
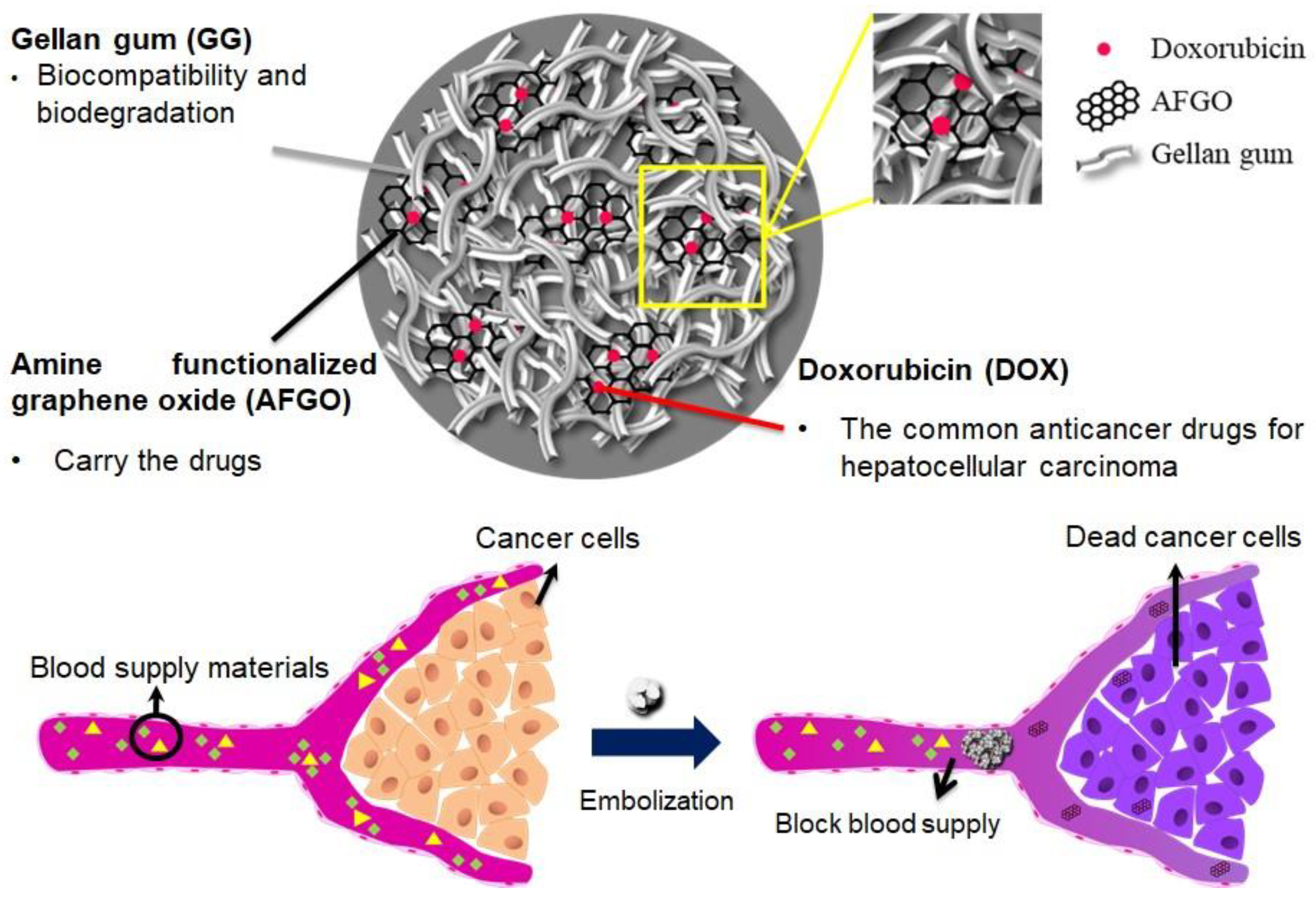
A Four-Step Cascade Drug-Release Management Strategy for Transcatheter Arterial Chemoembolization (TACE) Therapeutic Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
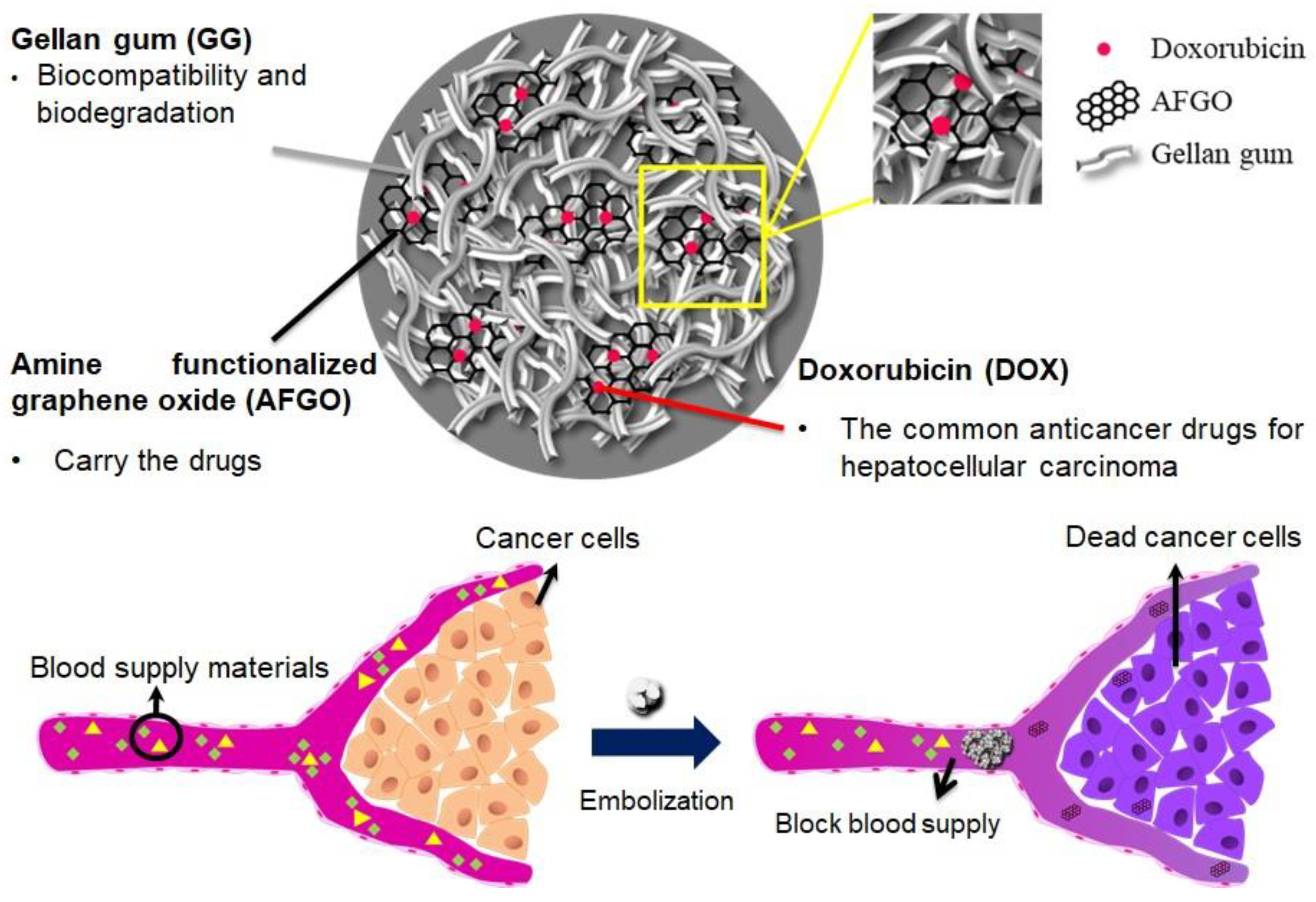
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Amine-Functionalized Graphene Oxide (AFGO) Synthesis
2.3. Preparation of Gellan Gum (GG), GG/AFGO and GG/AFGO–Dox Microsphere
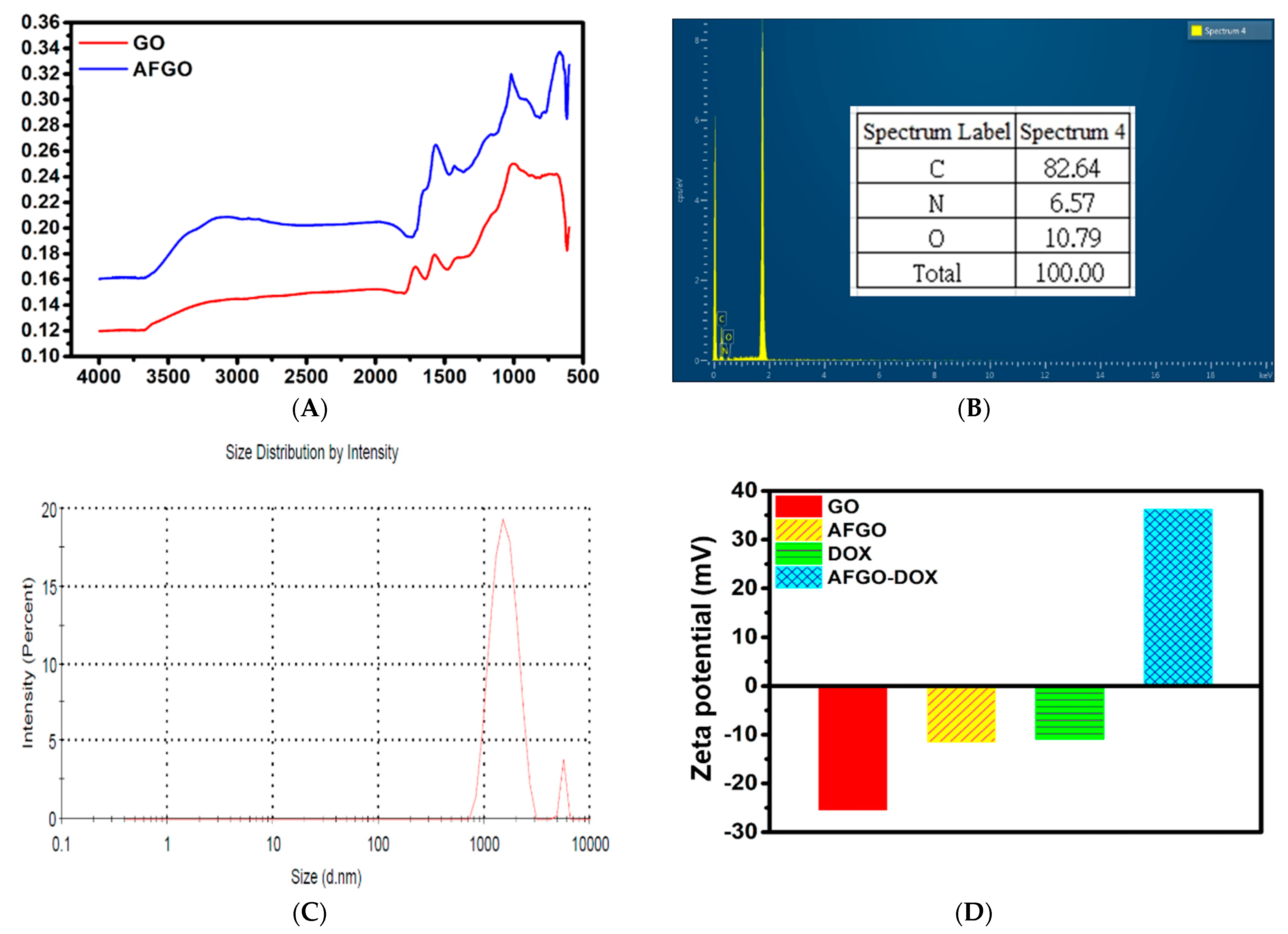
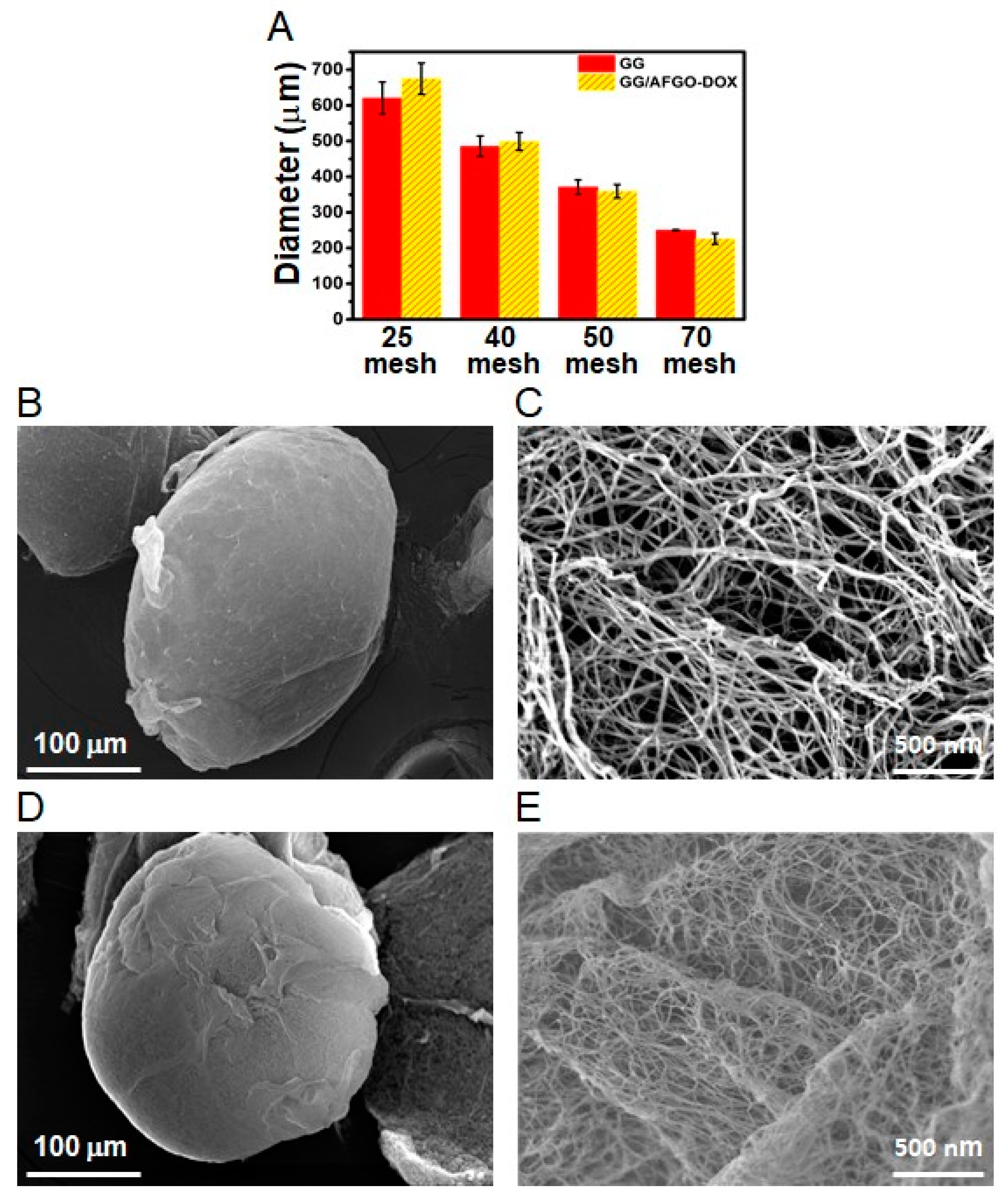
2.4. Characterization of GO, AFGO, GG/AFGO Microsphere and GG/AFGO–Dox Microsphere
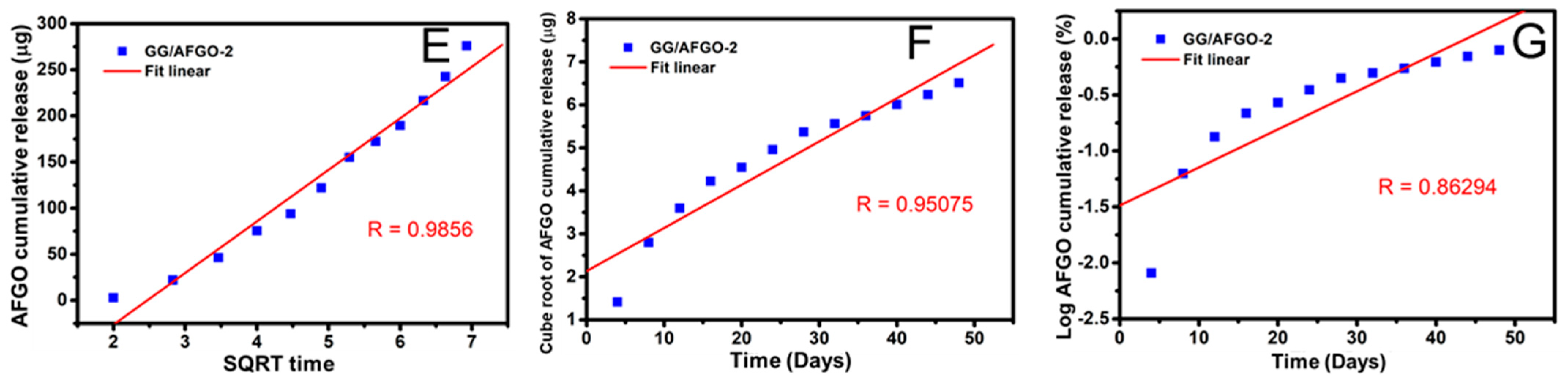
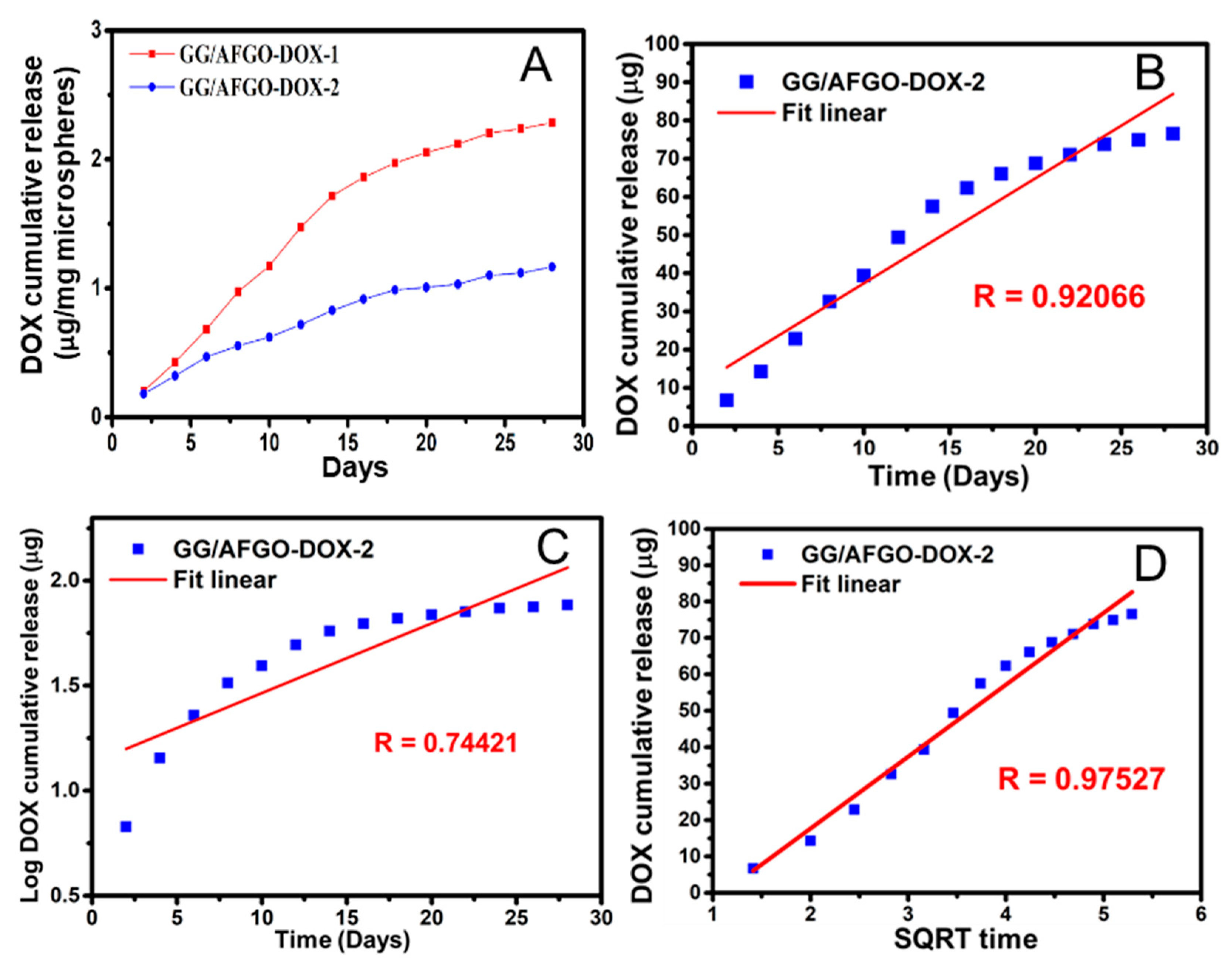
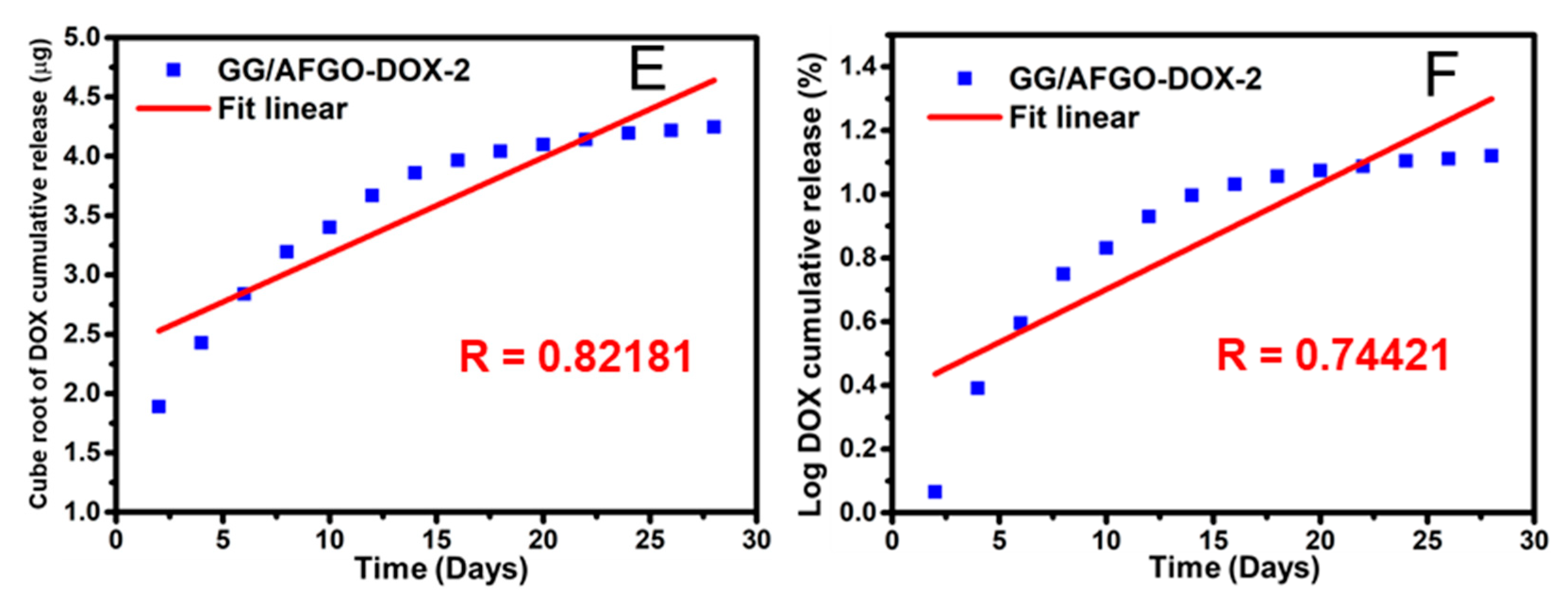
2.5. Release and Kinetic Model of AFGO and DOX in GG/AFGO–Dox Microsphere
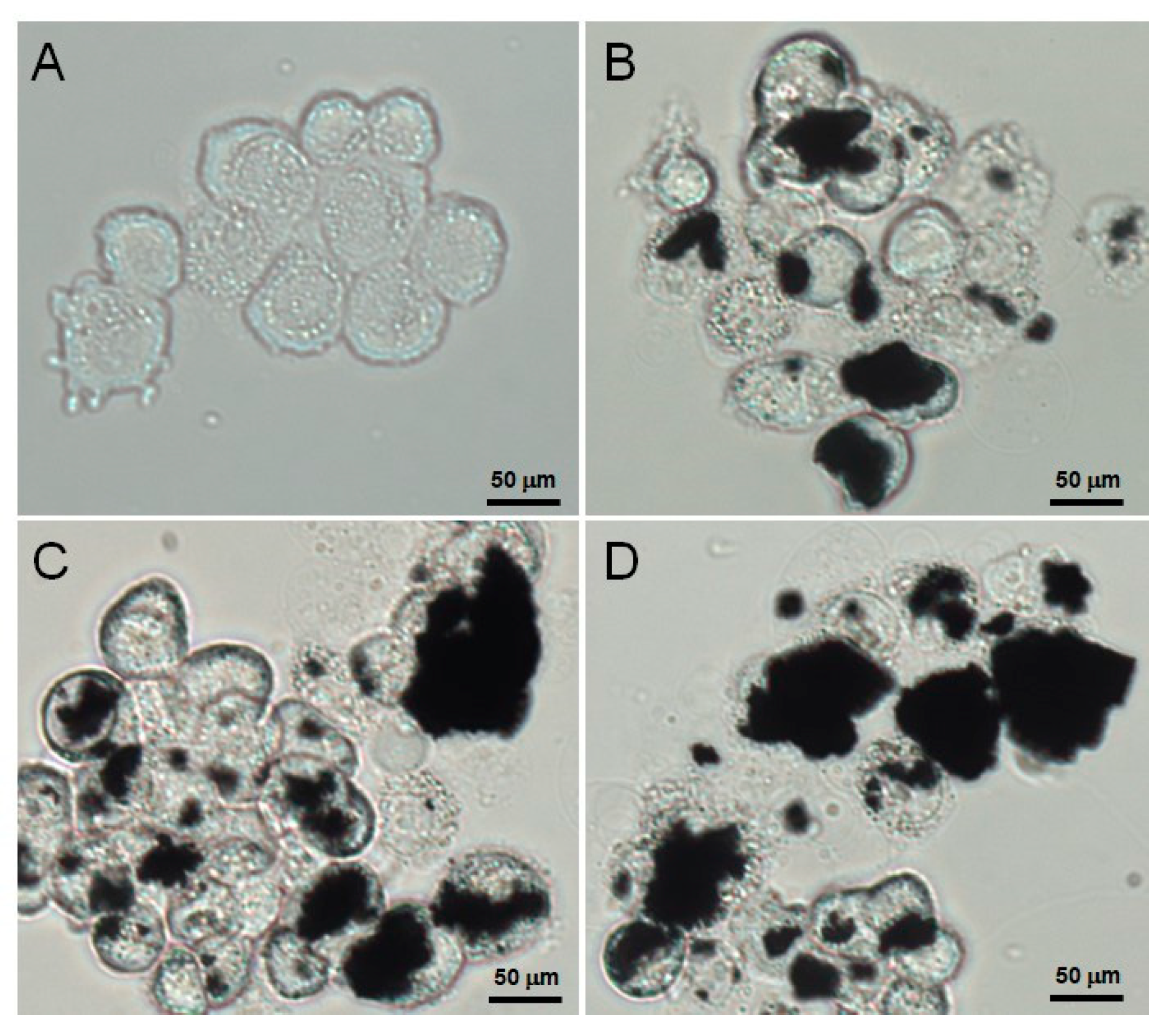
2.6. HepG2 Uptake of AFGO
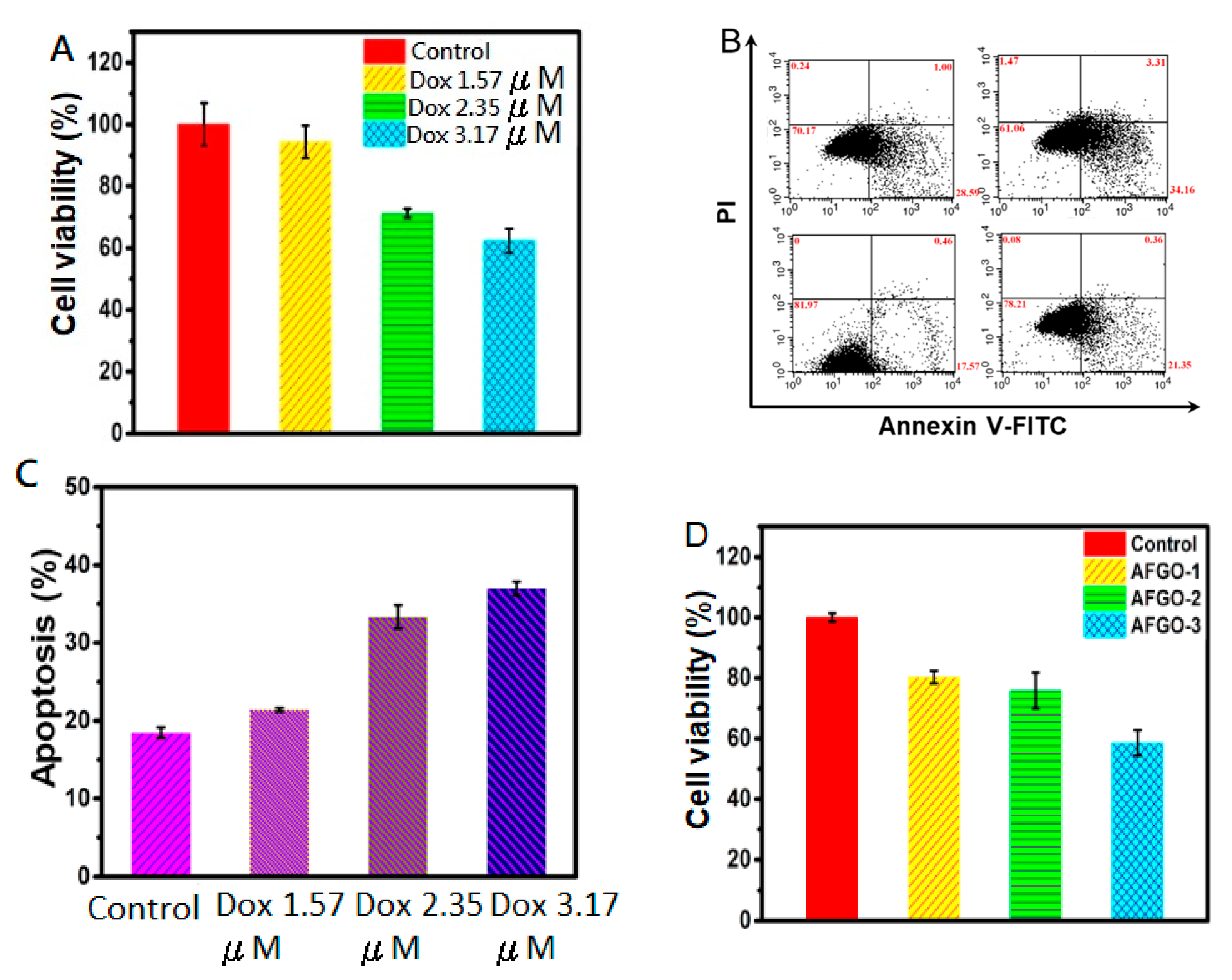
2.7. In Vitro Cell Viability and Apoptosis Assay
2.8. Vessel Embolization of Microsphere In Vivo
2.9. Statistical Analysis
3. Results and Discussion
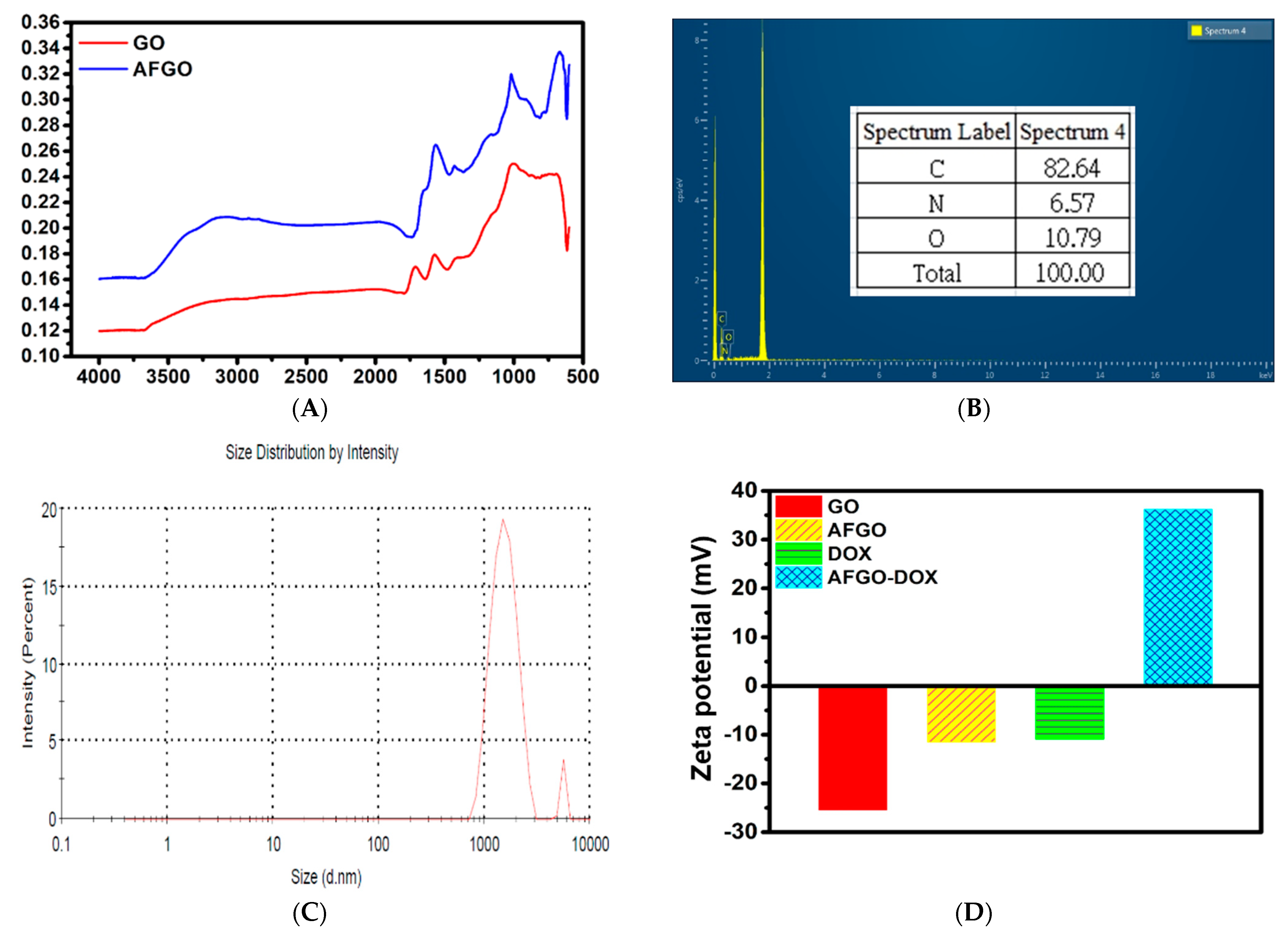
3.1. Synthesis and Characterization of Amine-Functionalized Graphene Oxide (AFGO) and Amine-Functionalized Graphene Oxide–Doxorubicin (AFGO–Dox)
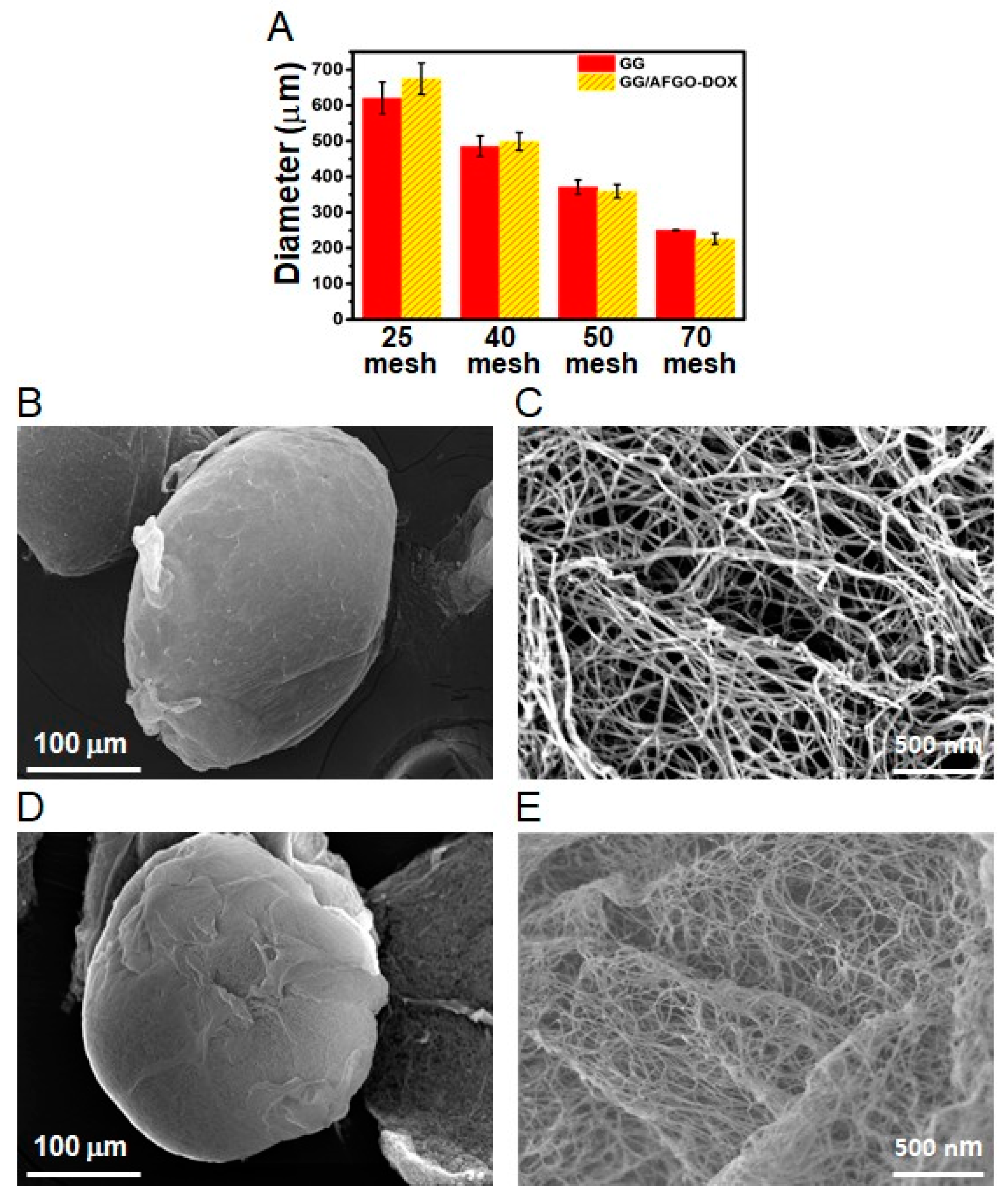
3.2. Synthesis and Characterization of GG and GG/AFGO–Dox Microspheres
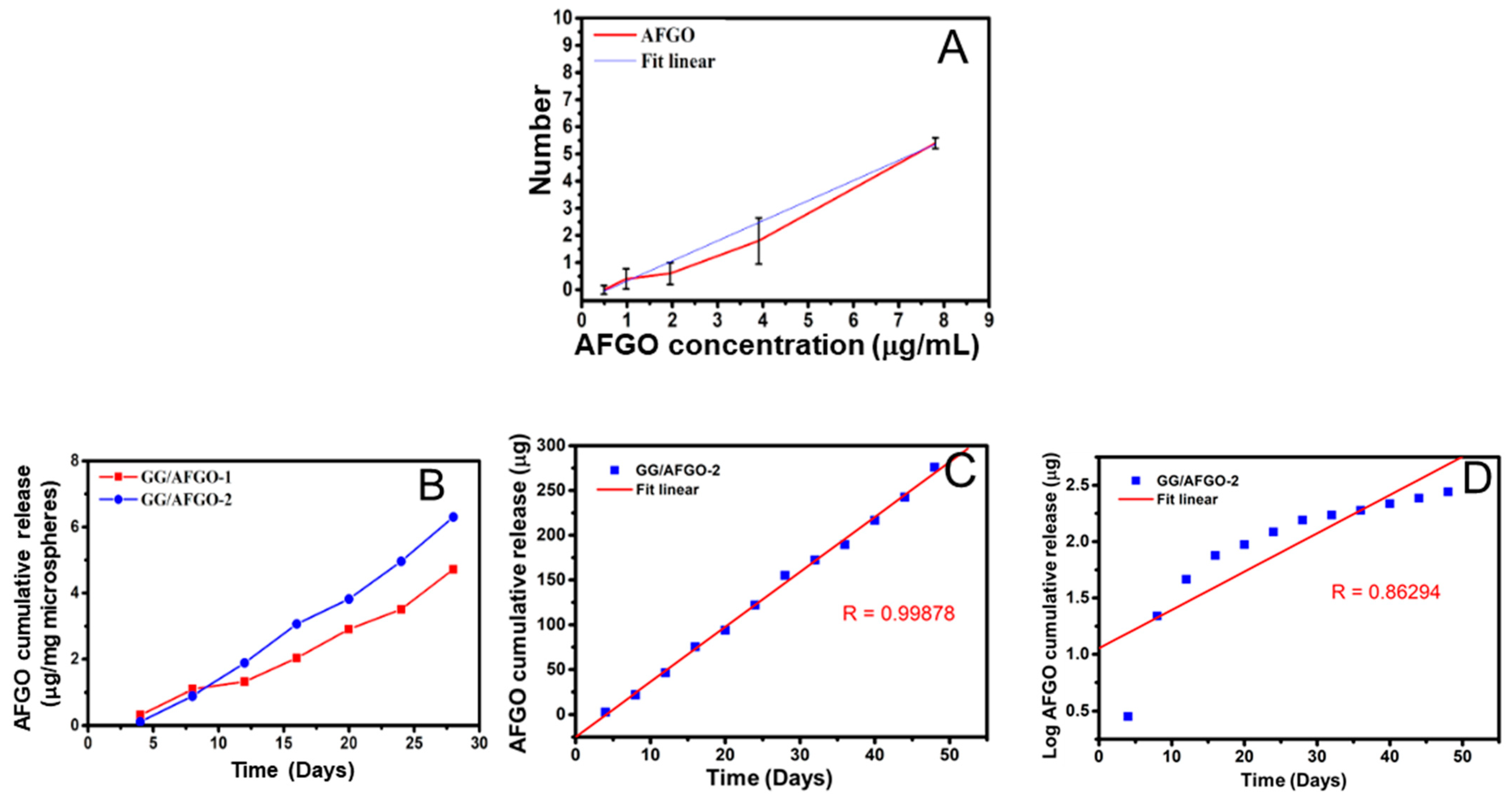
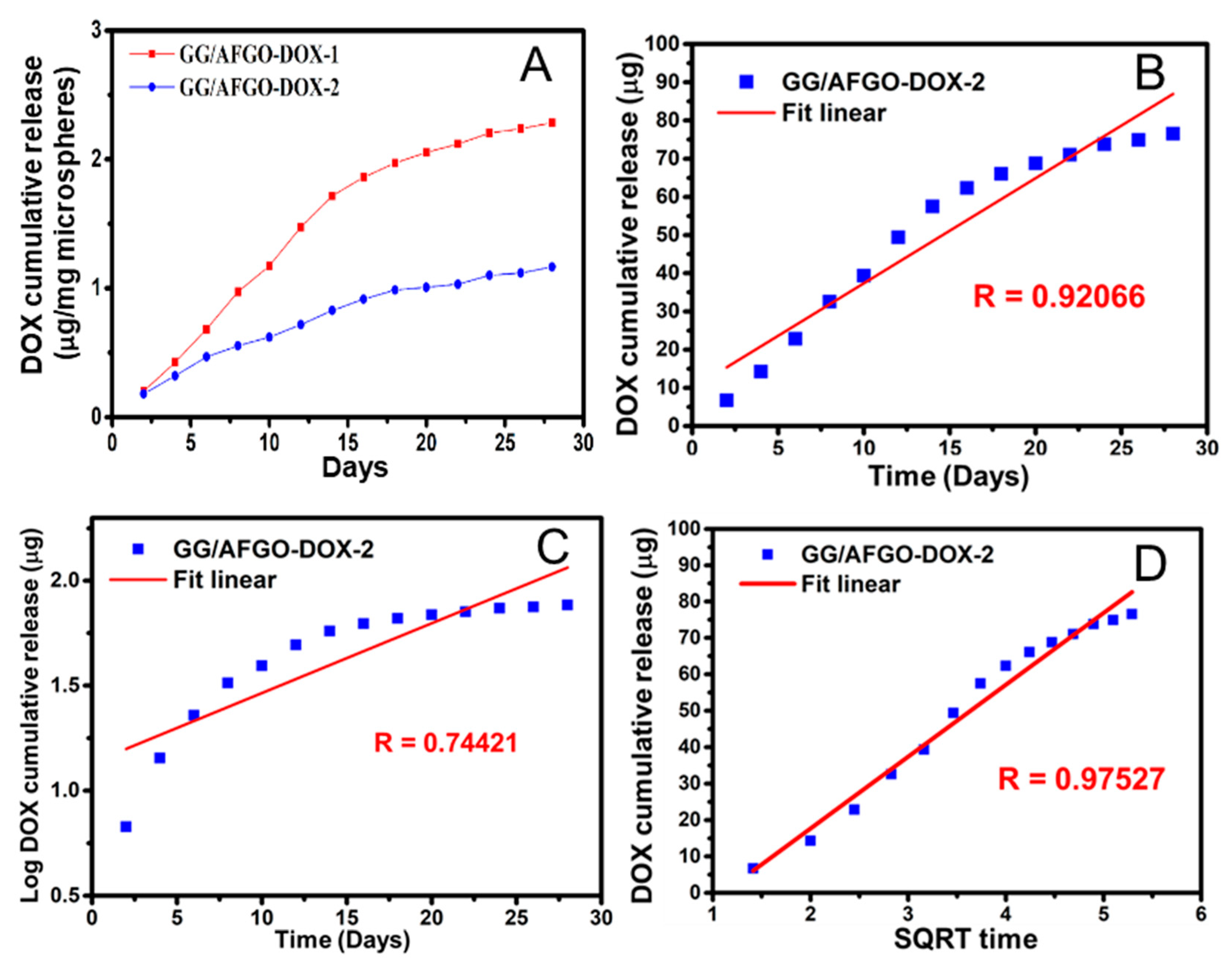
3.3. AFGO and Dox Release and Kinetic Model in GG/AFGO–Dox Microspheres
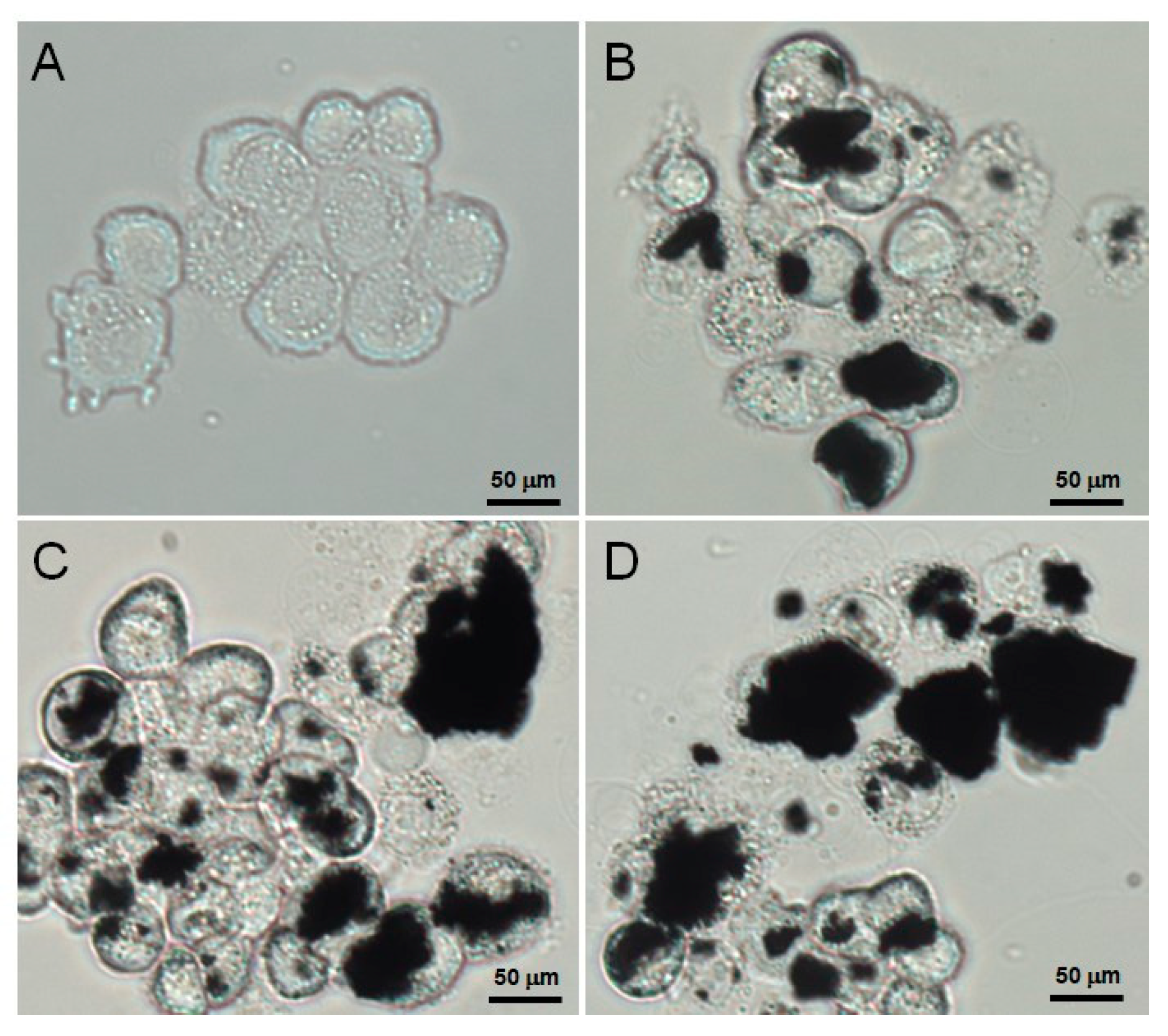
3.4. Verification of the Cellular Uptake of AFGO
