
Impact of Enzymatic Degradation on the Material Properties of Poly(Ethylene Terephthalate)

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Production and Purification
2.2. PET Sample Preparation
2.3. Sample Incubation with PETase
2.4. Ultra-High Performance Liquid Chromatography (UHPLC)
2.5. Optical and Topographical Characterization
2.6. Thermal Characterization
2.7. Mechanical Characterization
3. Results and Discussion
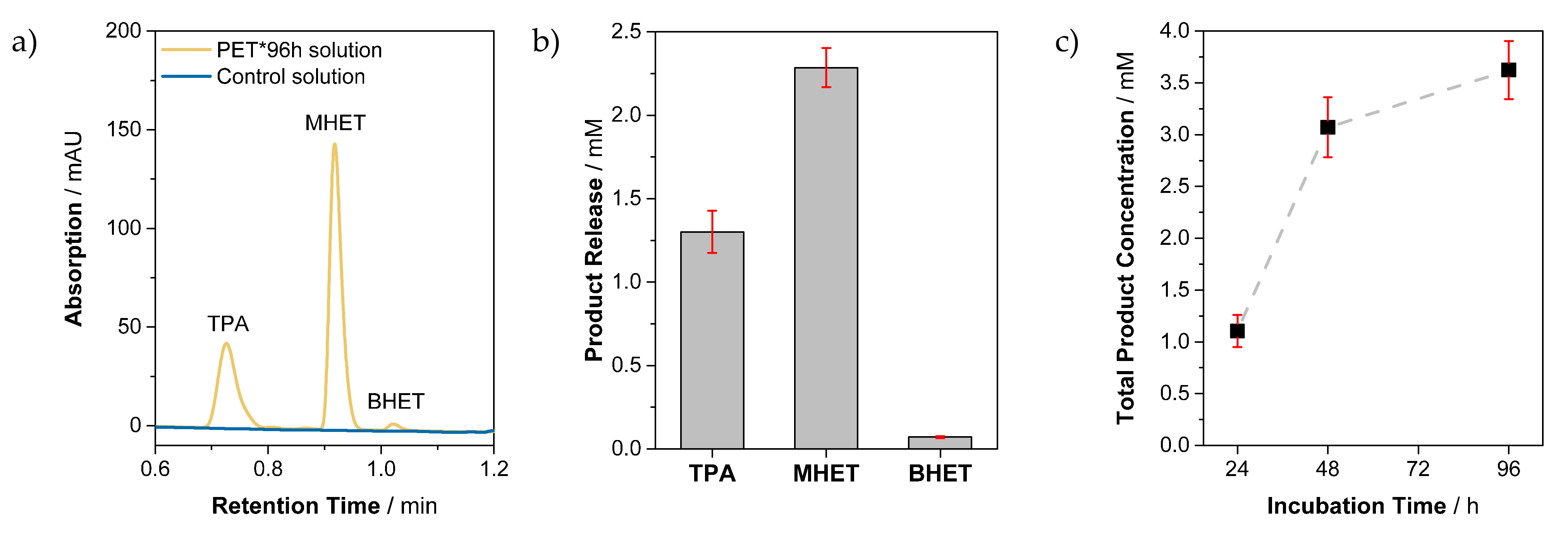
3.1. Analysis of Soluble Products
3.2. Characterization of PET Material Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, R.C.; Olsen, Y.; Mitchell, R.P.; Davis, A.; Rowland, S.J.; John, A.W.G.; McGonigle, D.; Russell, A.E. Lost at Sea: Where Is All the Plastic? Science 2004, 304, 838. [Google Scholar] [CrossRef]
- Weinstein, J.E.; Crocker, B.K.; Gray, A.D. From macroplastic to microplastic: Degradation of high-density polyethylene, polypropylene, and polystyrene in a salt marsh habitat. Environ. Toxicol. Chem. 2016, 35, 1632–1640. [Google Scholar] [CrossRef]
- Waldman, W.R.; Rillig, M.C. Microplastic Research Should Embrace the Complexity of Secondary Particles. Environ. Sci. Technol. 2020, 54, 7751–7753. [Google Scholar] [CrossRef] [PubMed]
- Khoironi, A.; Hadiyanto, H.; Anggoro, S.; Sudarno, S. Evaluation of polypropylene plastic degradation and microplastic identification in sediments at Tambak Lorok coastal area, Semarang, Indonesia. Mar. Pollut. Bull. 2020, 151, 110868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hamidian, A.H.; Tubić, A.; Zhang, Y.; Fang, J.K.; Wu, C.; Lam, P.K. Understanding plastic degradation and microplastic formation in the environment: A review. Environ. Pollut. 2021, 274, 116554. [Google Scholar] [CrossRef] [PubMed]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.M.; Scott, S.L.; Suh, S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. [Google Scholar] [CrossRef] [Green Version]
- Julienne, F.; Delorme, N.; Lagarde, F. From macroplastics to microplastics: Role of water in the fragmentation of polyethylene. Chemosphere 2019, 236, 124409. [Google Scholar] [CrossRef]
- Kalogerakis, N.; Karkanorachaki, K.; Kalogerakis, G.C.; Triantafyllidi, E.I.; Gotsis, A.D.; Partsinevelos, P.; Fava, F. Microplastics Generation: Onset of Fragmentation of Polyethylene Films in Marine Environment Mesocosms. Front. Mar. Sci. 2017, 4, 84. [Google Scholar] [CrossRef]
- Gewert, B.; Plassmann, M.M.; MacLeod, M. Pathways for degradation of plastic polymers floating in the marine environment. Environ. Sci. Process. Impacts 2015, 17, 1513–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, F.; Kawabata, T.; Oda, M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R.-J.; Schrader, H.; Profe, J.; Dresler, K.; Deckwer, W.-D. Enzymatic Degradation of Poly(ethylene terephthalate): Rapid Hydrolyse using a Hydrolase fromT. fusca. Macromol. Rapid Commun. 2005, 26, 1400–1405. [Google Scholar] [CrossRef]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.-J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a Novel Cutinase Homolog with Polyethylene Terephthalate-Degrading Activity from Leaf-Branch Compost by Using a Metagenomic Approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 2016, 351, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Danso, D.; Schmeisser, C.; Chow, J.; Zimmermann, W.; Wei, R.; Leggewie, C.; Li, X.; Hazen, T.; Streit, W.R. New Insights into the Function and Global Distribution of Polyethylene Terephthalate (PET)-Degrading Bacteria and Enzymes in Marine and Terrestrial Metagenomes. Appl. Environ. Microbiol. 2018, 84, e02773-17. [Google Scholar] [CrossRef] [Green Version]
