
Exohedral Functionalization of Fullerene by Substituents Controlling of Molecular Organization for Spontaneous C60 Dimerization in Liquid Crystal Solutions and in a Bulk Controlled by a Potential

 ,
,  ,
,  , ,
, ,  ,
, 
Abstract
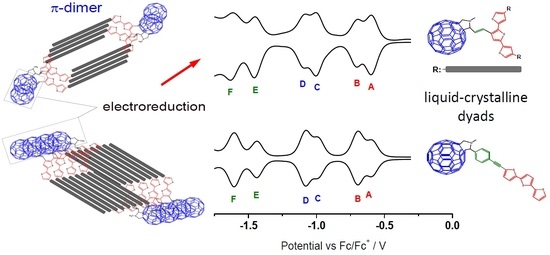
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. Electrochemical and Spectroelectrochemical Measurements
2.4. Theoretical Calculations
3. Results and Discussion
3.1. Electroreduction of Terthiophene-C60 Dyads
3.2. Electroreduction of C60-Pendant Polymer with Poly(Terthiophene)’s Scaffold
3.3. DFT Calculation
3.4. ESR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pei, C.; Wang, L. Recent Progress on High-Pressure and High-Temperature Studies of Fullerenes and Related Materials. Matter Radiat. Extrem. 2019, 4, 028201. [Google Scholar] [CrossRef] [Green Version]
- Ono, S.; Toda, Y.; Onoe, J. Unified Understanding of the Electron-Phonon Coupling Strength for Nanocarbon Allotropes. Phys. Rev. B 2014, 90, 155435. [Google Scholar] [CrossRef]
- Sundqvist, B. Polymeric Fullerene Phases Formed under Pressure. In Fullerene-Based Materials. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2004; pp. 85–126. [Google Scholar]
- Murata, Y.; Kato, N.; Fujiwara, K.; Komatsu, K. Solid-State [4 + 2] Cycloaddition of Fullerene C 60 with Condensed Aromatics Using a High-Speed Vibration Milling Technique. J. Org. Chem. 1999, 64, 3483–3488. [Google Scholar] [CrossRef]
- Manzetti, S. Molecular and Crystal Assembly inside the Carbon Nanotube: Encapsulation and Manufacturing Approaches. Adv. Manuf. 2013, 1, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Goclon, J.; Winkler, K.; Margraf, J.T. Theoretical Investigation of Interactions between Palladium and Fullerene in Polymer. RSC Adv. 2017, 7, 2202–2210. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, A.; de Bettencourt-Dias, A.; Winkler, K.; Balch, A.L. Redox-Active Films Formed by Electrochemical Reduction of Solutions of C60 and Platinum Complexes. J. Mater. Chem. 2002, 12, 2116–2122. [Google Scholar] [CrossRef]
- Winkler, K.; de Bettencourt-Dias, A.; Balch, A.L. Electrochemical Studies of C60/Pd Films Formed by the Reduction of C 60 in the Presence of Palladium(II) Acetate Trimer. Effects of Varying C60/Pd(II) Ratios in the Precursor Solutions. Chem. Mater. 2000, 12, 1386–1392. [Google Scholar] [CrossRef]
- Grądzka, E.; Wysocka-Żołopa, M.; Winkler, K. In Situ Conductance Studies of Two-Component C60-Pd Polymer. J. Phys. Chem. C 2014, 118, 14061–14072. [Google Scholar] [CrossRef]
- Zou, Y.J.; Zhang, X.W.; Li, Y.L.; Wang, B.; Yan, H.; Cui, J.Z.; Liu, L.M.; Da, D.A. Bonding Character of the Boron-Doped C60 Films Prepared by Radio Frequency Plasma Assisted Vapor Deposition. J. Mater. Sci. 2002, 37, 1043–1047. [Google Scholar] [CrossRef]
- Goze, C.; Rachdi, F.; Hajji, L.; Núñez-Regueiro, M.; Marques, L.; Hodeau, J.-L.; Mehring, M. High-resolution 13C NMR studies of high-pressure-polymerized C 60: Evidence for the [2 + 2] cycloaddition structure in the rhombohedral two-dimensional C60 polymer. Phys. Rev. B 1996, 54, R3676–R3678. [Google Scholar] [CrossRef]
