
Novel Multifunctional Epoxy (Meth)Acrylate Resins and Coatings Preparation via Cationic and Free-Radical Photopolymerization




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Multifunctional Epoxy (Meth)Acrylate Resins
2.3. Characterization Methods
2.4. Preparation of Coating Compositions and Cured Films
2.5. Characteristics of the Photopolymerization Process and Properties of Cured Coatings
3. Results and Discussion
3.1. The Approach to the Development of the Prepolymers Synthesis
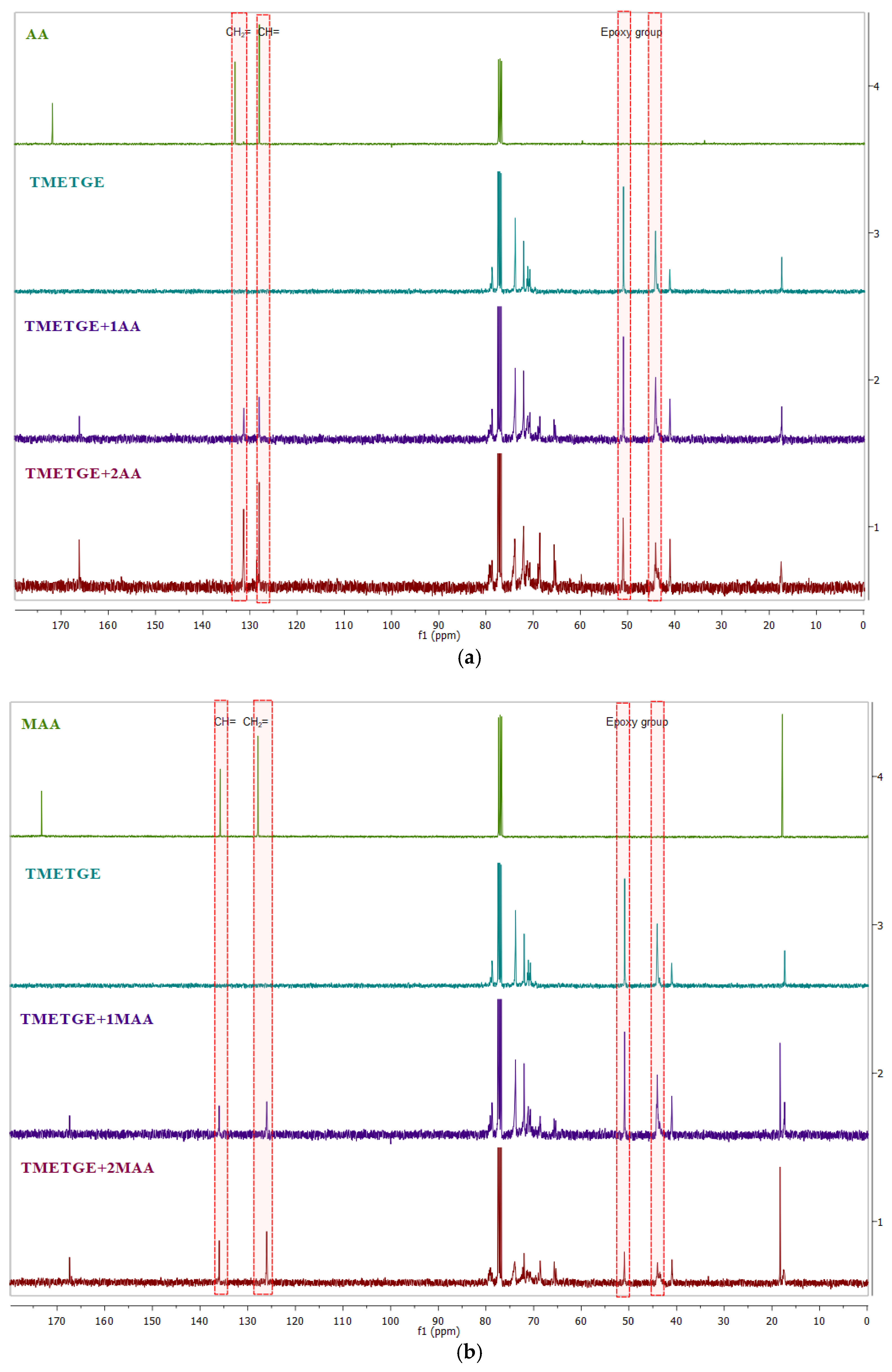
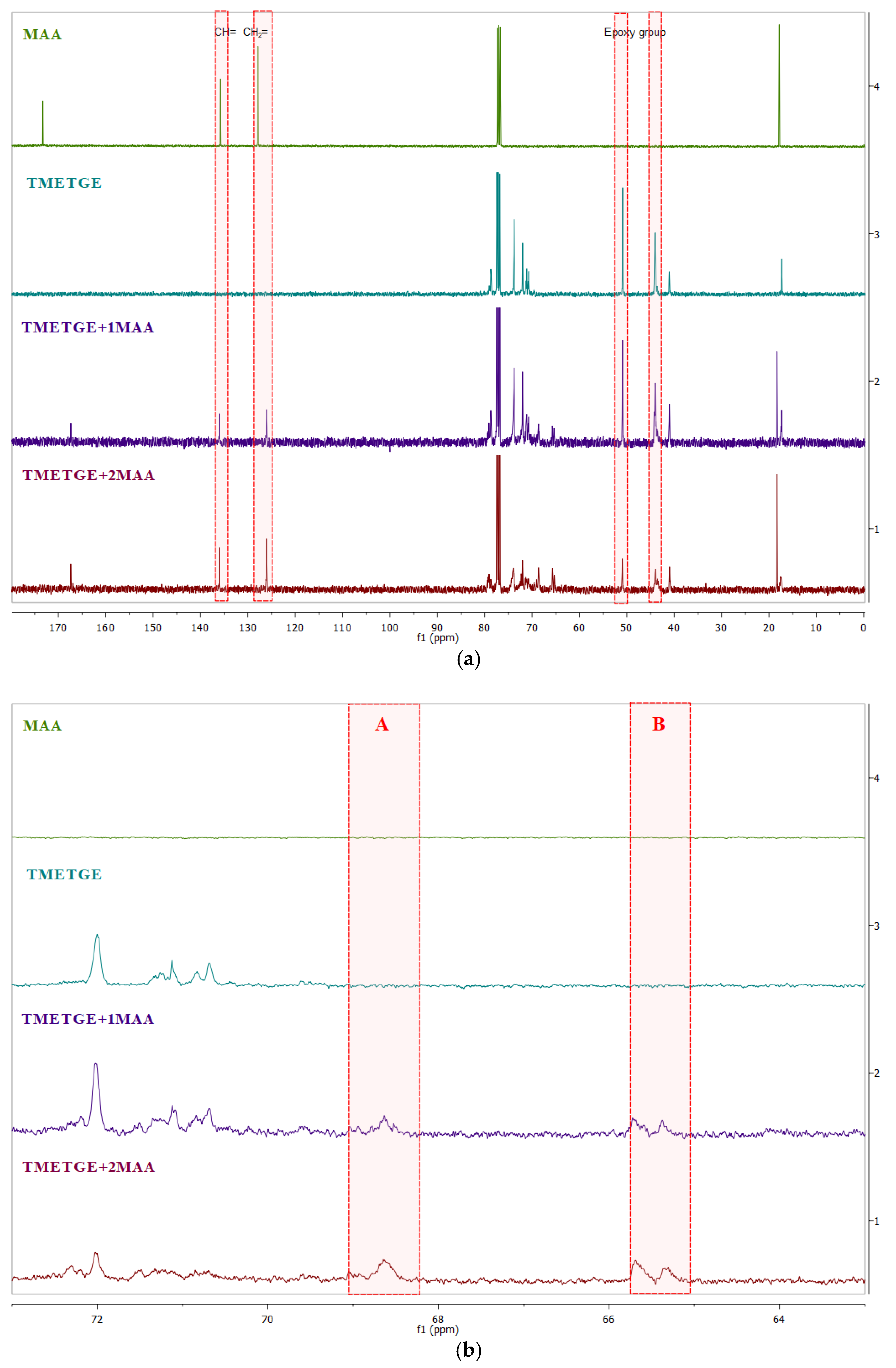
3.2. Prepolymer Characterization
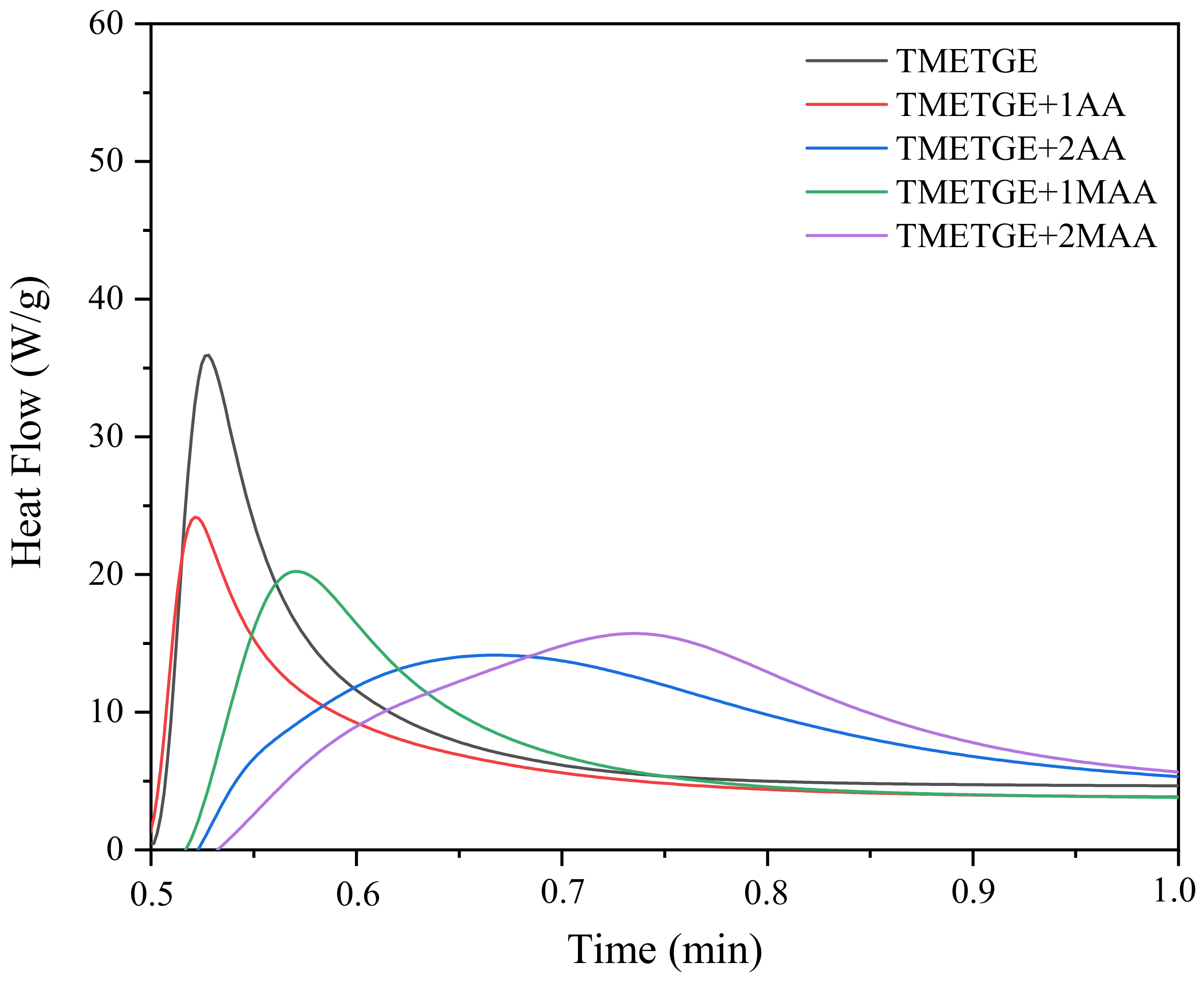
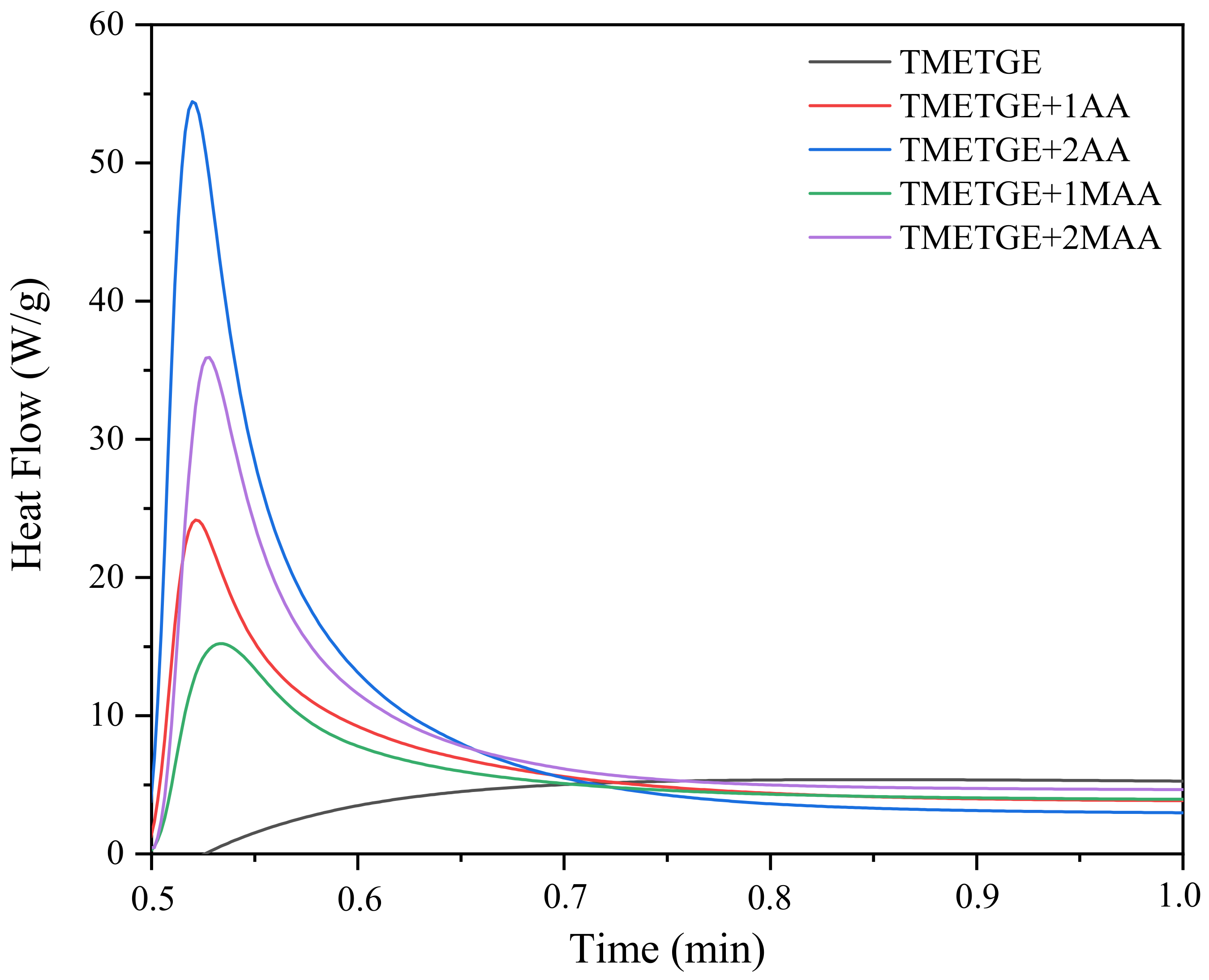
