1. Introduction
Polyimides (PI) are an essential class of polymers with a set of valuable properties. In addition to heat, thermal, radiation, and chemical resistance, good film-forming properties, high mechanical strength, and excellent insulating properties, polyimides demonstrate good biocompatibility in vitro and in vivo, as well as low toxicity [
1,
2,
3]. Multicomponent copolymers, in which PI blocks are combined with blocks of biocompatible aliphatic polymers, have great potential for the development of materials for tissue engineering.
Although atom transfer radical polymerization (ATRP) has been successfully used to synthesize many types of copolymers, it remains a challenge to synthesize copolymers of different architectures using monomers that polymerize through fundamentally different mechanisms, such as ATRP and ring opening polymerization (ROP). To solve this problem, one can use initiators containing two types of functional groups capable of initiating each of these processes in parallel and independently [
4,
5]. Second approach is introduction of a functional group that initiates the polymerization of the second monomer at the stage of initiation or termination of the first monomer polymerization (post-modification) [
6].
Thus, in [
7], graft copolymers with poly(2-hydroxyethyl methacrylate) (PHEMA) backbone and block copolymer side chains containing blocks of poly(ε-caprolactone) PCL and poly(butyl acrylate) (PBA) were synthesized. Copolymers of PCL with poly(octadecyl methacrylate) (POMA) and poly(N,N-dimethylamino-2-ethyl methacrylate) (PDMAEMA) PCL-
b-PODMA-
b-PDMAEMA or PCL-
b-(PODMA-
co-PDMAEMA) were obtained in [
5], in which the PODMA block had a "pseudo-brush" structure due to long aliphatic tails. Another example of a successful combination of the different methods of controlled polymerization is the synthesis of a three-armed star-shaped polymer with arms of different structures, for which different methods were used [
4]. Thus, the PCL arm was obtained using ROP, the polystyrene (PS) arm was obtained by the NMP, and the poly(tert-butyl acrylate) (PTBA) was obtained by the ATRP.
The ROP is effectively combined with other CRP methods in the synthesis of various copolymers. Thus, in [
8], the authors describe the preparation of triblock copolymers with a central PCL block and peripheral PDMAEMA blocks using a combination of ROP with RAFT. Synthesized by ROP PCL with hydroxyl end groups was functionalized using 4-cyanopentanoic acid dithionaphthalenoate (CPADN) and used as a macro-RAFT agent for polymerization of DMAEMA to obtain the targeted triblock copolymers. An efficient method for the synthesis of linear di- and triblock copolymers with PCL blocks with narrow molecular weight distribution is to carry out ROP on mono- or difunctional polymer initiators. ROP of CL on the corresponding polymer initiators with hydroxyl chain ends provides copolymers in which PCL blocks are covalently attached to a block of polydimethylsiloxane [
9], polyisobutylene [
10], and poly(ethylene oxide) [
11].
Grafting of polyester chains with narrow molecular weight distribution, in particular, PCL for the production of molecular brushes is also of great interest. The method of click chemistry is often used to graft PCL chains [
12,
13]. For example, in [
12], the surface modification of the nanodispersed cellulose systems by grafting PCL chains using click-chemistry is described. However, to obtain molecular brushes with polyester side chains, ROP is easier to perform [
14]. Therefore, numerous articles are devoted to the synthesis of molecular brushes with homopolymer PCL side chains or block copolymer side chains with a PCL block [
12,
13]. Carrying out polymerization of CL on multifunctional polymer initiators with varying numbers and positions of initiating groups leads to the production of PCL blocks with narrow molecular weight distribution, which are usually grafted to the carbochain backbone. To our knowledge, there are no data on the introduction of PCL blocks into the side chains of molecular brushes with a polyarylene backbone.
