
Composites of a Polypropylene Random Copolymer and Date Stone Flour: Crystalline Details and Mechanical Response


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Chemical Modification of Date Stone
2.3. Preparation of Composites and Films
2.4. Scanning Electron Microscopy
2.5. X-ray Diffraction Experiments
2.6. Differential Scanning Calorimetry
2.7. Microhardness
3. Results
3.1. Scanning Electron Microscopy
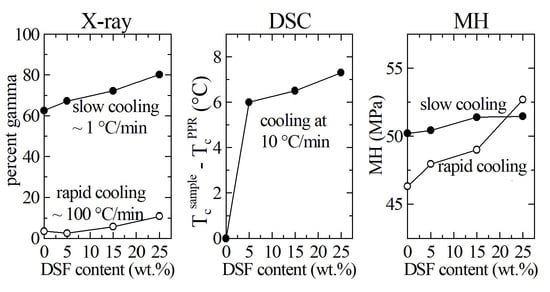
3.2. X-ray Diffraction
3.3. DSC Results
3.4. Microhardness Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- La Mantia, F.P.; Morreale, M. Green composites: A brief review. Compos. Part A 2011, 42, 579–588. [Google Scholar] [CrossRef]
- Scaffaro, R.; Maio, A.; Gulino, E.F.; Alaimo, G.; Morreale, M. Green Composites Based on PLA and Agricultural or Marine Waste Prepared by FDM. Polymers 2021, 13, 1361. [Google Scholar] [CrossRef]
- Bogoeva-Gaceva, G.; Avella, M.; Malinconico, M.; Buzarovska, A.; Grozdanov, A.; Gentile, G.; Errico, M.E. Natural fiber eco-composites. Polym. Compos. 2007, 28, 98–107. [Google Scholar] [CrossRef]
- Vaisanen, T.; Haapala, A.; Lappalainen, R.; Tomppo, L. Utilization of agricultural and forest industry waste and residues in natural fiber-polymer composites: A review. Waste Manag. 2016, 54, 62–73. [Google Scholar] [CrossRef]
- Koronis, G.; Silva, A.; Fontul, M. Green composites: A review of adequate materials for automotive applications. Compos. Part B 2013, 44, 120–127. [Google Scholar] [CrossRef]
- Ramamoorthy, S.K.; Skrifvars, M.; Persson, A. A Review of Natural Fibers Used in Biocomposites: Plant, Animal and Regenerated Cellulose Fibers. Polym. Rev. 2015, 55, 107–162. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Ramesh, M.; Palanikumar, K.; Reddy, K.H. Plant fibre based bio-composites: Sustainable and renewable green materials. Renew. Sustain. Energ. Rev. 2017, 79, 558–584. [Google Scholar] [CrossRef]
- Hamma, A.; Kaci, M.; Pegoretti, A. Polypropylene/date stone flour composites: Effects of filler contents and EBAGMA compatibilizer on morphology, thermal, and mechanical properties. J. Appl. Polym. Sci. 2013, 128, 4314–4321. [Google Scholar] [CrossRef]
- Gachter, R.; Muller, H. Plastics Additives, 3rd ed.; Gachter, R., Muller, H., Eds.; Hanser Verlag: Munich, Germany, 1990. [Google Scholar]
- Cerrada, M.L.; Benavente, R.; Pérez, E. Effect of short glass fiber on structure and mechanical behavior of an ethylene-1-octene copolymer. Macromol. Chem. Phys. 2001, 202, 2686–2695. [Google Scholar] [CrossRef]
- Serrano-Garcia, W.; Jayathilaka, W.A.D.M.; Chinnappan, A.; Tran Thang, Q.; Baskar, C.; Thomas, S.W.; Ramakrishna, S. Nanocomposites for electronic applications that can be embedded for textiles and wearables. Sci. China Technol. Sci. 2019, 62, 895–902. [Google Scholar] [CrossRef]
- Tran, T.Q.; Lee, J.K.Y.; Chinnappan, A.; Jayathilaka, W.A.D.M.; Ji, D.; Kumar, V.V.; Ramakrishna, S. Strong, lightweight, and highly conductive CNT/Au/Cu wires from sputtering and electroplating methods. J. Mater. Sci. Technol. 2020, 40, 99–106. [Google Scholar] [CrossRef]