- Fechine, G.; Souto-Maior, R.M.; Rabello, M.S. Structural changes during photodegradation of poly(ethylene terephthalate). J. Mater. Sci. 2002, 37, 4979–4984. [Google Scholar] [CrossRef]
- Fechine, G.; Rabello, M.; Maior, R.S.; Catalani, L. Surface characterization of photodegraded poly(ethylene terephthalate). The effect of ultraviolet absorbers. Polymer 2004, 45, 2303–2308. [Google Scholar] [CrossRef]
- Ioakeimidis, C.; Fotopoulou, K.N.; Karapanagioti, H.K.; Geraga, M.; Zeri, C.; Papathanassiou, E.; Galgani, F.; Papatheodorou, G. The degradation potential of PET bottles in the marine environment: An ATR-FTIR based approach. Sci. Rep. 2016, 6, 23501. [Google Scholar] [CrossRef] [PubMed]
- Meides, N.; Menzel, T.; Poetzschner, B.; Löder, M.G.J.; Mansfeld, U.; Strohriegl, P.; Altstaedt, V.; Senker, J. Reconstructing the Environmental Degradation of Polystyrene by Accelerated Weathering. Environ. Sci. Technol. 2021, 55, 7930–7938. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.S.; Edge, M.; Mohammadian, M.; Jones, K. Hydrolytic degradation of poly(ethylene terephthalate): Importance of chain scission versus crystallinity. Eur. Polym. J. 1991, 27, 1373–1378. [Google Scholar] [CrossRef]
- Edge, M.; Hayes, M.; Mohammadian, M.; Allen, N.; Jewitt, T.; Brems, K.; Jones, K. Aspects of poly(ethylene terephthalate) degradation for archival life and environmental degradation. Polym. Degrad. Stab. 1991, 32, 131–153. [Google Scholar] [CrossRef]
- Sammon, C.; Yarwood, J.; Everall, N. An FT–IR study of the effect of hydrolytic degradation on the structure of thin PET films. Polym. Degrad. Stab. 2000, 67, 149–158. [Google Scholar] [CrossRef]
- Arhant, M.; Le Gall, M.; Le Gac, P.-Y.; Davies, P. Impact of hydrolytic degradation on mechanical properties of PET—Towards an understanding of microplastics formation. Polym. Degrad. Stab. 2019, 161, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Fayolle, B.; Audouin, L.; Verdu, J. A critical molar mass separating the ductile and brittle regimes as revealed by thermal oxidation in polypropylene. Polymer 2004, 45, 4323–4330. [Google Scholar] [CrossRef]
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- OECD. Improving Plastics Management: Trends, policy responses, and the role of international co-operation and trade. OECD Environ. Policy Pap. 2018, 12, 20. [Google Scholar] [CrossRef]
- Austin, H.P.; Allen, M.D.; Donohoe, B.S.; Rorrer, N.; Kearns, F.; Silveira, R.L.; Pollard, B.C.; Dominick, G.; Duman, R.; El Omari, K.; et al. Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc. Natl. Acad. Sci. USA 2018, 115, E4350–E4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altstädt, V. The Influence of Molecular Variables on Fatigue Resistance in Stress Cracking Environments. In Intrinsic Molecular Mobility and Toughness of Polymers II; Springer: Berlin/Heidelberg, Germany, 2005; pp. 105–152. [Google Scholar]
- Liu, H.-Y.; Wang, G.; Mai, Y.-W. Cyclic fatigue crack propagation of nanoparticle modified epoxy. Compos. Sci. Technol. 2012, 72, 1530–1538. [Google Scholar] [CrossRef]
- Runt, J.; Gallagher, K.P. The influence of microstructure on fatigue crack propagation in polyoxymethylene. J. Mater. Sci. 1991, 26, 792–798. [Google Scholar] [CrossRef]
- Ramirez, A.; Manson, J.A.; Hertzberg, R.W. Fatigue crack propagation in amorphous poly(ethylene terephthalate). Polym. Eng. Sci. 1982, 22, 975–981. [Google Scholar] [CrossRef]
- Altstaedt, V.; Keiter, S.; Renner, M.; Schlarb, A. Environmental Stress Cracking of Polymers Monitored by Fatigue Crack Growth Experiments. Macromol. Symp. 2004, 214, 31–46. [Google Scholar] [CrossRef]