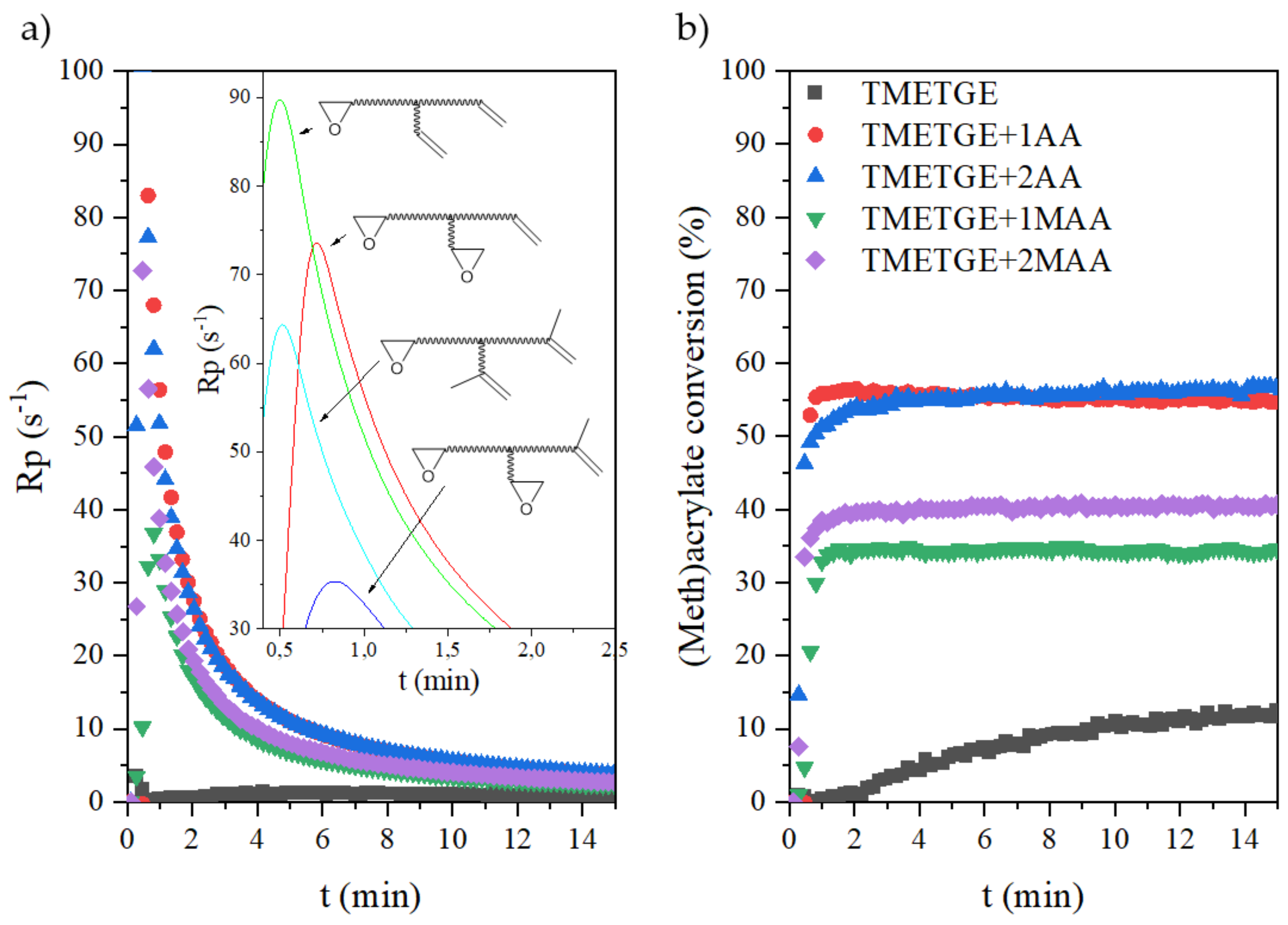
3.3. The Photocuring of the Epoxy (Meth)Acrylate Prepolymers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UV Curable Resins & Formulated Products Market Size | Share | Forecast. Available online: https://www.verifiedmarketresearch.com/product/uv-curable-resins-formulated-products-market (accessed on 27 April 2021).