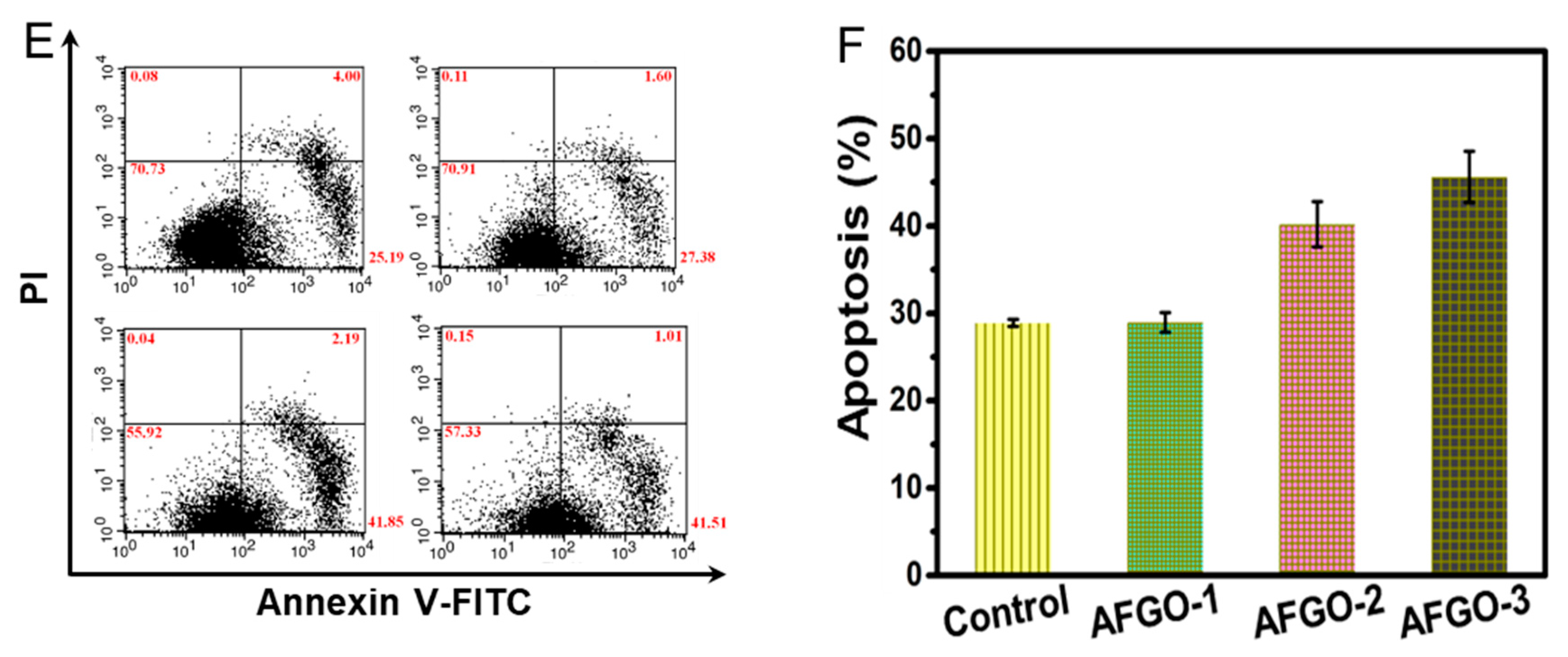
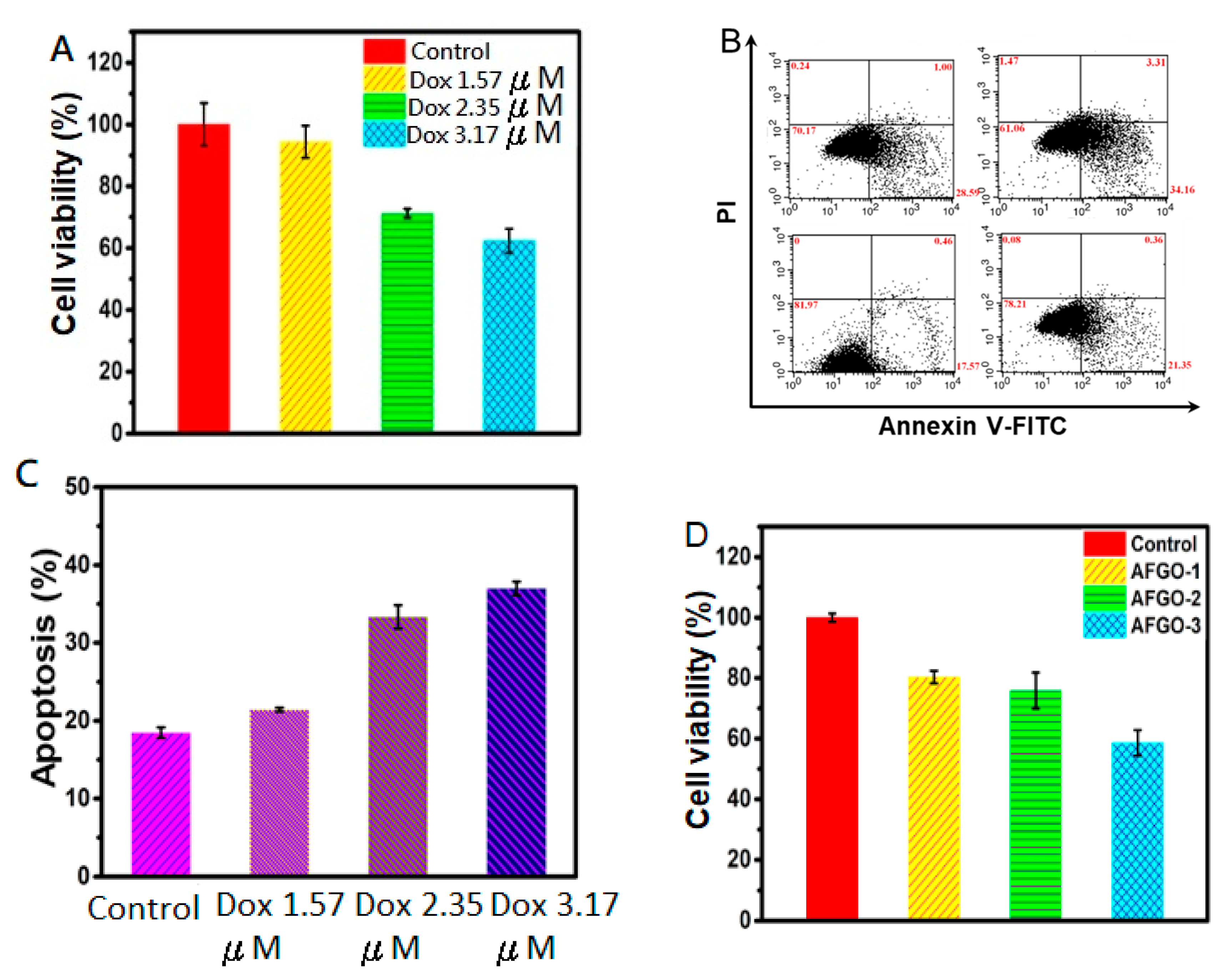
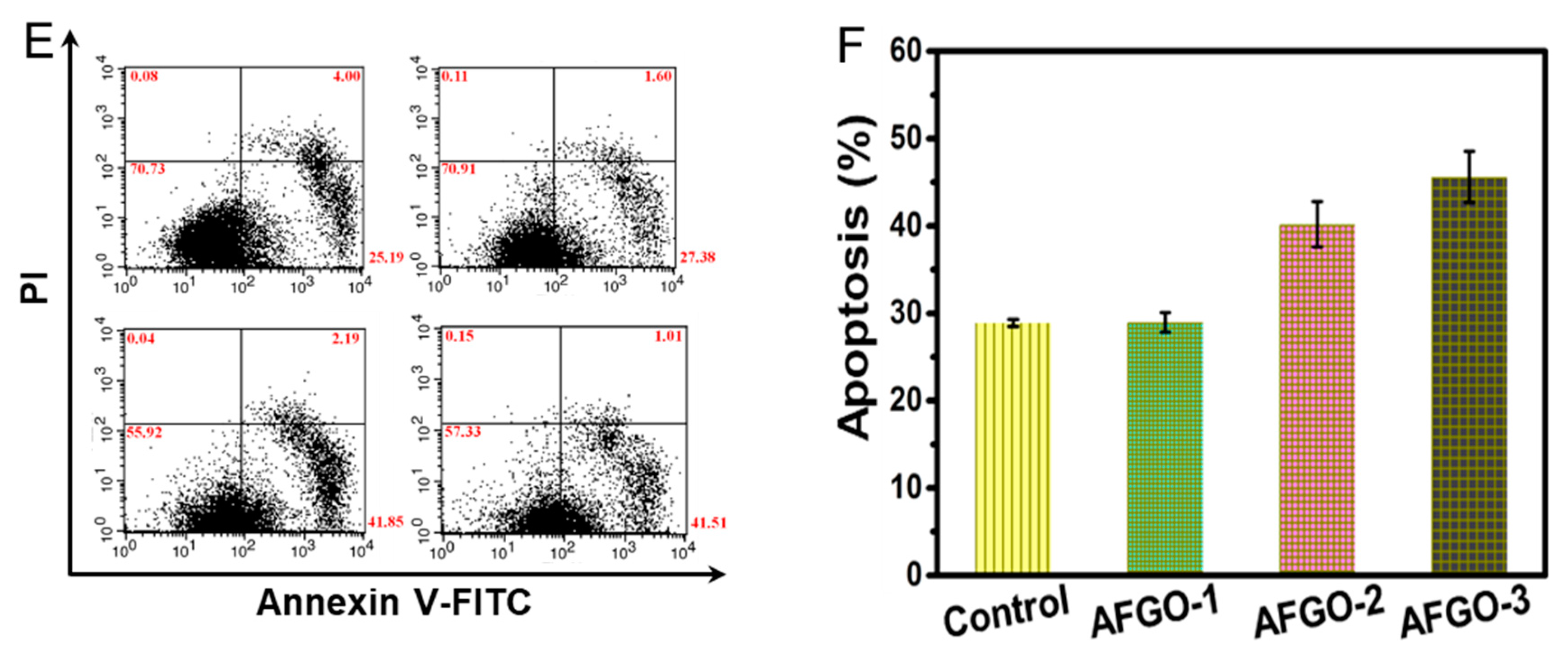
3.5. Cell Viability and Apoptosis of GG/AFGO–Dox
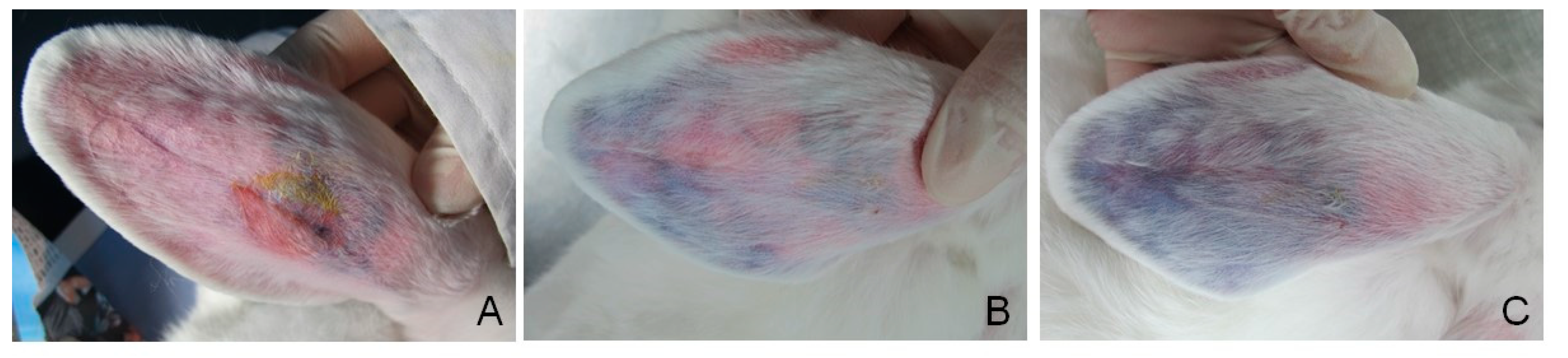
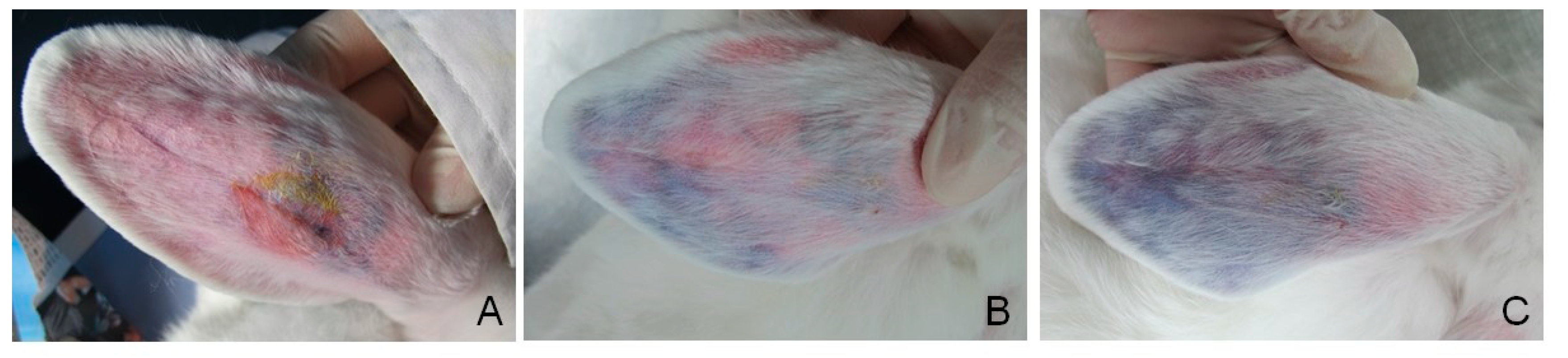
3.6. Vessel Embolization of GG/AFGO In Vivo
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Stirm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Voronin, D.V.; Abalymov, A.A.; Svenskaya, Y.I.; Lomova, M.V. Key Points in remote-controlled drug delivery: From the carrier design to clinical trials. Int. J. Mol. Sci. 2021, 22, 9149. [Google Scholar] [CrossRef]
- Xia, W.; Tao, Z.; Zhu, B.; Zhang, W.; Liu, C.; Chen, S.; Song, M. Targeted delivery of drugs and genes using polymer nanocarriers for cancer therapy. Int. J. Mol. Sci. 2021, 22, 9118. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef]
- Stoff, J.A. Selected office based anticancer treatment strategies. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Ahmadpour, S.T.; Desquiret-Dumas, V.; Yikilmaz, U.; Dartier, J.; Domingo, I.; Wetterwald, C.; Orre, C.; Gueguen, N.; Brisson, L.; Mahéo, L.; et al. Doxorubicin-induced autophago lysosome formation is partly prevented by mitochondrial ROS elimination in DOX-resistant breast cancer cells. Int. J. Mol. Sci. 2021, 22, 9283. [Google Scholar] [CrossRef] [PubMed]
- Mikušová, V.; Mikuš, P. Advances in chitosan-based nanoparticles for drug delivery. Int. J. Mol. Sci. 2021, 22, 9652. [Google Scholar] [CrossRef]
- Sun, T.; Dasgupta, A.; Zhao, Z.; Nurunnabi, M.; Mitragotra, S. Physical triggering strategies for drug delivery. Adv. Drug Deliv. Rev. 2021, 158, 36–62. [Google Scholar] [CrossRef]