- Álvarez-Murga, M.; Hodeau, J.L. Structural Phase Transitions of C60 under High-Pressure and High-Temperature. Carbon 2015, 82, 381–407. [Google Scholar] [CrossRef]
- Wang, G.; Li, Y.; Huang, Y. Structures and Electronic Properties of Peanut-Shaped Dimers and Carbon Nanotubes. J. Phys. Chem. B 2005, 109, 10957–10961. [Google Scholar] [CrossRef]
- Yamanaka, S.; Kubo, A.; Inumaru, K.; Komaguchi, K.; Kini, N.S.; Inoue, T.; Irifune, T. Electron conductive three-dimensional polymer of cuboidal C60. Phys. Rev. Lett. 2006, 96, 076602. [Google Scholar] [CrossRef] [Green Version]
- Pei, C.; Feng, M.; Yang, Z.; Yao, M.; Yuan, Y.; Li, X.; Hu, B.; Shen, M.; Chen, B.; Sundqvist, B.; et al. Quasi 3D Polymerization in C60 Bilayers in a Fullerene Solvate. Carbon 2017, 124, 499–505. [Google Scholar] [CrossRef]
- Bourque, A.J.; Engmann, S.; Fuster, A.; Snyder, C.R.; Richter, L.J.; Geraghty, P.B.; Jones, D.J. Morphology of a Thermally Stable Small Molecule OPV Blend Comprising a Liquid Crystalline Donor and Fullerene Acceptor. J. Mater. Chem. A 2019, 7, 16458–16471. [Google Scholar] [CrossRef]
- Fujitsuka, M.; Luo, C.; Ito, O.; Murata, Y.; Komatsu, K. Triplet Properties and Photoinduced Electron-Transfer Reactions of C 120, the [2 + 2] Dimer of Fullerene C60. J. Phys. Chem. A 1999, 103, 7155–7160. [Google Scholar] [CrossRef]
- Liu, F.-L.; Zhao, X.-X. Two Intact C60 Cages Linked by Six Carbon–Carbon Single Bonds. J. Mol. Struct. THEOCHEM 2007, 804, 117–121. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Lopatin, D.V.; Rodaev, V.V.; Lyubovskaya, R.N. Fullerene Complexes with Divalent Metal Dithiocarbamates: Structures, Magnetic Properties, and Photoconductivity. Russ. Chem. Bull. 2007, 56, 2145–2161. [Google Scholar] [CrossRef]
- Yamauchi, S.; Funayama, T.; Ohba, Y.; Paul, P.; Reed, C.A.; Fujiwara, K.; Komatsu, K. Direct Observation of Localized Excitation in the Lowest Excited Triplet State of Fullerene Dimers C120 and C120O by Means of Time-Resolved Electron Paramagnetic Resonance. Chem. Phys. Lett. 2002, 363, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Gromov, A.; Lebedkin, S.; Hull, W.E.; Krätschmer, W. Isomers of the Dimeric Fullerene C120O2. J. Phys. Chem. A 1998, 102, 4997–5005. [Google Scholar] [CrossRef]
- Chorro, M.; Cambedouzou, J.; Iwasiewicz-Wabnig, A.; Noé, L.; Rols, S.; Monthioux, M.; Sundqvist, B.; Launois, P. Discriminated Structural Behaviour of C60 and C70 Peapods under Extreme Conditions. Europhys. Lett. EPL 2007, 79, 56003. [Google Scholar] [CrossRef]
- Pekker, S.; Kováts, É.; Oszlányi, G.; Bényei, G.; Klupp, G.; Bortel, G.; Jalsovszky, I.; Jakab, E.; Borondics, F.; Kamarás, K.; et al. Rotor–Stator Molecular Crystals of Fullerenes with Cubane. Nat. Mater. 2005, 4, 764–767. [Google Scholar] [CrossRef]