- Chatani, S.; Kloxin, C.J.; Bowman, C.N. The power of light in polymer science: Photochemical processes to manipulate polymer formation, structure, and properties. Polym. Chem. 2014, 5, 2187–2201. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, N.; Yeow, J.; Judzewitsch, P.; Xu, J.; Boyer, C. Seeing the light: Advancing materials chemistry through photopolymerization. Angew. Chem. Int. Ed. 2019, 58, 5170–5189. [Google Scholar] [CrossRef]
- Dietlin, C.; Schweizer, S.; Xiao, P.; Zhang, J.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; Lalevée, J. Photopolymerization upon LEDs: New photoinitiating systems and strategies. Polym. Chem. 2015, 6, 3895–3912. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Mozelewska, K.; Czech, Z. Influence of the UV crosslinking method on the properties of acrylic adhesive. Int. J. Adhes. Adhes. 2020, 102, 102652. [Google Scholar] [CrossRef]
- Gziut, K.; Kowalczyk, A.; Schmidt, B. Free-radical bulk-photopolymerization process as a method of obtaining thermally curable structural self-adhesive tapes and effect of used type I photoinitiators. Polymers 2020, 12, 2191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, H.; Yang, H.; Xiong, L.; Zhou, J.; Huang, S.; Zhao, C.; Zhong, J.; Fan, X. UV-curable self-healing polyurethane coating based on thiol-ene and Diels-Alder double click reactions. Prog. Org. Coat. 2019, 137, 105282. [Google Scholar] [CrossRef]
- Wagner, A.; Mühlberger, M.; Paulik, C. Photoinitiator-free photopolymerization of acrylate-bismaleimide mixtures and their application for inkjet printing. J. Appl. Polym. Sci. 2019, 136, 47789. [Google Scholar] [CrossRef]
- Lorusso, E.; Ali, W.; Hildebrandt, M.; Mayer-Gall, T.; Gutmann, J.S. Hydrogel functionalized polyester fabrics by UV-induced photopolymerization. Polymers 2019, 11, 1329. [Google Scholar] [CrossRef] [Green Version]
- Cadenaro, M.; Maravic, T.; Comba, A.; Mazzoni, A.; Fanfoni, L.; Hilton, T.; Ferracane, J.; Breschi, L. The role of polymerization in adhesive dentistry. Dent. Mater. 2019, 35, e1–e22. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Simon-Masseron, A.; Lalevée, J. Radical photoinitiation with LEDs and applications in the 3D printing of composites. Chem. Soc. Rev. 2021, 50, 3824–3841. [Google Scholar] [CrossRef]
- Khudyakov, I.V. Fast photopolymerization of acrylate coatings: Achievements and problems. Prog. Org. Coat. 2018, 121, 151–159. [Google Scholar] [CrossRef]
- Sangermano, M.; Roppolo, I.; Chiappone, A. New horizons in cationic photopolymerization. Polymers 2018, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Ong, G.; Kasi, R.; Subramaniam, R. A review on plant extracts as natural additives in coating applications. Prog. Org. Coat. 2021, 151, 106091. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated polymerization: Advances, challenges, and opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- UV Curable Resin Market Size, Share and Global Trends by 2026 | AMR. Available online: https://www.alliedmarketresearch.com/uv-curable-resin-market (accessed on 27 April 2021).