- Delicque, J.; Guiu, B.; Boulin, M.; Schwanz, H.; Piron, L.; Cassinotto, C. Liver chemoembolization of hepatocellular carcinoma using TANDEM® microspheres. Future Oncol. 2018, 14, 2761–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schicho, A.; Pereira, P.L.; Haimerl, M.; Niessen, C.; Michalik, K.; Beyer, L.P.; Stroszczynski, C.; Wiggermann, P. Transarterial chemoembolization (TACE) with degradable starch microspheres (DSM) in hepatocellular carcinoma (HCC): Multi-center results on safety and efficacy. Oncotarget 2017, 8, 72613–72620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammer, J.; Malagari, K.; Vogl, T.; Pilleul, F.; Denys, A.; Watkinson, A.; Pitton, M.; Sergent, G.; Pfammatter, T.; Terraz, S.; et al. Prospective randomized study of doxorubicin-eluting-bead embolization in the treatment of hepatocellular carcinoma: Results of the PRECISION V study. Cardiovasc. Interv. Radiol. 2010, 33, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.B.; Sharma, H. Role of Transcatheter Intra-arterial Therapies for Hepatocellular Carcinoma. J. Clin. Exp. Hepatol. 2014, 4, S112–S121. [Google Scholar] [CrossRef] [Green Version]
- Erinjeri, J.P.; Fine, G.C.; Adema, G.J.; Ahmed, M.; Chapiro, J.; Brok, M.; Duran, R.; Hunt, S.J.; Johnson, T.; Ricke, J.; et al. Immunotherapy and the interventional oncologist: Challenges and opportunities—A society of interventional oncology white paper. Radiology 2019, 292, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinto, A.M.; Nutu, O.; Manso, R.S.R.; Alonso, I.J.; Pulido, J.; Municio, A.M.; García-Sesma, A.; Segurola, C.L.; Caballero, J.M.; Romero, L.C. Complications of transarterial chemoembolization (TACE) in the treatment of liver tumors. Cirugia Es-Panola 2018, 96, 560–567. [Google Scholar]