- Bubenchikov, A.; Bubenchikov, M.; Mamontov, D.; Lun-Fu, A. Md-Simulation of Fullerene Rotations in Molecular Crystal Fullerite. Crystals 2019, 9, 496. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, S.; Hara, T.; Yokomae, T.; Okino, F.; Touhara, H.; Kataura, H.; Watanuki, T.; Ohishi, Y. Pressure-Polymerization of C60 Molecules in a Carbon Nanotube. Chem. Phys. Lett. 2006, 418, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.H.; Scuseria, G.E. Theoretical Predictions for a Two-Dimensional Rhombohedral Phase of Solid C60. Phys. Rev. Lett. 1995, 74, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Kuzmin, A.V.; Khasanov, S.S.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Design, Crystal Structures and Magnetic Properties of Anionic Salts Containing Fullerene C60 and Indium(Iii) Bromide Phthalocyanine Radical Anions. Dalton Trans 2014, 43, 13061–13069. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Saito, G.; Otsuka, A.; Lyubovskaya, R.N. Dimerization of C60•− in Multi-Component Ionic Complexes with Bis(Ethylenedithio)Tetrathiafulvalene: (Cation+)2·ET·(C60•−)2. J. Mater. Chem. 2007, 17, 4171. [Google Scholar] [CrossRef]
- Brumm, H.; Jansen, M. Synthese Und Einkristallstrukturanalyse von [M(NH3)6]C60 · 6 NH3 (M = Co2+, Zn2+). Z. Für Anorg. Allg. Chem. 2001, 627, 1433–1435. [Google Scholar] [CrossRef]
- Gorishnyi, M.P.; Koval’chuk, O.V.; Koval’chuk, T.N.; Verbitsky, A.B.; Vovk, V.E. Optical and Photoelectric Properties of Heterostructures of Fullerene C 60 with Phthalocyanines and Tetracyanoquinodimethane (TCNQ). Mol. Cryst. Liq. Cryst. 2011, 535, 49–56. [Google Scholar] [CrossRef]
- Konarev, D.V.; Lyubovskaya, R.N. Donor–Acceptor Complexes and Radical Ionic Salts Based on Fullerenes. Russ. Chem. Rev. 1999, 68, 19–38. [Google Scholar] [CrossRef]
- Armaroli, N.; Marconi, G.; Echegoyen, L.; Bourgeois, J.-P.; Diederich, F. Charge-Transfer Interactions in Face-to-Face Porphyrin-Fullerene Systems: Solvent-Dependent Luminescence in the Infrared Spectral Region. Chem. Eur. J. 2000, 6, 1629–1645. [Google Scholar] [CrossRef]
- Cui, C.-X.; Liu, Y.-J.; Zhang, Y.-P.; Qu, L.-B.; Zhang, Z.-P. Theoretical Study on the Reactivity and Regioselectivity of the Diels–Alder Reaction of Fullerene: Effects of Charges and Encapsulated Lanthanum on the Bis-Functionalization of C70. J. Phys. Chem. A 2017, 121, 523–531. [Google Scholar] [CrossRef]
- Morton, J.R.; Preston, K.F.; Krusic, P.J.; Hill, S.A.; Wasserman, E. ESR Studies of the Reaction of Alkyl Radicals with Fullerene (C60). J. Phys. Chem. 1992, 96, 3576–3578. [Google Scholar] [CrossRef]
- Chamberlain, T.W.; Davies, E.S.; Khlobystov, A.N.; Champness, N.R. Multi-Electron-Acceptor Dyad and Triad Systems Based on Perylene Bisimides and Fullerenes. Chem. Eur. J. 2011, 17, 3759–3767. [Google Scholar] [CrossRef]
- Bobrowska, D.M.; Plonska-Brzezinska, M.E. Endohedral and Exohedral Single-Layered Fullerenes. In Synthesis and Applications of Nanocarbons; Wiley: Hoboken, NJ, USA, 2020; pp. 25–62. [Google Scholar]
- Fukuzumi, S.; Nakanishi, I.; Suenobu, T.; Kadish, K.M. Electron-Transfer Properties of C60 and Tert -Butyl-C 60 Radical. J. Am. Chem. Soc. 1999, 121, 3468–3474. [Google Scholar] [CrossRef]
- Morton, J.R.; Preston, K.F. The Hyperfine Interactions of Free Radical Adducts of C60. In Physics and Chemistry of the Fullerenes; Springer: Dordrecht, The Netherlands, 1994; pp. 141–168. [Google Scholar]