- Launikitis, M.B. Vinyl ester resins. In Handbook of Composites; Springer: Boston, MA, USA, 1982; pp. 38–49. [Google Scholar]
- Kandelbauer, A.; Tondi, G.; Zaske, O.C.; Goodman, S.H. Unsaturated polyesters and vinyl esters. In Handbook of Thermoset Plastics; Elsevier Inc.: Amsterdam, The Netherlands, 2014; pp. 111–172. ISBN 9781455731077. [Google Scholar]
- Jaswal, S.; Gaur, B. New trends in vinyl ester resins. Rev. Chem. Eng. 2014, 30, 567–581. [Google Scholar] [CrossRef]
- Scott, T.F.; Cook, W.D.; Forsythe, J.S. Photo-DSC cure kinetics of vinyl ester resins. I. Influence of temperature. Polymer 2002, 43, 5839–5845. [Google Scholar] [CrossRef]
- Nowak, D.; Ortyl, J.; Kamińska-Borek, I.; Kukuła, K.; Topa, M.; Popielarz, R. Photopolymerization of hybrid monomers: Part I: Comparison of the performance of selected photoinitiators in cationic and free-radical polymerization of hybrid monomers. Polym. Test. 2017, 64, 313–320. [Google Scholar] [CrossRef]
- Park, Y.J.; Lim, D.H.; Kim, H.J.; Park, D.S.; Sung, I.K. UV- and thermal-curing behaviors of dual-curable adhesives based on epoxy acrylate oligomers. Int. J. Adhes. Adhes. 2009, 29, 710–717. [Google Scholar] [CrossRef]
- Yildiz, Z.; Gungor, A.; Onen, A.; Usta, I. Synthesis and characterization of dual-curable epoxyacrylates for polyester cord/rubber applications. J. Ind. Text. 2016, 46, 596–610. [Google Scholar] [CrossRef]
- Su, Y.C.; Cheng, L.P.; Cheng, K.C.; Don, T.M. Synthesis and characterization of UV-and thermo-curable difunctional epoxyacrylates. Mater. Chem. Phys. 2012, 132, 540–549. [Google Scholar] [CrossRef]
- Konuray, O.; Di Donato, F.; Sangermano, M.; Bonada, J.; Tercjak, A.; Fernández-Francos, X.; Serra Albet, R.A.; Ramis, X. Dual-curable stereolithography resins for superior thermomechanical properties. Express Polym. Lett. 2020, 14, 881–894. [Google Scholar] [CrossRef]
- Flores, M.; Tomuta, A.M.; Fernández-Francos, X.; Ramis, X.; Sangermano, M.; Serra, A. A new two-stage curing system: Thiol-ene/epoxy homopolymerization using an allyl terminated hyperbranched polyester as reactive modifier. Polymer 2013, 54, 5473–5481. [Google Scholar] [CrossRef]