- Chen, Y.P.; Zhang, J.L.; Zou, Y.; Wu, Y.L. Recent Advances on Polymeric Beads or Hydrogels as Embolization Agents for Improved Transcatheter Arterial Chemoembolization (TACE). Front. Chem. 2019, 7, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wu, F.; Duan, M.; Zhang, G. Drug-eluting bead transarterial chemoembolization (TACE) vs conventional TACE in treating hepatocellular carcinoma patients with multiple conventional TACE treatments history: A comparison of efficacy and safety. Medicine 2019, 98, e15314. [Google Scholar] [CrossRef] [PubMed]
- Idee, J.M.; Guiu, B. Use of Lipiodol as a drug-delivery system for transcatheter arterial chemoembolization of hepatocellular carcinoma: A review. Crit. Rev. Oncol. Hematol. 2013, 88, 530–549. [Google Scholar] [CrossRef]
- Woo, H.Y.; Heo, J. Transarterial chemoembolization using drug eluting beads for the treatment of hepatocellular carcinoma: Now and future. Clin. Mol. Hepatol. 2015, 21, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.A.; Pelage, T.P.; Ettles, D.F. Fibroid Calcification after Uterine Artery Embolization: Ultrasonographic Appearance and Pathology. J. Vasc. Interv. Radiol. 2001, 12, 443–446. [Google Scholar] [CrossRef]
- Lee, M.W.; Tsai, H.F.; Wen, S.M.; Huang, C.H. Photocrosslinkable gellan gum film as an anti-adhesion barrier. Carbohydr. Polym. 2012, 90, 1132–1138. [Google Scholar] [CrossRef]