- Keizer, P.N.; Morton, J.R.; Preston, K.F.; Krusic, P.J. Electron Paramagnetic Resonance Spectra of R–C60 Radicals Part 2: Hindered Rotation in Alkyl- and Silyl-C60 Radicals. J. Chem. Soc. Perkin Trans. 2 1993, 6, 1041–1045. [Google Scholar] [CrossRef]
- Martín-Gomis, L.; Seetharaman, S.; Herrero, D.; Karr, P.A.; Fernández-Lázaro, F.; D’Souza, F.; Sastre-Santos, Á. Distance-Dependent Electron Transfer Kinetics in Axially Connected Silicon Phthalocyanine-Fullerene Conjugates. ChemPhysChem 2020, 21, 2254–2262. [Google Scholar] [CrossRef] [PubMed]
- Dabbagh, H.A.; Zamani, M.; Mortaji, H. Conformational Stability and Rotational Energy Barrier of RC60–C60R Dimers: Hyperconjugation versus Steric Effect. J. Iran. Chem. Soc. 2012, 9, 205–223. [Google Scholar] [CrossRef]
- Sastre-Santos, Á.; Parejo, C.; Martín-Gomis, L.; Ohkubo, K.; Fernández-Lázaro, F.; Fukuzumi, S. C60 Dimers Connected through Pleiadene Bridges: Fullerenes Talking to Each Other. J. Mater. Chem. 2011, 21, 1509–1515. [Google Scholar] [CrossRef]
- Sánchez, L.; Rispens, M.T.; Hummelen, J.C. A Supramolecular Array of Fullerenes by Quadruple Hydrogen Bonding. Angew. Chem. Int. Ed. 2002, 41, 838–840. [Google Scholar] [CrossRef]
- Konarev, D.V.; Kuzmin, A.V.; Khasanov, S.S.; Goryunkov, A.A.; Brotsman, V.A.; Ioffe, I.N.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Electronic Communication between S= 1/2 Spins in Negatively-charged Double-caged Fullerene C60 Derivative Bonded by Two Single Bonds and Pyrrolizidine Bridge. Chem. Asian J. 2019, 14, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, Y.; Tanoue, K.; Mitani, T.; Iwasa, Y.; Izuoka, A.; Sugawara, T.; Yagi, T. High Yield Selective Synthesis of C60 Dimers. Chem. Commun. 1998, 1411–1412. [Google Scholar] [CrossRef]
- de la Torre, G.; Vázquez, P.; Agulló-López, F.; Torres, T. Role of Structural Factors in the Nonlinear Optical Properties of Phthalocyanines and Related Compounds. Chem. Rev. 2004, 104, 3723–3750. [Google Scholar] [CrossRef] [PubMed]
- Nisic, F.; Colombo, A.; Dragonetti, C.; Cominetti, A.; Pellegrino, A.; Perin, N.; Po, R.; Tacca, A. Novel Terthiophene-Substituted Fullerene Derivatives as Easily Accessible Acceptor Molecules for Bulk-Heterojunction Polymer Solar Cells. Int. J. Photoenergy 2014, 2014, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lincker, F.; Bourgun, P.; Stoeckli-Evans, H.; Saez, I.M.; Goodby, J.W.; Deschenaux, R. Optically Active Liquid-Crystalline Fullerodendrimers from Enantiomerically Pure Fulleropyrrolidines. Chem. Commun. 2010, 46, 7522. [Google Scholar] [CrossRef] [Green Version]
- Vergara, J.; Barberá, J.; Serrano, J.L.; Ros, M.B.; Sebastián, N.; de la Fuente, R.; López, D.O.; Fernández, G.; Sánchez, L.; Martín, N. Liquid-Crystalline Hybrid Materials Based on [60]Fullerene and Bent-Core Structures. Angew. Chem. Int. Ed. 2011, 50, 12523–12528. [Google Scholar] [CrossRef]
- Plonska, M.E.; de Bettencourt-Dias, A.; Balch, A.L.; Winkler, K. Electropolymerization of 2‘-Ferrocenylpyrrolidino-[3‘,4‘;1,2][C 60]Fullerene in the Presence of Palladium Acetate. Formation of an Electroactive Fullerene-Based Film with a Covalently Attached Redox Probe. Chem. Mater. 2003, 15, 4122–4131. [Google Scholar] [CrossRef]
