, Barmpalexis P. Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications. Polymers. 2021; 13(1):146. https://doi.org/10.3390/polym13010146
Chicago/Turabian StylePalamidi, Artemis, Afroditi Kapourani, Evi Christodoulou, Panagiotis A. Klonos, Konstantinos N. Kontogiannopoulos, Apostolos Kyritsis, Dimitrios N. Bikiaris, and Panagiotis Barmpalexis. 2021. "Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications" Polymers 13, no. 1: 146. https://doi.org/10.3390/polym13010146
APA StylePalamidi, A., Kapourani, A., Christodoulou, E., Klonos, P. A., Kontogiannopoulos, K. N., Kyritsis, A., Bikiaris, D. N., & Barmpalexis, P. (2021). Low Molecular Weight Oligomers of Poly(alkylene succinate) Polyesters as Plasticizers in Poly(vinyl alcohol) Based Pharmaceutical Applications. Polymers, 13(1), 146. https://doi.org/10.3390/polym13010146