- Alawar, A.; Hamed, A.M.; Al-Kaabi, K. Characterization of treated date palm tree fiber as composite reinforcement. Compos. Part B 2009, 40, 601–606. [Google Scholar] [CrossRef]
- Kaddami, H.; Dufresne, A.; Khelifi, B.; Bendahou, A.; Taourirte, M.; Raihane, M.; Issartel, N.; Sautereau, H.; Gerard, J.-F.; Sami, N. Short palm tree fibers—Thermoset matrices composites. Compos. Part A 2006, 37, 1413–1422. [Google Scholar] [CrossRef]
- Al-Khanbashi, A.; Al-Kaabi, K.; Hammami, A. Date palm fibers as polymeric matrix reinforcement: Fiber characterization. Polym. Compos. 2005, 26, 486–497. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Sautereau, H.; Raihane, M.; Erchiqui, F.; Dufresne, A. Short palm tree fibers polyolefin composites: Effect of filler content and coupling agent on physical properties. Macromol. Mater. Eng. 2008, 293, 140–148. [Google Scholar] [CrossRef]
- Karian, H.G. Handbook of Polypropylene and Polypropylene Composites, 2nd ed.; Karian, H.G., Ed.; Marcel Dekker: New York, NY, USA, 2009. [Google Scholar]
- Arranz-Andrés, J.; Suárez, I.; Benavente, R.; Pérez, E. Characterization and properties of ethylene-propylene copolymers synthesized with homogeneous and supported metallocene catalyst in the whole range of compositions. Macromol. Res. 2011, 19, 351–363. [Google Scholar] [CrossRef]
- Pérez, E.; Gómez-Elvira, J.M.; Benavente, R.; Cerrada, M.L. Tailoring the formation rate of the mesophase in random propylene-co-1-pentene copolymers. Macromolecules 2012, 45, 6481–6490. [Google Scholar] [CrossRef]
- Danyadi, L.; Renner, K.; Szabo, Z.; Nagy, G.; Moczo, J.; Pukanszky, B. Wood flour filled PP composites: Adhesion, deformation, failure. Polym. Adv. Technol. 2006, 17, 967–974. [Google Scholar] [CrossRef]
- Vardai, R.; Lummerstorfer, T.; Pretschuh, C.; Jerabek, M.; Gahleitner, M.; Pukanszky, B.; Renner, K. Impact modification of PP/wood composites: A new approach using hybrid fibers. Express Polym. Lett. 2019, 13, 223–234. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, H.; Njuguna, J.; Abhyankar, H. Recent Development of Flax Fibres and Their Reinforced Composites Based on Different Polymeric Matrices. Materials 2013, 6, 5171–5198. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Mamun, A.A.; Volk, J. Physical, chemical and surface properties of wheat husk, rye husk and soft wood and their polypropylene composites. Compos. Part A 2010, 41, 480–488. [Google Scholar] [CrossRef]
- Peltola, H.; Paakkonen, E.; Jetsu, P.; Heinemann, S. Wood based PLA and PP composites: Effect of fibre type and matrix polymer on fibre morphology, dispersion and composite properties. Compos. Part A 2014, 61, 13–22. [Google Scholar] [CrossRef]
- Bouza, R.; Marco, C.; Ellis, G.; Martin, Z.; Gomez, M.A.; Barral, L. Analysis of the isothermal crystallization of polypropylene/wood flour composites. J. Therm. Anal. Calorim. 2008, 94, 119–127. [Google Scholar] [CrossRef]
- Huang, L.; Wu, Q.; Li, S.; Ou, R.; Wang, Q. Toughness and crystallization enhancement in wood fiber-reinforced polypropylene composite through controlling matrix nucleation. J. Mater. Sci. 2018, 53, 6542–6551. [Google Scholar] [CrossRef]