- Rio, Y.; Accorsi, G.; Nierengarten, H.; Rehspringer, J.-L.; Hönerlage, B.; Kopitkovas, G.; Chugreev, A.; Van Dorsselaer, A.; Armaroli, N.; Nierengarten, J.-F. Fullerodendrimers with Peripheral Triethyleneglycol Chains: Synthesis, Mass Spectrometric Characterization, and Photophysical Properties. New J. Chem. 2002, 26, 1146–1154. [Google Scholar] [CrossRef]
- Donnio, B.; Buathong, S.; Bury, I.; Guillon, D. Liquid Crystalline Dendrimers. Chem. Soc. Rev. 2007, 36, 1495. [Google Scholar] [CrossRef]
- Kimura, M.; Saito, Y.; Ohta, K.; Hanabusa, K.; Shirai, H.; Kobayashi, N. Self-Organization of Supramolecular Complex Composed of Rigid Dendritic Porphyrin and Fullerene. J. Am. Chem. Soc. 2002, 124, 5274–5275. [Google Scholar] [CrossRef]
- Felder-Flesch, D.; Guillon, D.; Donnio, B. Fullerene-Containing Liquid Crystals. In Handbook of Liquid Crystals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 1–45. [Google Scholar]
- Bellani, S.; Ghadirzadeh, A.; Meda, L.; Savoini, A.; Tacca, A.; Marra, G.; Meira, R.; Morgado, J.; Di Fonzo, F.; Antognazza, M.R. Hybrid Organic/Inorganic Nanostructures for Highly Sensitive Photoelectrochemical Detection of Dissolved Oxygen in Aqueous Media. Adv. Funct. Mater. 2015, 25, 4531–4538. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Song, F.; Palkar, A.; Echegoyen, L.; Bonifazi, D.; Diederich, F. Photophysical and Electrochemical Properties Ofmeso, Meso-Linked Oligoporphyrin Rods with Appended Fullerene Terminals. ChemPhysChem 2005, 6, 732–743. [Google Scholar] [CrossRef]
- Homma, T.; Harano, K.; Isobe, H.; Nakamura, E. Nanometer-Sized Fluorous Fullerene Vesicles in Water and on Solid Surfaces. Angew. Chem. Int. Ed. 2010, 49, 1665–1668. [Google Scholar] [CrossRef]
- Cominetti, A.; Pellegrino, A.; Longo, L.; Po, R.; Tacca, A.; Carbonera, C.; Salvalaggio, M.; Baldrighi, M.; Meille, S.V. Polymer Solar Cells Based on Poly(3-Hexylthiophene) and Fullerene: Pyrene Acceptor Systems. Mater. Chem. Phys. 2015, 159, 46–55. [Google Scholar] [CrossRef]
- Cravino, A.; Zerza, G.; Neugebauer, H.; Maggini, M.; Bucella, S.; Menna, E.; Svensson, M.; Andersson, M.R.; Brabec, C.J.; Sariciftci, N.S. Electrochemical and Photophysical Properties of a Novel Polythiophene with Pendant Fulleropyrrolidine Moieties: Toward “Double Cable” Polymers for Optoelectronic Devices. J. Phys. Chem. B 2002, 106, 70–76. [Google Scholar] [CrossRef]
- Lu, F.; Nakanishi, T. Alkyl-π Engineering in State Control toward Versatile Optoelectronic Soft Materials. Sci. Technol. Adv. Mater. 2015, 16, 014805. [Google Scholar] [CrossRef]
- Nakanishi, T.; Ariga, K.; Michinobu, T.; Yoshida, K.; Takahashi, H.; Teranishi, T.; Möhwald, H.; Kurth, D.G. Cover Picture: Flower-Shaped Supramolecular Assemblies: Hierarchical Organization of a Fullerene Bearing Long Aliphatic Chains (Small 12/2007). Small 2007, 3, 1981. [Google Scholar] [CrossRef]
- Czichy, M.; Wagner, P.; Grządziel, L.; Krzywiecki, M.; Szwajca, A.; Łapkowski, M.; Żak, J.; Officer, D.L. Electrochemical and Photoelectronic Studies on C60-Pyrrolidine-Functionalised Poly(Terthiophene). Electrochim. Acta 2014, 141, 51–60. [Google Scholar] [CrossRef]