At the same time, works on combining aromatic PIs with aliphatic polyesters, in particular with PCL, have been going on for more than two decades. With simple mixing of homopolymer PI and PCL, for example, through a common solvent, phase separation of polymers is observed at the macroscopic level [
15]. A more uniform phase system of PI and PCL is obtained by synthesizing chemically bonded PI and PCL blocks [
15,
16,
17,
18,
19,
20]. Until recently, the covalent bonding of these polymers was carried out either by stepwise polycondensation, leading to the production of linear alternative segmented (or multisegmented) block copolymers [
18,
19], or by cross-linking between PI and PCL chains in a polymer mixture or between PI and PCL layers in a multilayer coating [
16,
17,
20]. A valuable feature of such copolymers is microphase separation [
17], which gives them new interesting, for example, membrane properties. Thus, based on multisegmented linear block copolymers containing segments of polyurethanimide and PCL, effective diffusion membranes have been developed for the separation of binary mixtures of organic liquids [
17]. However, the preparation of such segmented block copolymers is very laborious. Another method for combining PI and PCL by preparing block copolymers with sequentially attached PI and PCL blocks was proposed in [
21]. In [
22] the synthesis of linear block copolyimide with PCL and PI blocks based on 3,3’-dioxybenzidine was proposed using ROP of CL on a difunctional polyimide initiator. The obtained copolyimide was intended to improve the dispersion of carbon nanotubes in low-boiling organic solvents.
It should be noted that the introduction of PCL blocks into multiblock copolymers is of considerable interest from the point of view of further applications of these copolymers, since PCL blocks are capable of undergoing alkaline [
23] and plasma [
24] etching and biodegradation. The synthesis of new initiators and ROP of monomers and macromonomers containing functional groups on such initiators is a promising strategy for the preparation of macromolecules of complex architecture. In recent years, due to the development of the ROP method, it has been successfully used to obtain copolymers of different chemical structures and architecture.
Earlier, our research group reported the synthesis of triblock copolymers based on polyimide with external blocks of poly(ε-caprolactone) (PCL) [
25], as well as grafted pentablock copolymers of linear-brush topology with external PCL and PMMA blocks and an internal brush-type block PI-
g-PMMA [
26]. This work is devoted to the development of methods for the synthesis of previously undescribed molecular brushes with a PI backbone and PCL side chains. To obtain such copolymers, a combination of various synthesis methods was used, including polycondensation, ATRP, ROP, and Cu (I) catalyzed azide-alkyne Huisgen cycloaddition (CuAAC) [
27].
2. Materials and Methods
2.1. Materials
N-methyl-2-pyrrolidone (N-MP, 98%, Aldrich, St. Louis, MO, USA), toluene (analytical grade) methylene chloride, chloroform, THF, DMF (all reagent grade, Vekton, Voronezh, Russia), trimethylamine (≥99%, Aldrich, Overijse, Belgium), and pyridine (99%, Acros Organics, NJ, USA) were purified through standard techniques. 3,3′,4,4′-(1,3-diphenoxybenzene)tetracarboxylic dianhydride (99%, Ambinter Stock Screening Collections, China), 2,4-diaminophenol dihydrochloride (98%, Lancaster, Eastgate, White Lund, Morecambe, England), and potassium iodide (≥99.5%, Aldrich, St. Louis, MO, USA) were dried at elevated temperature in a vacuum prior to synthesis. 2-Hydroxyethyl methacrylate (HEMA) (99%, Acros Organics, UK) and ε-caprolactone (99%, Aldrich, St. Louis, MO, USA) were distilled twice under vacuum. Copper(I) chloride (99%, Aldrich, St. Louis, MO, USA) was stirred in glacial acetic acid overnight, filtered, and washed with argon-purged methanol (reagent grade, Vekton, Russia). Sn(II) 2-ethyl hexanoate (Sn(EH)2) (~95%, Aldrich, Japan) was distilled under vacuum. 2-Bromoisobutyryl bromide (98%, Aldrich, St. Louis, MO, USA), 2,2’-bipyridine (bpy) (≥99%, Aldrich, St. Louis, MO, USA), sodium azide (NaN3) (≥99.5%, Aldrich, St. Louis, MO, USA), propargyl alcohol (99%, Aldrich, Steinheim, Germany), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (99%, Aldrich, Steinheim, Germany), copper(I) bromide (CuBr) (98%, Aldrich, St. Louis, MO, USA), and LiBr (≥99%, Aldrich, St. Louis, MO, USA) were used without additional purification.