- Shin, Y.W.; Uozumi, T.; Terano, M.; Nitta, K.H. Synthesis and characterization of ethylene-propylene random copolymers with isotactic propylene sequence. Polymer 2001, 42, 9611–9615. [Google Scholar] [CrossRef]
- Gahleitner, M.; Jaaskelainen, P.; Ratajski, E.; Paulik, C.; Reussner, J.; Wolfschwenger, J.; Neissl, W. Propylene-ethylene random copolymers: Comonomer effects on crystallinity and application properties. J. Appl. Polym. Sci. 2005, 95, 1073–1081. [Google Scholar] [CrossRef]
- Yu, L.; Wu, T.; Chen, T.; Yang, F.; Xiang, M. Polypropylene random copolymer in pipe application: Performance improvement with controlled molecular weight distribution. Thermochim. Acta 2014, 578, 43–52. [Google Scholar] [CrossRef]
- Caveda, S.; Pérez, E.; Blázquez-Blázquez, E.; Peña, B.; van Grieken, R.; Suárez, I.; Benavente, R. Influence of structure on the properties of polypropylene copolymers and terpolymers. Polym. Test. 2017, 62, 23–32. [Google Scholar] [CrossRef]
- Bodirlau, R.; Spiridon, I.; Teaca, C.A. Chemical investigation of wood tree species in temperate forest in East-Northern Romania. Bioresources 2007, 2, 41–57. [Google Scholar] [CrossRef]
- Madhu, P.; Sanjay, M.R.; Jawaid, M.; Siengchin, S.; Khan, A.; Pruncu, C.I. A new study on effect of various chemical treatments on Agave Americana fiber for composite reinforcement: Physico-chemical, thermal, mechanical and morphological properties. Polym. Test. 2020, 85, 106437. [Google Scholar] [CrossRef]
- Hong, C.K.; Hwang, I.; Kim, N.; Park, D.H.; Hwang, B.S.; Nah, C. Mechanical properties of silanized jute-polypropylene composites. J. Ind. Eng. Chem. 2008, 14, 71–76. [Google Scholar] [CrossRef]
- Mansel, S.; Pérez, E.; Benavente, R.; Pereña, J.M.; Bello, A.; Röll, W.; Kirsten, R.; Beck, S.; Brintzinger, H.H. Synthesis and properties of elastomeric poly(propylene). Macromol. Chem. Phys. 1999, 200, 1292–1297. [Google Scholar] [CrossRef][Green Version]
- Prieto, O.; Pereña, J.M.; Benavente, R.; Pérez, E.; Cerrada, M.L. Viscoelastic relaxation mechanisms of conventional polypropylene toughened by a plastomer. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1878–1888. [Google Scholar] [CrossRef]
- Krache, R.; Benavente, R.; López-Majada, J.M.; Perena, J.M.; Cerrada, M.L.; Pérez, E. Competition between α, β, and γ polymorphs in a β-nucleated metallocenic isotactic polypropylene. Macromolecules 2007, 40, 6871–6878. [Google Scholar] [CrossRef]
- Bond, E.B.; Spruiell, J.E.; Lin, J.S. A WAXD/SAXS/DSC study on the melting behavior of Ziegler-Natta and metallocene catalyzed isotactic polypropylene. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 3050–3064. [Google Scholar] [CrossRef]
- Fonseca, C.; Pereña, J.M.; Benavente, R.; Cerrada, M.L.; Bello, A.; Pérez, E. Microhardness and thermal study of the annealing effects in vinyl alcohol-ethylene copolymers. Polymer 1995, 36, 1887–1892. [Google Scholar] [CrossRef]