- Tsai, W.C.; Tsai, H.F.; Wong, Y.A.; Hong, J.Y.; Chang, S.J.; Lee, M.W. Preparation and characterization of gellan gum/glucosamine/clioquinol film as oral cancer treatment patch. Mater. Sci. Eng. C 2018, 82, 317–322. [Google Scholar] [CrossRef]
- Hsu, M.F.; Tyan, Y.S.; Chien, Y.C.; Lee, M.W. Hyaluronic acid-based nano-sized drug carrier-containing Gellan gum microspheres as potential multifunctional embolic agent. Sci. Rep. 2018, 8, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Osmałek, T.; Froelich, A.; Tasarek, S. Application of gellan gum in pharmacy and medicine. Int. J. Pharm. 2014, 466, 328–340. [Google Scholar] [CrossRef]
- Silva-Correia, J.; Oliveira, J.M.; Caridade, S.G.; Oliveira, J.T.; Sousa, R.A.; Mano, J.F.; Reis, R.L. Gellan gum-based hydrogels for intervertebral disc tissue-engineering applications. J. Tissue Eng. Regen Med. 2011, 5, e97–e107. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, D.; Kim, J.; Kim, T.I.; Kim, W.J. Photothermally triggered cytosolic drug delivery via endosome disruption us-ing a functionalized reduced graphene oxide. ACS Nano 2013, 7, 6735–6746. [Google Scholar] [CrossRef]
- Zhou, J.; Han, T.; Ma, H.; Yan, T.; Pang, X.; Li, Y.; Wei, Q. A novel electro chemiluminescent immunosensor based on the quenching effect of aminated graphene on nitrogen-doped carbon quantum dots. Anal. Chim. Acta 2015, 889, 82–89. [Google Scholar] [CrossRef]
- Björk, J.; Hanke, F.; Palma, C.A.; Samorì, P.; Cecchini, M.; Persson, M. Adsorption of aromatic and anti-aromatic systems on graphene through π−π stacking. J. Phys. Chem. Lett. 2010, 1, 3407–3412. [Google Scholar] [CrossRef]