- Fernández-Francos, X.; Konuray, A.O.; Belmonte, A.; De La Flor, S.; Serra, À.; Ramis, X. Sequential curing of off-stoichiometric thiol-epoxy thermosets with a custom-tailored structure. Polym. Chem. 2016, 7, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Hu, J.; Wang, F.; Huang, Q.; Peng, C.; Xu, Z. Synthesizing promising epoxy acrylate prepolymers applied in ultraviolet cured adhesives based on esterification reaction. Mater. Res. Express 2018, 5, 065321. [Google Scholar] [CrossRef]
- Srivastava, A.; Pal, N.; Agarwal, S.; Rai, J.S.P. Kinetics and mechanism of esterification of epoxy resin with methacrylic acid in the presence of tertiary amines. Adv. Polym. Technol. 2005, 24, 1–13. [Google Scholar] [CrossRef]
- Bajpai, M.; Shukla, V.; Kumar, A. Film performance and UV curing of epoxy acrylate resins. Prog. Org. Coat. 2002, 44, 271–278. [Google Scholar] [CrossRef]
- Duran, H.; Meng, S.; Kim, N.; Hu, J.; Kyu, T.; Natarajan, L.V.; Tondiglia, V.P.; Bunning, T.J. Kinetics of photopolymerization-induced phase separation and morphology development in mixtures of a nematic liquid crystal and multifunctional acrylate. Polymer 2008, 49, 534–545. [Google Scholar] [CrossRef]
- Yin, B.; Zhang, J. A novel photocurable modified epoxy resin for high heat resistance coatings. Colloid Polym. Sci. 2020, 298, 1303–1312. [Google Scholar] [CrossRef]
- Wu, Z.; Li, S.; Liu, M.; Wang, Z.; Liu, X. Liquid oxygen compatible epoxy resin: Modification and characterization. RSC Adv. 2015, 5, 11325–11333. [Google Scholar] [CrossRef]
- Ying, L.; Wang, P.; Kang, E.T.; Neoh, K.G. Synthesis and characterization of poly(acrylic acid)-graft-poly(vinylidene fluoride) copolymers and pH-sensitive membranes. Macromolecules 2002, 35, 673–679. [Google Scholar] [CrossRef]
- Feng, L.; Yang, H.; Dong, X.; Lei, H.; Chen, D. pH-sensitive polymeric particles as smart carriers for rebar inhibitors delivery in alkaline condition. J. Appl. Polym. Sci. 2018, 135, 45886. [Google Scholar] [CrossRef]