- Baltá-Calleja, F.J. Microhardness relating to crystalline polymers. Adv. Polym. Sci. 1985, 66, 117–148. [Google Scholar]
- Natta, G.; Corradini, P. Structure and properties of isotactic polypropylene. Stereoregul. Polym. Stereospecif. Polym. 1960, 15, 40–51. [Google Scholar] [CrossRef]
- Miller, R.L. On the existence of near-range order in isotactic polypropylenes. Polymer 1960, 1, 135–143. [Google Scholar] [CrossRef]
- Turner-Jones, A.; Aizlewood, J.M.; Beckett, D.R. Crystalline forms of isotactic polypropylene. Makromol. Chem. 1964, 75, 134–158. [Google Scholar] [CrossRef]
- Brückner, S.; Meille, S.V.; Petraccone, V.; Pirozzi, B. Polymorphism in isotactic polypropylene. Prog. Polym. Sci. 1991, 16, 361–404. [Google Scholar] [CrossRef]
- Lotz, B.; Wittmann, J.C.; Lovinger, A.J. Structure and morphology of poly(propylenes): A molecular analysis. Polymer 1996, 37, 4979–4992. [Google Scholar] [CrossRef]
- Varga, J. Supermolecular structure of isotactic polypropylene. J. Mater. Sci. 1992, 27, 2557–2579. [Google Scholar] [CrossRef]
- Polo-Corpa, M.J.; Benavente, R.; Velilla, T.; Quijada, R.; Perez, E.; Cerrada, M.L. Development of the mesomorphic phase in isotactic propene/higher alpha-olefin copolymers at intermediate comonomer content and its effect on properties. Eur. Polym. J. 2010, 46, 1345–1354. [Google Scholar] [CrossRef][Green Version]
- Poon, B.; Rogunova, M.; Hiltner, A.; Baer, E.; Chum, S.P.; Galeski, A.; Piorkowska, E. Structure and properties of homogeneous copolymers of propylene and 1-hexene. Macromolecules 2005, 38, 1232–1243. [Google Scholar] [CrossRef]
- García-Peñas, A.; Gómez-Elvira, J.M.; Barranco-García, R.; Pérez, E.; Cerrada, M.L. Trigonal δ form as a tool for tuning mechanical behavior in poly(propylene-co-1-pentene-co-1-heptene) terpolymers. Polymer 2016, 99, 112–121. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, C.; Tang, C.; Zhang, Q.; Du, R.; Fu, Q.; Li, L. Rheologically determined negative influence of increasing nucleating agent content on the crystallization of isotactic polypropylene. Polymer 2009, 50, 696–706. [Google Scholar] [CrossRef]
- Horvath, Z.; Menyhard, A.; Doshev, P.; Gahleitner, M.; Friel, D.; Varga, J.; Pukanszky, B. Improvement of the impact strength of ethylene-propylene random copolymers by nucleation. J. Appl. Polym. Sci. 2016, 133, 43823. [Google Scholar] [CrossRef]
- Hosier, I.L.; Alamo, R.G.; Esteso, P.; Isasi, J.R.; Mandelkern, L. Formation of the alpha and gamma polymorphs in random metallocene-propylene copolymers. Effect of concentration and type of comonomer. Macromolecules 2003, 36, 5623–5636. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Pérez, E.; Benavente, R.; Ressia, J.; Sarmoria, C.; Vallés, E.M. Gamma polymorph and branching formation as inductors of resistance to electron beam irradiation in metallocene isotactic polypropylene. Polym. Degrad. Stab. 2010, 95, 462–469. [Google Scholar] [CrossRef]
- Barranco-García, R.; López-Majada, J.M.; Martínez, J.C.; Gómez-Elvira, J.M.; Pérez, E.; Cerrada, M.L. Confinement of iPP crystallites within mesoporous SBA-15 channels in extruded iPP-SBA-15 nanocomposites studied by Small Angle X-ray scattering. Micropor. Mesopor. Mat. 2018, 272, 209–216. [Google Scholar] [CrossRef]