- Karki, N.; Tiwari, H.; Tewari, C.; Rana, A.; Pandey, N.; Basak, S.; Sahoo, N.G. Functionalized graphene oxide as a vehicle for targeted drug delivery and bioimaging applications. J. Mater. Chem. B 2020, 8, 8116–8148. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, H.; von dem Bussche, A.; Creighton, M.; Hurt, R.H.; Kane, A.B.; Gao, H. Graphene microsheets enter cells through spontaneous membrane penetration at edge asperities and corner sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12295–12300. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.J.; Zhang, Z.H.; Cai, R.; Chen, X.; Liu, Y.N.; Rao, W.; Yao, S.Z. Molecularly imprinted electrochemical sensor based on amine group modified graphene covalently linked electrode for 4-nonylphenol detection. Talanta 2013, 115, 222–227. [Google Scholar] [CrossRef]
- Pruna, A.; Cárcel, A.; Benedito, A.; Giménez, E. Hydrothermal-Freeze-Casting of Poly(amidoamine)-Modified Graphene Aerogels towards CO2 Adsorption. Int. J. Mol. Sci. 2021, 22, 9333. [Google Scholar] [CrossRef]
- Fang, F.; Kong, L.; Huang, J.; Wu, S.; Zhang, K.; Wang, X.; Sun, B.; Jin, Z.; Wang, J.; Huang, X.J.; et al. Removal of cobalt ions from aqueous solution by an amination graphene oxide nanocomposite. J. Hazard. Mater. 2014, 270, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Xia, J.G.; Yang, Z.Q.; Zhou, W.Z.; Chen, W.H.; Qi, C.J.; Gu, J.P.; Wang, Q. Transarterial chemoembolization with medium-sized doxorubicin-eluting Callisphere is safe and effective for patients with hepatocellular carcinoma. Sci. Rep. 2020, 10, 4434. [Google Scholar] [CrossRef]