- González, M.G.; Cabanelas, J.C.; Baselga, J. Applications of FTIR on epoxy resins—Identification, monitoring the curing process, phase separation and water uptake. In Infrared Spectroscopy—Materials Science, Engineering and Technology; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Park, C.H.; Lee, S.W.; Park, J.W.; Kim, H.J. Preparation and characterization of dual curable adhesives containing epoxy and acrylate functionalities. React. Funct. Polym. 2013, 73, 641–646. [Google Scholar] [CrossRef]
- Arbain, N.H.; Salimon, J. Synthesis and characterization of ester trimethylolpropane based jatropha curcas oil as biolubricant base stocks. J. Sci. Technol. 2010, 2, 47–58. [Google Scholar]
- Danso, R.; Hoedebecke, B.; Whang, K.; Sarrami, S.; Johnston, A.; Flipse, S.; Wong, N.; Rawls, H.R. Development of an oxirane/acrylate interpenetrating polymer network (IPN) resin system. Dent. Mater. 2018, 34, 1459–1465. [Google Scholar] [CrossRef]

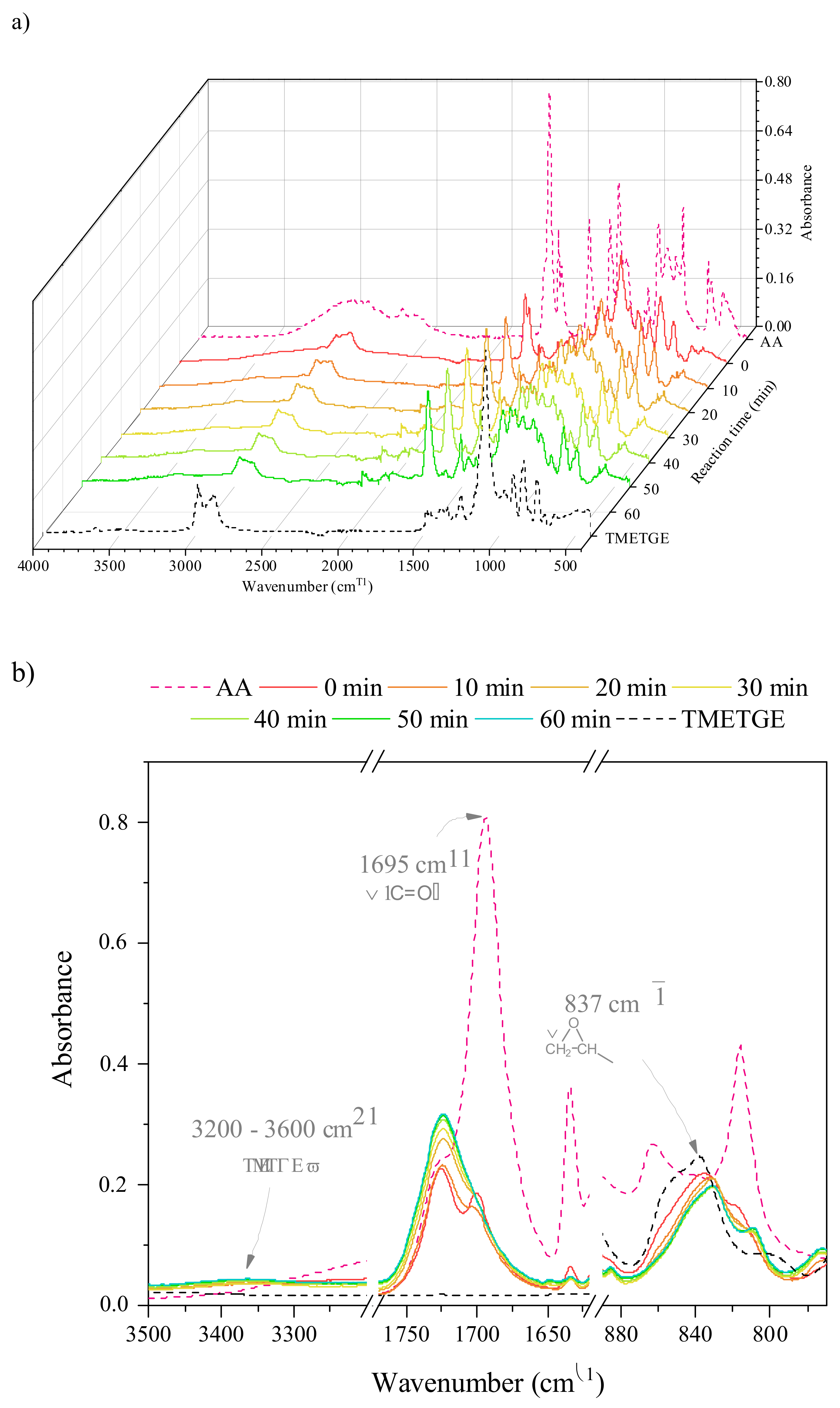
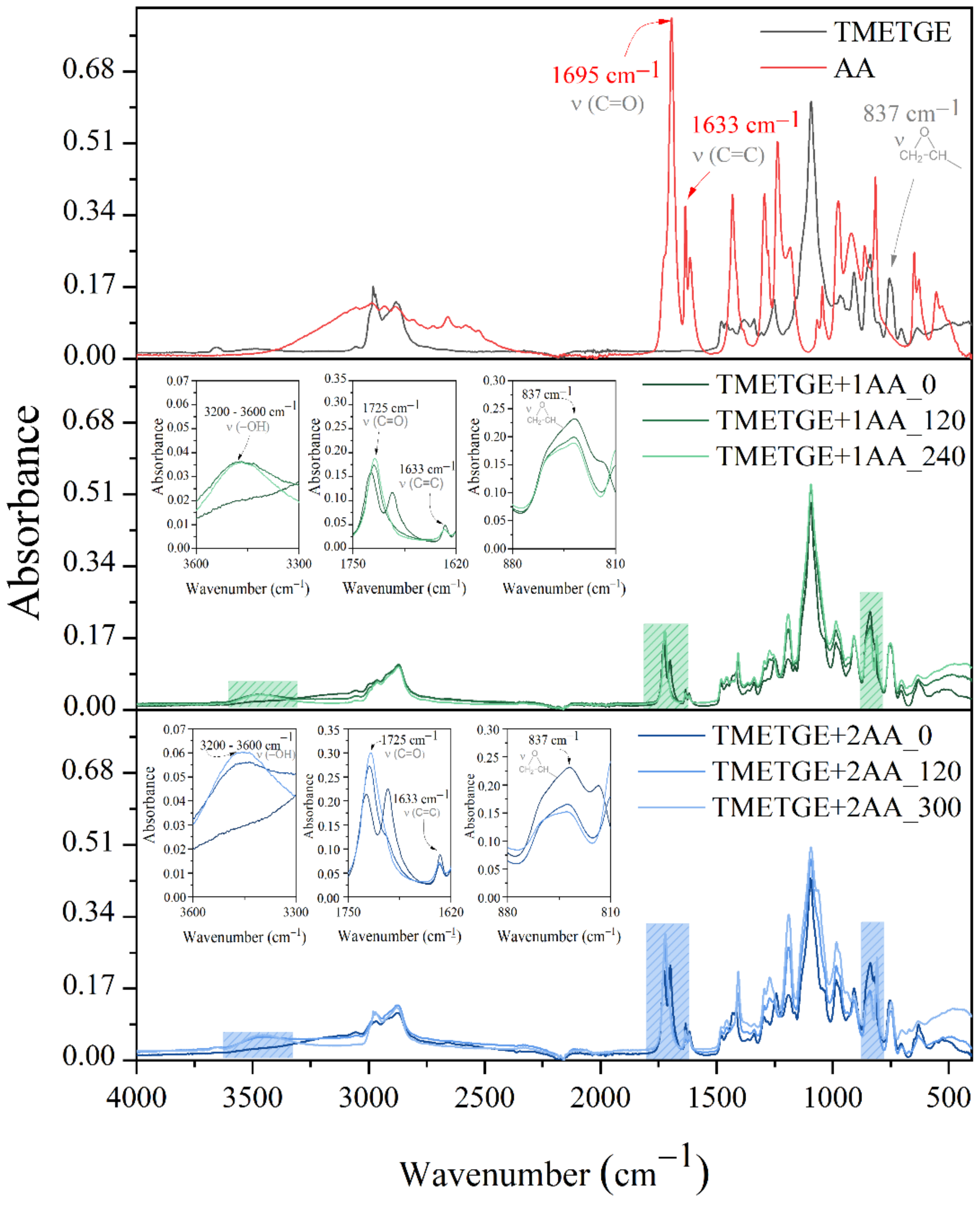

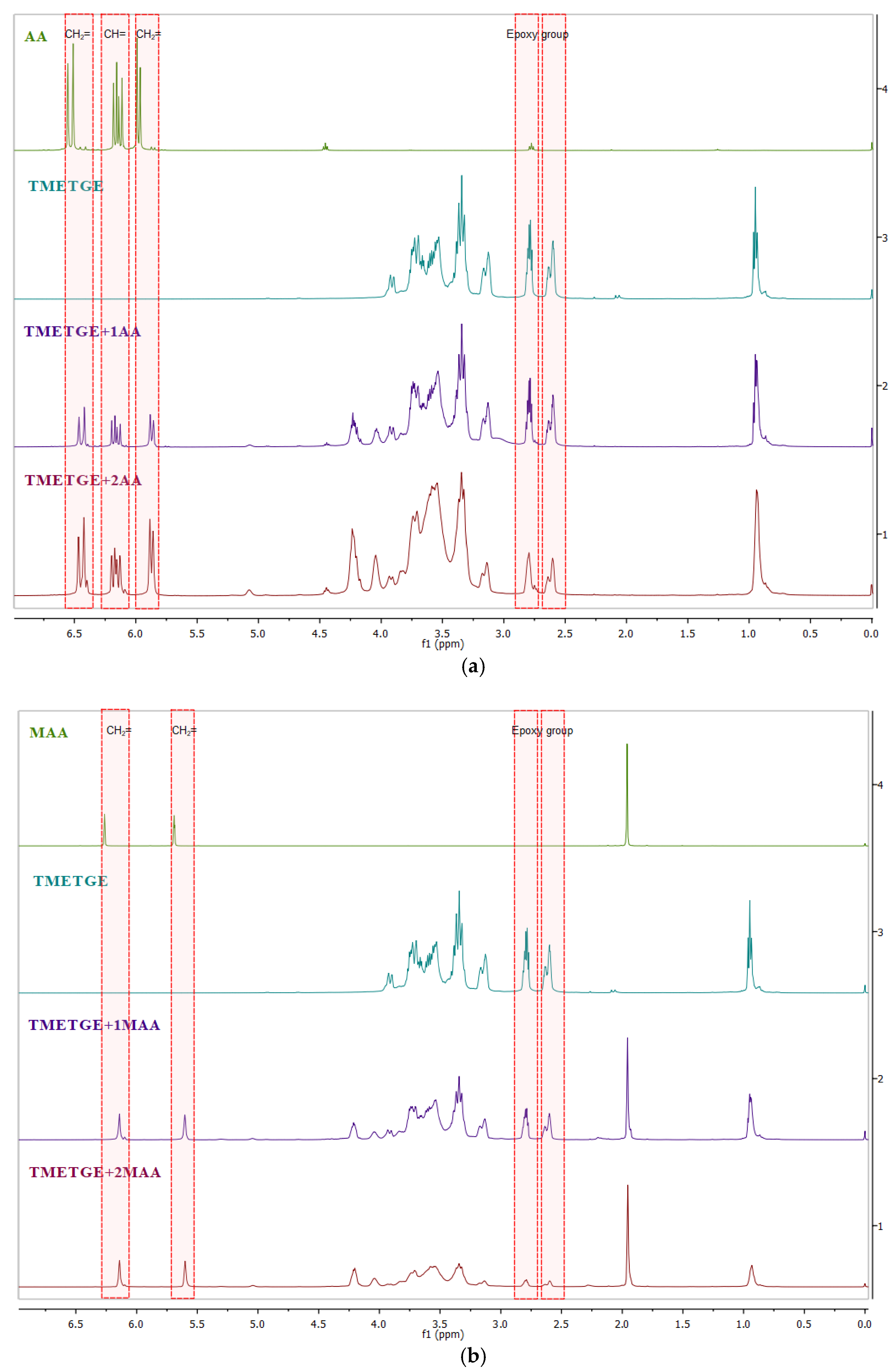
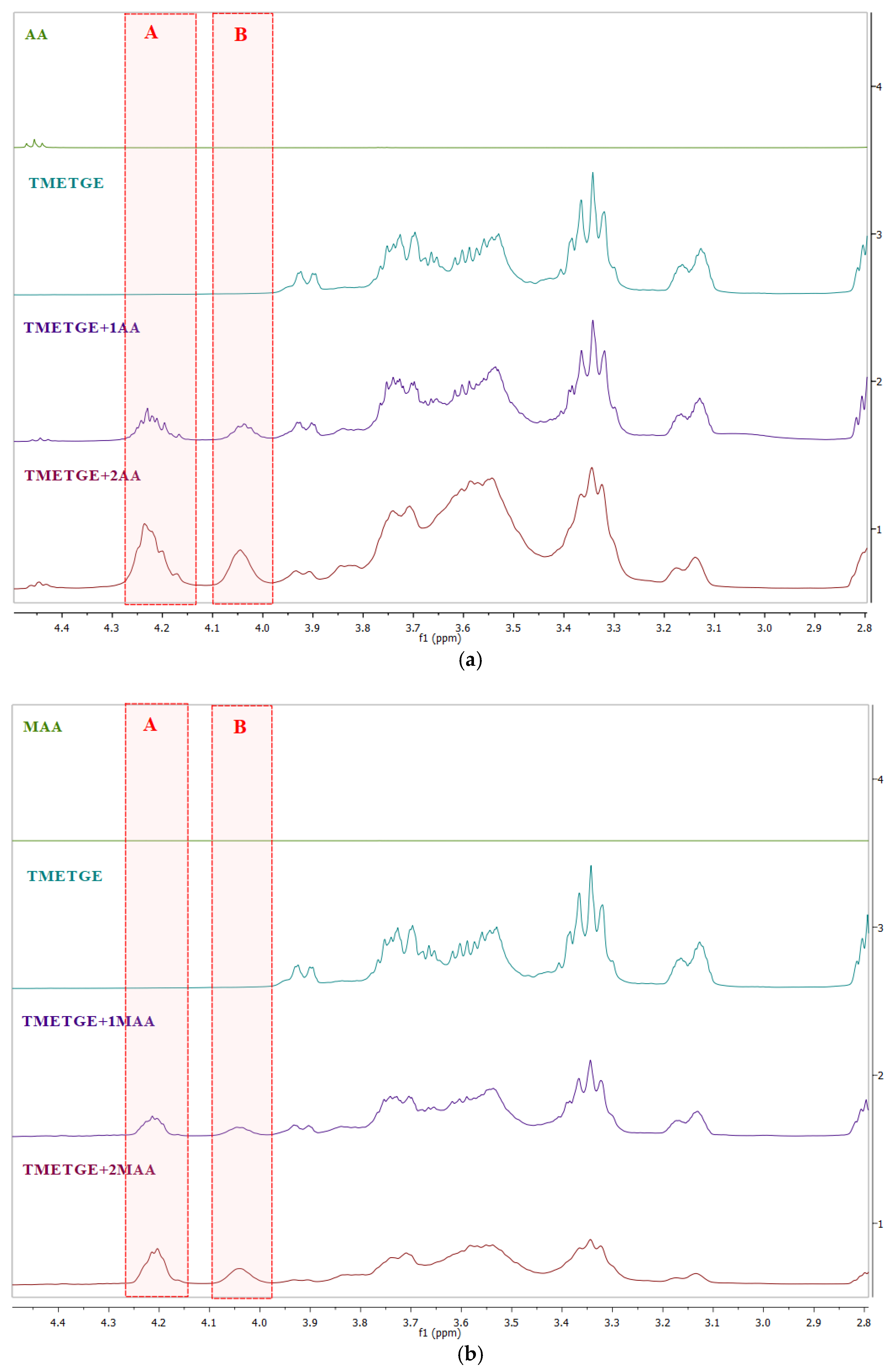


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Time (min) | AA | MAA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NV (%) | PAVs (mgKOH/g) | AAC (%) | EE (g/mol) | EGC (%) | η (mPa·s) | NV (%) | PAVs (mgKOH/g) | MAAC (%) | EE (g/mol) | EGC (%) | η (mPa·s) | |
| TMETGE | ||||||||||||
| 98.7 | 158.774 | 336 | 98.7 | 158.774 | 336 | |||||||
| TMETGE + 1AA | TMETGE + 1MAA | |||||||||||
| 0 | 87.27 | 106.1 | 0.0 | 165.334 | 0.1 | 84.18 | 111.7 | 0.0 | 190.055 | 0.2 | ||
| 120 | 96.34 | 19.9 | 81.2 | 205.801 | 19.7 | 98.16 | 9.1 | 91.9 | 252.121 | 22.7 | ||
| 240 | 98.56 | 2.2 | 97.9 | 233.678 | 29.3 | 1189 | 98.56 | 5.3 | 95.3 | 276.795 | 29.6 | 1344 |
| TMETGE + 2AA | TMETGE + 2MAA | |||||||||||
| 0 | 79.25 | 210.8 | 0.0 | 165.131 | 0.0 | 74.71 | 222.3 | 0.0 | 225.026 | 0.3 | ||