- Lorenzo, V.; Perena, J.M.; Fatou, J.M. Relationships between mechanical-properties and microhardness of polyethylenes. Angew. Makromol. Chem. 1989, 172, 25–35. [Google Scholar] [CrossRef]
- Baltá-Calleja, F.J.; Rueda, D.R.; Porter, R.S.; Mead, W.T. New aspects of the microhardness of ultraoriented polyethylene. J. Mater. Sci. 1980, 15, 765–772. [Google Scholar] [CrossRef]
- Engel, P.A.; Derwin, M.D. Indentation Test for Polymer-Film-Coated Computer Board Substrate. In Microindentation Techniques in Materials Science and Engineering; Blau, P.T., Lawn, B.R., Eds.; Special Technical Publication 889; American Society for Testing and Materials: Philadelphia, PA, USA, 1986; pp. 272–285. [Google Scholar]
- Cerrada, M.L.; Benavente, R.; Pérez, E. Crystalline structure and viscoelastic behavior in composites of a metallocenic ethylene-1-octene copolymer and glass fiber. Macromol. Chem. Phys. 2002, 203, 718–726. [Google Scholar] [CrossRef]
- López-Majada, J.M.; Palza, H.; Guevara, J.L.; Quijada, R.; Martínez, M.C.; Benavente, R.; Pereña, J.M.; Pérez, E.; Cerrada, M.L. Metallocene copolymers of propene and 1-hexene: The influence of the comonomer content and thermal history on the structure and mechanical properties. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 1253–1267. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose | Lignin | Hemicellulose | Ash | Extractable | Water |
|---|---|---|---|---|---|
| 42.6 | 16.3 | 19.2 | 2.2 | 14.8 | 4.9 |
| Sample Code | Composition (wt.%) | ||
|---|---|---|---|
| PPR | DSF | Millad | |
| PPR | 100 | 0 | 0 |
| PPR-D5 | 95 | 5 | 0 |
| PPR-D15 | 85 | 15 | 0 |
| PPR-D25 | 75 | 25 | 0 |
| PPRN | 99.8 | 0 | 0.2 |
| PPRN-D5 | 94.8 | 5 | 0.2 |
| PPRN-D15 | 84.8 | 15 | 0.2 |
| PPRN-D25 | 74.8 | 25 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benarab, A.; Blázquez-Blázquez, E.; Krache, R.; Benavente, R.; Cerrada, M.L.; Pérez, E. Composites of a Polypropylene Random Copolymer and Date Stone Flour: Crystalline Details and Mechanical Response. Polymers 2021, 13, 2957. https://doi.org/10.3390/polym13172957
Benarab A, Blázquez-Blázquez E, Krache R, Benavente R, Cerrada ML, Pérez E. Composites of a Polypropylene Random Copolymer and Date Stone Flour: Crystalline Details and Mechanical Response. Polymers. 2021; 13(17):2957. https://doi.org/10.3390/polym13172957
Chicago/Turabian StyleBenarab, Amina, Enrique Blázquez-Blázquez, Rachida Krache, Rosario Benavente, María L. Cerrada, and Ernesto Pérez. 2021. "Composites of a Polypropylene Random Copolymer and Date Stone Flour: Crystalline Details and Mechanical Response" Polymers 13, no. 17: 2957. https://doi.org/10.3390/polym13172957
APA StyleBenarab, A., Blázquez-Blázquez, E., Krache, R., Benavente, R., Cerrada, M. L., & Pérez, E. (2021). Composites of a Polypropylene Random Copolymer and Date Stone Flour: Crystalline Details and Mechanical Response. Polymers, 13(17), 2957. https://doi.org/10.3390/polym13172957