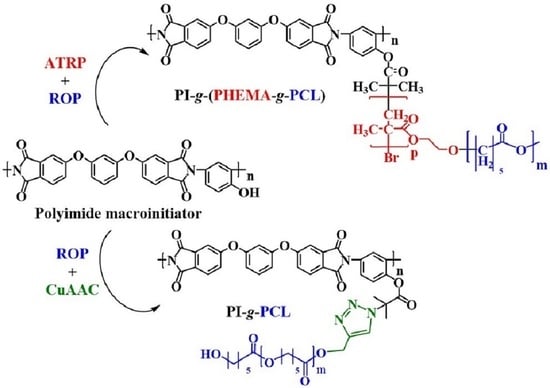
2.2. Synthesis of PI-g-PHEMA as Branched Multicenter Macroinitiator with Hydroxyl Groups in Each Repeating Unit of the Side Chains of the Grafted Copolyimide
The polyimide multifunctional macroinitiator with initiating groups in almost every monomer unit was obtained by polycondensation of dianhydride of 3,3′,4,4′-(1,3-diphenoxybenzene)-tetracarboxylic acid and 2,4-diaminophenol, its phenol groups being further modified by 2-bromoisobutyryl bromide. The initial PI1 and the related PI2 multifunctional macroinitiator (Figure 1, see Result and Discussion section) were synthesized using procedures described in our previous publications [
28,
29,
30].
To obtain a brush-type macroinitiator with hydroxyl groups in the side chains, graft copolyimide PI-g-PHEMA was synthesized by polymerization of 2-hydroxyethyl methacrylate (HEMA) on PI2 macroinitiator (Figure 1) by the ATRP. A typical synthesis procedure was as follows. A weighed portion of PI2 (0.075 g, 0.11 mmol per initiating group) and 2,2′-bipyridine (0.052 g, 0.33 mmol) was placed into a 25 mL Schlenk flask equipped with a magnetic stirrer, then DMF (16.5 mL, 0.21 mol) and HEMA (3.65 mL, 0.03 mol) were added with the syringe. The solvent, monomer, and syringes were purged with argon. The flask was sealed with a rubber septum, and the mixture was stirred until the powder was completely dissolved. Then, three freeze−pump−thaw cycles were carried out (evacuation for 15 min), after which the flask was filled with argon. After opening the septum in a stream of argon, CuCl (0.011 g, 0.11 mmol) was added to the reaction mixture, after which the flask was closed again with the septum. Three more freeze−pump−thaw cycles (evacuation for 15 min) of the reaction mixture were carried out. The flask was filled with argon and thermostated in an oil bath, and placed on a magnetic stirrer with a temperature controller, at 30 °C.
After a given reaction time, the reaction mixture was quickly cooled to room temperature and diluted two times with THF. To remove copper salts from the mixture, reaction solution was passed through a column filled with Al2O3, then concentrated using a rotary evaporator, and the polymerization product was precipitated into a water/methanol mixture with a volume ratio of 1/6. The filtered powder was dried under vacuum at 50 °C.
2.3. Synthesis of Graft Copolymers PI-g-(PHEMA-g-PCL)
The synthesis of the copolymer was carried out by ROP of CL in bulk on PI-g-PHEMA. A typical synthesis procedure is as follows: 0.05 g (0.23 mmol per initiating group) of PI-g-PHEMA was added to a 25-mL Schlenk flask equipped with a magnetic stirrer, and three freeze−pump−thaw cycles were carried out (evacuation for 15 min), after which the flask was filled with argon. Then, 1.2 mL (0.011 mol) of ε-caprolactone was introduced in an argon flow and thermostated at 130 °C using an oil bath. After complete dissolution of the macroinitiator, 0.05 mL (0.15 mmol) of Sn(Oct)2 was introduced into the reaction in an argon flow. Upon completion of polymerization, the reaction mixture was cooled to room temperature and diluted with methylene chloride. The resulting solution was passed through a silica gel column to purify the product from catalyst and monomer impurities. The solution was then concentrated on a rotary evaporator and the product was precipitated into cooled petroleum ether. The polymer was dried under vacuum at 30 °C.