| 120 | 93.06 | 59.6 | 71.8 | 258.223 | 36.0 | 94.22 | 42.2 | 81.0 | 389.916 | 42.5 | ||
| 240 | 97.51 | 19.9 | 90.2 | 521.127 | 57.0 | |||||||
| 300 | 98 | 15.6 | 96.3 | 404.444 | 59.2 | 4081 | 4291 | |||||
| Sample | Hmax (1) (W/g) | Rpmax (2) (s−1) | C (3) (%) | Tack-Free Time (s) | Hardness (s) | Adhesion | Gloss (GU) | Yellowness Index | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | R | C | R | C | R | C | R | C | R | C | R | C | R | C | R | |
| TMETGE | 36 | 7 | 3,37 | 34 | 11 | 74 | 0 | 154 | 4.3 | |||||||
| TMETGE + 1AA | 24 | 24 | 2,08 | 73 | 13 | 57 | 12 | 15 | 50 | 41 | 0 | 1.5 | 169 | 140 | 11.5 | 10.1 |
| TMETGE + 2AA | 15 | 54 | 1,92 | 89 | 5 | 57 | 16 | 13 | 35 | 23 | 0,5 | 1 | 153 | 120 | 10.6 | 7.1 |
| TMETGE + 1MAA | 21 | 15 | 1,92 | 35 | 5 | 34 | 12 | 10 | 132 | 27 | 0 | 1.5 | 159 | 123 | 3.6 | 3.5 |
| TMETGE + 2MAA | 16 | 36 | 1,81 | 64 | 4 | 41 | 22 | 10 | 115 | 14 | 0,5 | 3 | 145 | 119 | 3.9 | 2.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bednarczyk, P.; Irska, I.; Gziut, K.; Ossowicz-Rupniewska, P. Novel Multifunctional Epoxy (Meth)Acrylate Resins and Coatings Preparation via Cationic and Free-Radical Photopolymerization. Polymers 2021, 13, 1718. https://doi.org/10.3390/polym13111718
Bednarczyk P, Irska I, Gziut K, Ossowicz-Rupniewska P. Novel Multifunctional Epoxy (Meth)Acrylate Resins and Coatings Preparation via Cationic and Free-Radical Photopolymerization. Polymers. 2021; 13(11):1718. https://doi.org/10.3390/polym13111718
Chicago/Turabian StyleBednarczyk, Paulina, Izabela Irska, Konrad Gziut, and Paula Ossowicz-Rupniewska. 2021. "Novel Multifunctional Epoxy (Meth)Acrylate Resins and Coatings Preparation via Cationic and Free-Radical Photopolymerization" Polymers 13, no. 11: 1718. https://doi.org/10.3390/polym13111718
APA StyleBednarczyk, P., Irska, I., Gziut, K., & Ossowicz-Rupniewska, P. (2021). Novel Multifunctional Epoxy (Meth)Acrylate Resins and Coatings Preparation via Cationic and Free-Radical Photopolymerization. Polymers, 13(11), 1718. https://doi.org/10.3390/polym13111718