- Kothmann, M.; Zeiler, R.; de Anda, A.R.; Brückner, A.; Altstädt, V. Fatigue crack propagation behaviour of epoxy resins modified with silica-nanoparticles. Polymer 2015, 60, 157–163. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Kawai, F. The Current State of Research on PET Hydrolyzing Enzymes Available for Biorecycling. Catalysts 2021, 11, 206. [Google Scholar] [CrossRef]
- Son, H.F.; Cho, I.J.; Joo, S.; Seo, H.; Sagong, H.-Y.; Choi, S.Y.; Lee, S.Y.; Kim, K.-J. Rational Protein Engineering of Thermo-Stable PETase from Ideonella sakaiensis for Highly Efficient PET Degradation. ACS Catalysts 2019, 9, 3519–3526. [Google Scholar] [CrossRef]
- Groeninckx, G.; Reynaers, H. Morphology and melting behavior of semicrystalline poly(ethylene terephthalate). II. Annealed PET. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 1325–1341. [Google Scholar] [CrossRef]
- Mancini, S.D.; Zanin, M. Recyclability of PET from virgin resin. Mater. Res. 1999, 2, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Güçlü, G.; Kasgöz, A.; Özbudak, S.; Özgümüs, S.; Orbay, M. Glycolysis of poly(ethylene terephthalate) wastes in xylene. J. Appl. Polym. Sci. 1998, 69, 2311–2319. [Google Scholar] [CrossRef]
- Baliga, S.; Wong, W.T. Depolymerization of poly(ethylene terephthalate) recycled from post-consumer soft-drink bottles. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 2071–2082. [Google Scholar] [CrossRef]
- Krehula, L.K.; Hrnjak-Murgić, Z.; Jelenčić, J.; Andričić, B. Evaluation of Poly(ethylene-terephthalate) Products of Chemical Recycling by Differential Scanning Calorimetry. J. Polym. Environ. 2009, 17, 20–27. [Google Scholar] [CrossRef]
- Imran, M.; Kim, B.-K.; Han, M.; Cho, B.G.; Kim, D.H. Sub- and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis(2-hydroxyethyl) terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693. [Google Scholar] [CrossRef]
- Fecker, T.; Galaz-Davison, P.; Engelberger, F.; Narui, Y.; Sotomayor, M.; Parra, L.P.; Ramírez-Sarmiento, C.A. Active Site Flexibility as a Hallmark for Efficient PET Degradation by I. sakaiensis PETase. Biophys. J. 2018, 114, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.-L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nat. Cell Biol. 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Pecorini, T.; Hertzberg, R. The fracture toughness and fatigue crack propagation behaviour of annealed PET. Polymer 1993, 34, 5053–5062. [Google Scholar] [CrossRef]
- Yeh, J.T.; Runt, J. Fatigue crack-propagation in annealed poly(butylene terephthalate). J. Mater. Sci. 1989, 24, 2637–2642. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menzel, T.; Weigert, S.; Gagsteiger, A.; Eich, Y.; Sittl, S.; Papastavrou, G.; Ruckdäschel, H.; Altstädt, V.; Höcker, B. Impact of Enzymatic Degradation on the Material Properties of Poly(Ethylene Terephthalate). Polymers 2021, 13, 3885. https://doi.org/10.3390/polym13223885
Menzel T, Weigert S, Gagsteiger A, Eich Y, Sittl S, Papastavrou G, Ruckdäschel H, Altstädt V, Höcker B. Impact of Enzymatic Degradation on the Material Properties of Poly(Ethylene Terephthalate). Polymers. 2021; 13(22):3885. https://doi.org/10.3390/polym13223885
Chicago/Turabian StyleMenzel, Teresa, Sebastian Weigert, Andreas Gagsteiger, Yannik Eich, Sebastian Sittl, Georg Papastavrou, Holger Ruckdäschel, Volker Altstädt, and Birte Höcker. 2021. "Impact of Enzymatic Degradation on the Material Properties of Poly(Ethylene Terephthalate)" Polymers 13, no. 22: 3885. https://doi.org/10.3390/polym13223885