- Raja, R.; Liu, W.-S.; Hsiow, C.-Y.; Hsieh, Y.-J.; Rwei, S.-P.; Chiu, W.-Y.; Wang, L. Novel Fulleropyrrolidines Bearing π-Conjugated Thiophene Derivatives as Compatibilizing Group for Developing Highly Stable Polymer Solar Cells. Org. Electron. 2014, 15, 2223–2233. [Google Scholar] [CrossRef]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Allouche, A.-R. Gabedit-A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Moreira da Costa, L.; Stoyanov, S.R.; Gusarov, S.; Seidl, P.R.; Walkimar de M. Carneiro, J.; Kovalenko, A. Computational Study of the Effect of Dispersion Interactions on the Thermochemistry of Aggregation of Fused Polycyclic Aromatic Hydrocarbons as Model Asphaltene Compounds in Solution. J. Phys. Chem. A 2014, 118, 896–908. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Lu, L.; Hu, H.; Hou, H.; Wang, B. An Improved B3LYP Method in the Calculation of Organic Thermochemistry and Reactivity. Comput. Theor. Chem. 2013, 1015, 64–71. [Google Scholar] [CrossRef]
- Zandler, M.E.; D’Souza, F. The Remarkable Ability of B3LYP/3-21G(*) Calculations to Describe Geometry, Spectral and Electrochemical Properties of Molecular and Supramolecular Porphyrin–Fullerene Conjugates. Comptes Rendus Chim. 2006, 9, 960–981. [Google Scholar] [CrossRef]
- Popov, A.A.; Avdoshenko, S.M.; Cuniberti, G.; Dunsch, L. Dimerization of Radical-Anions: Nitride Clusterfullerenes versus Empty Fullerenes. J. Phys. Chem. Lett. 2011, 2, 1592–1600. [Google Scholar] [CrossRef]
- Lebedeva, M.A.; Chamberlain, T.W.; Davies, E.S.; Thomas, B.E.; Schröder, M.; Khlobystov, A.N. Tuning the Interactions between Electron Spins in Fullerene-Based Triad Systems. Beilstein J. Org. Chem. 2014, 10, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, K.; Fujiwara, K.; Tanaka, T.; Murata, Y. The Fullerene Dimer C120 and Related Carbon Allotropes. Carbon 2000, 38, 1529–1534. [Google Scholar] [CrossRef]
- Komatsu, K.; Fujiwara, K.; Murata, Y. The Fullerene Cross-Dimer C130: Synthesis and Properties. Chem. Commun. 2000, 1583–1584. [Google Scholar] [CrossRef]
- Balch, A.L.; Costa, D.A.; Fawcett, W.R.; Winkler, K. Electronic Communication in Fullerene Dimers. Electrochemical and Electron Paramagnetic Resonance Study of the Reduction of C120O. J. Phys. Chem. 1996, 100, 4823–4827. [Google Scholar] [CrossRef]
- Kutner, W.; Noworyta, K.; Marczak, R.; D’Souza, F. Electrochemical Quartz Crystal Microbalance Studies of Thin-Solid Films of Higher Fullerenes: C76, C78 and C84. Electrochim. Acta 2002, 47, 2371–2380. [Google Scholar] [CrossRef]
- Konarev, D.V.; Lyubovskaya, R.N.; Drichko, N.V.; Yudanova, E.I.; Shul’ga, Y.M.; Litvinov, A.L.; Semkin, V.N.; Tarasov, B.P. Donor–Acceptor Complexes of Fullerene C60 with Organic and Organometallic Donors. J. Mater. Chem. 2000, 10, 803–818. [Google Scholar] [CrossRef]
- Marumoto, K.; Takeuchi, N.; Ozaki, T.; Kuroda, S. ESR Studies of Photogenerated Polarons in Regioregular Poly(3-Alkylthiophene)–Fullerene Composite. Synth. Met. 2002, 129, 239–247. [Google Scholar] [CrossRef]