2.4. Introduction of Azide Groups into Polyimide
The functionalization of the polyimide with azide groups was carried out as follows. A weighed portion of PI2 macroinitiator (0.5 g, 0.66 mmol per initiating group) (Figure 1) and DMF (14.4 mL, 0.19 mol) was placed in a 25-mL round-bottom flask equipped with a magnetic stirrer, and the mixture was stirred until the powder was completely dissolved. Then NaN3 (0.214 g, 3.3 mmol) was added to the flask and purged with argon for 30 min. The mixture was stirred on a magnetic stirrer at room temperature overnight. The reacted polymer was precipitated into methanol. The filtered powder was dried under vacuum at 50 °C.
2.5. Introduction of Distant Hydroxyl Groups to Polyimide
To obtain PI4 initiator (Figure 8, see Result and Discussion section), an azide-alkyne cycloaddition was carried out between the azide groups of PI3 (Figure 8) and the acetylene groups of propargyl alcohol. The typical synthesis was as follows. A sample of PI3 macroinitiator (0.25 g, 0.32 mmol per initiating group) (Figure 8) was placed in a 25 mL Schlenk flask equipped with a magnetic stirrer. Propargyl alcohol (92.9 μL, 1.6 mmol) and PMDETA (66.7 μL, 0.32 mmol) were added, and then DMF (4 mL, 0.052 mol) was added using a syringe. The solvent and syringes were purged with argon. The flask was sealed with a rubber septum, and the mixture was stirred until the powder was completely dissolved. Then, three freeze−pump−thaw cycles (evacuation for 15 min) were carried out, after which the flask was filled with argon. After opening the septum, CuBr (0.0457 g, 0.32 mmol) was added to the reaction mixture in an argon flow, after which the flask was closed again with the septum, three more freeze−pump−thaw cycles (evacuation for 15 min) of the reaction mixture were carried out, the flask was filled with argon and thermostated in an oil bath, placed on a magnetic stirrer with a temperature regulator at 50 °C overnight. Then the reaction mixture was cooled to room temperature and the product was precipitated into methanol. Residual copper was disposed of by changing the precipitant and reprecipitation from DMF. The filtered powder was dried under vacuum at 50 °C.
2.6. Synthesis of Linear Homopolymer PCL with Terminal Alkyne Groups
The synthesis was carried out by ROP of CL in a solution in toluene using propargyl alcohol as an initiator. A typical synthesis procedure is as follows: Propargyl alcohol (51 μL, 0.89 mmol), ε-caprolactone (1.97 mL, 17.8 mmol), and toluene (2.52 mL, 23.7 mmol) were added to a 10 mL Schlenk flask equipped with a magnetic stirrer. Then three freeze−pump−thaw cycles (evacuation for 15 min) were carried out, after which the flask was filled with argon. Then, 0.08 mL (0.25 mmol) of Sn(Oct)2 was introduced in an argon flow into the reaction, after which the flask was closed again with a septum, three more freeze−pump−thaw cycles (evacuation for 15 min) of the reaction mixture were carried out, the flask was filled with argon and thermostated in the oil bath, placed on a magnetic stirrer with a temperature controller, at 100 °C for a specified time (3 h) and at 80 °C overnight. Upon completion of polymerization, the reaction mixture was cooled to room temperature and diluted with methylene chloride. The resulting solution was passed through a silica gel column to purify the product from catalyst and monomer impurities. The solution was then concentrated on a rotary evaporator and the product was precipitated into cooled petroleum ether. The polymer was dried under vacuum at 30 °C.