- Siddique, Y.H.; Khan, W.; Khanam, S.; Jyoti, S.; Naz, F.; Rahul Singh, B.R.; Naqvi, A.H. Toxic potential of synthesized graphene zinc oxide nanocomposite in the third instar larvae of transgenic Drosophila melanogaster (hsp70-lacZ) Bg9. Bio-Med. Res. Int. 2014, 2014, 382124. [Google Scholar] [CrossRef] [Green Version]
- Siddique, Y.H.; Fatima, A.; Jyoti, S.; Naz, F.; Rahul Khan, W.; Singh, B.R.; Naqvi, A.H. Evaluation of the toxic potential of graphene copper nanocomposite (GCNC) in the third instar larvae of transgenic Drosophila melanogaster (hsp70-lacZ) Bg9. PLoS ONE 2013, 8, e80944. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Kasiński, A.; Zielińska-Pisklak, M.; Kowalczyk, S.; Plichta, A.; Zgadzaj, A.; Oledzka, E.; Sobczak, M. Synthesis and Charac-terization of New Biodegradable Injectable Thermosensitive Smart Hydrogels for 5-Fluorouracil Delivery. Int. J. Mol. Sci. 2021, 22, 8330. [Google Scholar] [CrossRef]
- Nokhodchi, A.; Raja, S.; Patel, P.; Asare-Addo, K. The role of oral controlled release matrix tablets in drug delivery systems. Bioimpacts 2012, 2, 175–187. [Google Scholar] [PubMed] [Green Version]
- Keremidarska-Markova, M.; Hristova-Panusheva, K.; Andreeva, T.; Speranza, G.; Wang, D.; Krasteva, N. Cytotoxicity Evaluation of Ammonia-Modified Graphene Oxide Particles in Lung Cancer Cells and Embryonic Stem Cells. Adv. Condens. Matter. Phys. 2018, 18, 9571828. [Google Scholar] [CrossRef] [Green Version]
- Park, C.S.; Choi, K.S.; Shin, J.W.; Kim, S.Y. Inhibition of viability of the respiratory epithelial cells using functionalized graphene oxide. J. Nanosci. Nanotechnol. 2015, 15, 2060–2066. [Google Scholar] [CrossRef]