- Lobach, A.S.; Goldshleger, N.F.; Kaplunov, M.G.; Kulikov, A.V. Near-IR and ESR Studies of the Radical Anions of C60 and C70 in the System Fullerene-Primary Amine. Chem. Phys. Lett. 1995, 243, 22–28. [Google Scholar] [CrossRef]
- Tomita, S.; Andersen, J.U.; Cederquist, H.; Concina, B.; Echt, O.; Forster, J.S.; Hansen, K.; Huber, B.A.; Hvelplund, P.; Jensen, J.; et al. Lifetimes of C602− and C702− Dianions in a Storage Ring. J. Chem. Phys. 2006, 124, 024310. [Google Scholar] [CrossRef] [PubMed]
- Meletov, K.P.; Arvanitidis, J.; Christofilos, D.; Kourouklis, G.A.; Davydov, V.A. Raman Study of the Temperature-Induced Decomposition of the Fullerene Dimers C120. Chem. Phys. Lett. 2016, 654, 81–85. [Google Scholar] [CrossRef]
- Liu, H.; Gao, H.; Lin, J.; Hayat, T.; Alsaedi, A.; Tan, Z. Self-Assembled Bulk Heterojunctions from Integral Molecules with Nonconjugately Linked Donor and Acceptor Units for Photovoltaic Applications. Sustain. Energy Fuels 2020, 4, 3190–3210. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I/I’ | II/II’ | III/III’ | ||||
|---|---|---|---|---|---|---|
| C60/E1 | −0.98/−0.91 | −1.42/−1.31 | −1.97/−1.89 | |||
| 1/E1 | −1.05/−0.96 | −1.44/−1.36 | −2.01/−1.90 | |||
| A/A’ | B/B’ | C/C’ | D/D’ | E/E’ | F/F’ | |
| 2/E1 | −1.01/−0.95 | −1.10/−1.03 | −1.41/−1.36 | −1.50/−1.48 | −1.87/−1.81 | --- |
| 2/E2 | −1.03/−0.96 | −1.12/−1.08 | −1.41/−1.34 | −1.49/−1.46/ | −1.85/−1.79 | --- |
| 3/E1 | −1.08/−1.00 | −1.18/−1.12 | −1.48/−1.40 | −1.55/−1.48/ | −1.95/−1.89 | −2.01/−1.96 |
| 3/E2 | −1.10/−0.96 | −1.20/−1.16 | −1.45/−1.36 | −1.57/−1.48/ | −1.93/−1.86 | −2.05/−1.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czichy, M.; Colombo, A.; Wagner, P.; Janasik, P.; Dragonetti, C.; Raja, R.; Officer, D.L.; Wang, L. Exohedral Functionalization of Fullerene by Substituents Controlling of Molecular Organization for Spontaneous C60 Dimerization in Liquid Crystal Solutions and in a Bulk Controlled by a Potential. Polymers 2021, 13, 2816. https://doi.org/10.3390/polym13162816
Czichy M, Colombo A, Wagner P, Janasik P, Dragonetti C, Raja R, Officer DL, Wang L. Exohedral Functionalization of Fullerene by Substituents Controlling of Molecular Organization for Spontaneous C60 Dimerization in Liquid Crystal Solutions and in a Bulk Controlled by a Potential. Polymers. 2021; 13(16):2816. https://doi.org/10.3390/polym13162816
Chicago/Turabian StyleCzichy, Malgorzata, Alessia Colombo, Pawel Wagner, Patryk Janasik, Claudia Dragonetti, Rathinam Raja, David L. Officer, and Leeyih Wang. 2021. "Exohedral Functionalization of Fullerene by Substituents Controlling of Molecular Organization for Spontaneous C60 Dimerization in Liquid Crystal Solutions and in a Bulk Controlled by a Potential" Polymers 13, no. 16: 2816. https://doi.org/10.3390/polym13162816
APA StyleCzichy, M., Colombo, A., Wagner, P., Janasik, P., Dragonetti, C., Raja, R., Officer, D. L., & Wang, L. (2021). Exohedral Functionalization of Fullerene by Substituents Controlling of Molecular Organization for Spontaneous C60 Dimerization in Liquid Crystal Solutions and in a Bulk Controlled by a Potential. Polymers, 13(16), 2816. https://doi.org/10.3390/polym13162816