2.7. Synthesis of Grafted Copolyimides PI-g-PCL
To obtain the targeted copolymers, two approaches were used. The first approach consisted in ROP of CL on a macroinitiator with distant hydroxyl groups (“graft from”). A typical experiment was as follows. A 10 mL Schlenk flask equipped with a magnetic stirrer was charged with 0.05 g (0.054 mmol per initiating group) of PI4 macroinitiator (Figure 8), sealed with a rubber septum, and then 3 mL (26.9 mmol) of ε-caprolactone was introduced in an argon flow. The mixture was thermostated at 130 °C in an oil bath. After complete dissolution of the initiator, 0.123 mL (0.38 mmol) of Sn(Oct)2 was introduced into the flask in an argon flow, and the reaction mixture was thermostated for a preset time. The molar ratio of PI4/CL was 1/500. The amount of Sn(Oct)2 was 5 wt.% in relation to the monomer. Upon completion of polymerization, the reaction mixture was rapidly cooled to room temperature and diluted with methylene chloride. The resulting solution was passed through a silica gel column to purify the product from catalyst and monomer impurities. The solution was then concentrated using a rotary evaporator and the product was precipitated into cooled petroleum ether. The polymer was dried under vacuum at 30 °C.
The second approach was to carry out an azide-alkyne cycloaddition between the azide groups PI3 macroinitiator (Figure 8) and the alkyne groups of linear PCL. A typical reaction was carried out as follows. A weighed portion of PI3 macroinitiator (0.05 g, 0.064 mmol per initiating group) (Figure 8), PCL (0.447 g, 3.9 mmol per initiating group), and PMDETA (13 μL, 0.064 mmol) was placed in a 25-mL Schlenk flask equipped with a magnetic stirrer, then DMF (3.7 mL, 47.8 mmol) was added using a syringe. The solvent and syringes were purged with argon. The flask was sealed with a rubber septum, and the mixture was stirred until the powder was completely dissolved. Then, three freeze−pump−thaw cycles (evacuation for 15 min) were carried out, after which the flask was filled with argon. After opening the septum, CuBr (0.009 g, 0.064 mmol) was added to the reaction mixture in an argon flow, after which the flask was closed again with the septum, three more freeze−pump−thaw cycles (evacuation for 15 min) of the reaction mixture were carried out, the flask was filled with argon and thermostated in an oil bath placed on a magnetic stirrer with a temperature regulator, at 50 °C overnight. After that the reaction mixture was quickly cooled to room temperature and diluted two times with THF. To remove copper salts from the mixture, it was passed through a column filled with Al2O3, then concentrated using a rotary evaporator, and precipitated into cooled petroleum ether. The filtered powder was dried under vacuum at 30 °C.
2.8. Methods
1H NMR spectra were recorded on Bruker AC-400 (400.1 MHz), with the signals of the solvent (DMSO-d6, CDCl3) as the reference.
IR spectra were recorded using a Shimadzu IR Affinity-1S spectrophotometer in the ATR mode (multiple attenuated total internal reflection) with a 4 cm−1 resolution and 30 scans.
Molecular weights and dispersities (Ð) of the samples were estimated by size exclusion chromatography (SEC) on Agilent-1260 Infinity fitted with differential refractive index (RID), light scattering (LS), and viscometry (VS) detectors equipped with two Agilent PLgel MIXED-C columns (7.5 × 300 mm, 5 µm), 1 × 43 PLgel 5 M guard column (50 × 7.5 mm), and autosampler. The analysis was carried out at 50 °C using 0.1 M LiBr in DMF as eluent at a flow rate of 1.0 mL min−1. The salt was added to suppress the aggregation of macromolecules. For triple detection analysis the system was calibrated using PMMA standard with Mp = 2.16 × 103 g mol−1 with dn/dc=0.052. Before analysis, all samples were passed through a 0.45 µm Nylon filters. SEC data were analyzed using Agilent GPC/SEC Software version 1.2.
Thermogravimetric analysis (TGA) of obtained copolymers was performed using a TG 209 F1 Libra analyzer (NETZSCH, Germany) in the temperature range of 25–800 °C and heating rate of 10 deg min–1 under nitrogen. The weight of samples was 2–3 mg.
Melting temperatures and enthalpies of synthesized copolymers were determined by differential scanning calorimetry (DSC) using a DSC 204 F1 Phoenix device (NETZSCH, Germany) at a heating rate of 10 deg min–1 in the range from −30 to +120 °C under nitrogen. The weight of samples was 2.5–3.5 mg.
The wide-angle X-ray scattering (WAXS) was carried out on an upgraded DRON 2.0 diffractometer (Saint-Petersburg, Russia) with CuKα radiation with a 0.154 nm wavelength.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}