- Feng, X.; Chen, L.; Guo, W.; Zhang, Y.; Lai, X.; Shao, L.; Li, Y. Graphene oxide induces p62/SQSTM-dependent apoptosis through the impairment of autophagic flux and lysosomal dysfunction in PC12 cells. Acta Biomater. 2018, 81, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Tao, X.; Yang, Z.; Li, K.; Yang, H.; Li, A.; Cheng, R. Effects of the oxidation degree of graphene oxide on the adsorp-tion of methylene blue. J. Hazard. Mater. 2014, 268, 191–198. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-J.; Cheng, H.-W.; Chen, H.-Y.; Lee, M.-W. A Four-Step Cascade Drug-Release Management Strategy for Transcatheter Arterial Chemoembolization (TACE) Therapeutic Applications. Polymers 2021, 13, 3701. https://doi.org/10.3390/polym13213701
Hsieh Y-J, Cheng H-W, Chen H-Y, Lee M-W. A Four-Step Cascade Drug-Release Management Strategy for Transcatheter Arterial Chemoembolization (TACE) Therapeutic Applications. Polymers. 2021; 13(21):3701. https://doi.org/10.3390/polym13213701
Chicago/Turabian StyleHsieh, Ying-Jiun, Hung-Wei Cheng, Hung-Yu Chen, and Ming-Wei Lee. 2021. "A Four-Step Cascade Drug-Release Management Strategy for Transcatheter Arterial Chemoembolization (TACE) Therapeutic Applications" Polymers 13, no. 21: 3701. https://doi.org/10.3390/polym13213701
APA StyleHsieh, Y.-J., Cheng, H.-W., Chen, H.-Y., & Lee, M.-W. (2021). A Four-Step Cascade Drug-Release Management Strategy for Transcatheter Arterial Chemoembolization (TACE) Therapeutic Applications. Polymers, 13(21), 3701. https://doi.org/10.3390/polym13213701