
Exposure of Von Willebrand Factor Cleavage Site in A1A2A3-Fragment under Extreme Hydrodynamic Shear

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structures
2.2. The OPEP Model
2.3. The Lattice Boltzmann Molecular Dynamics Framework
2.4. All-Atom Molecular Dynamics
2.5. Kinetic Models for Unfolding
2.6. Maximum Likelihood Analysis of Unfolding Kinetics
3. Results
3.1. Validating the Unfolding of the A2 Domain
3.1.1. Single A2 Domain: Pulling with a Constant Force
3.1.2. Single A2 Domain: Structural Analysis of Force Unfolding
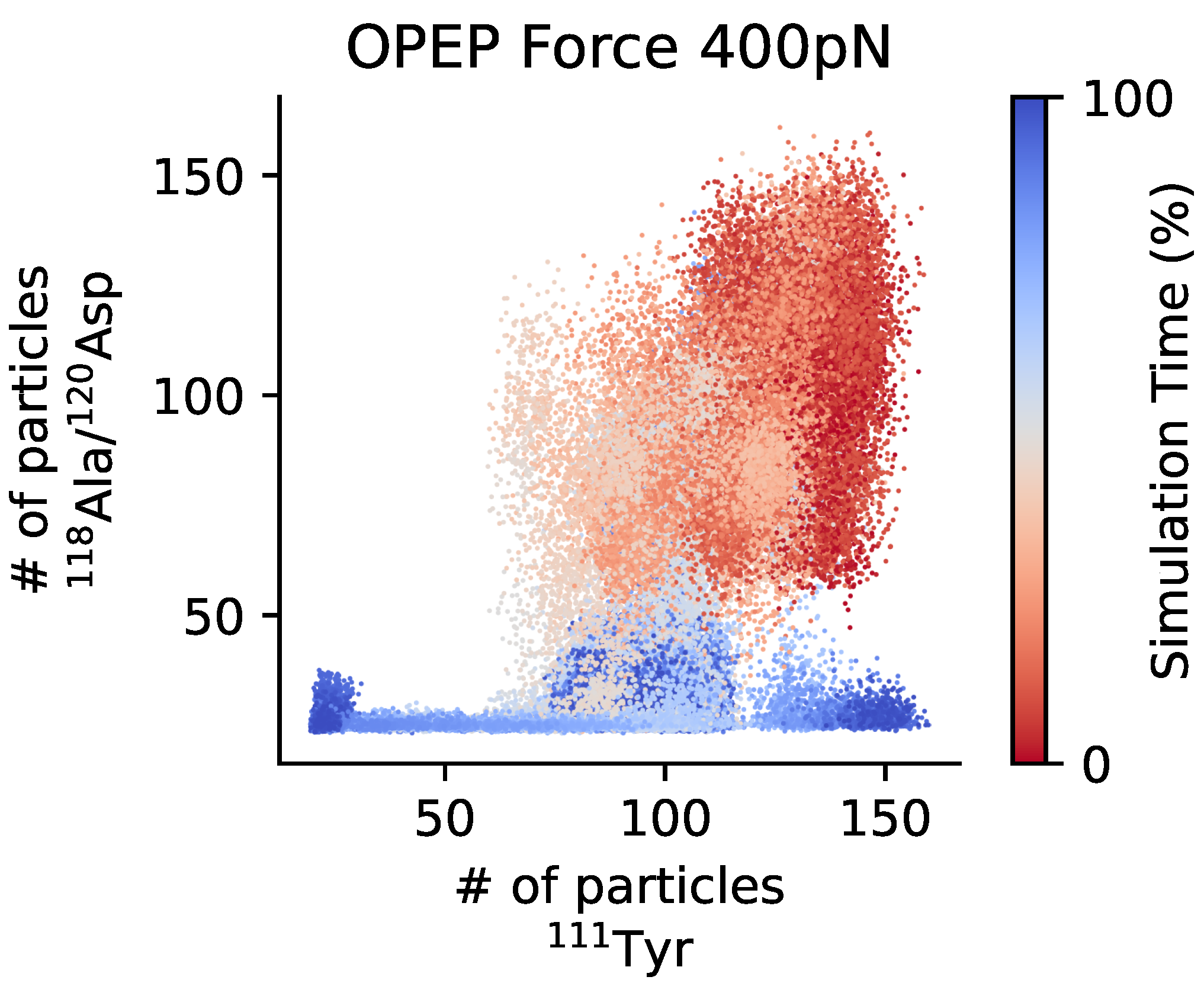
3.1.3. Exposure of the Cleavage Site
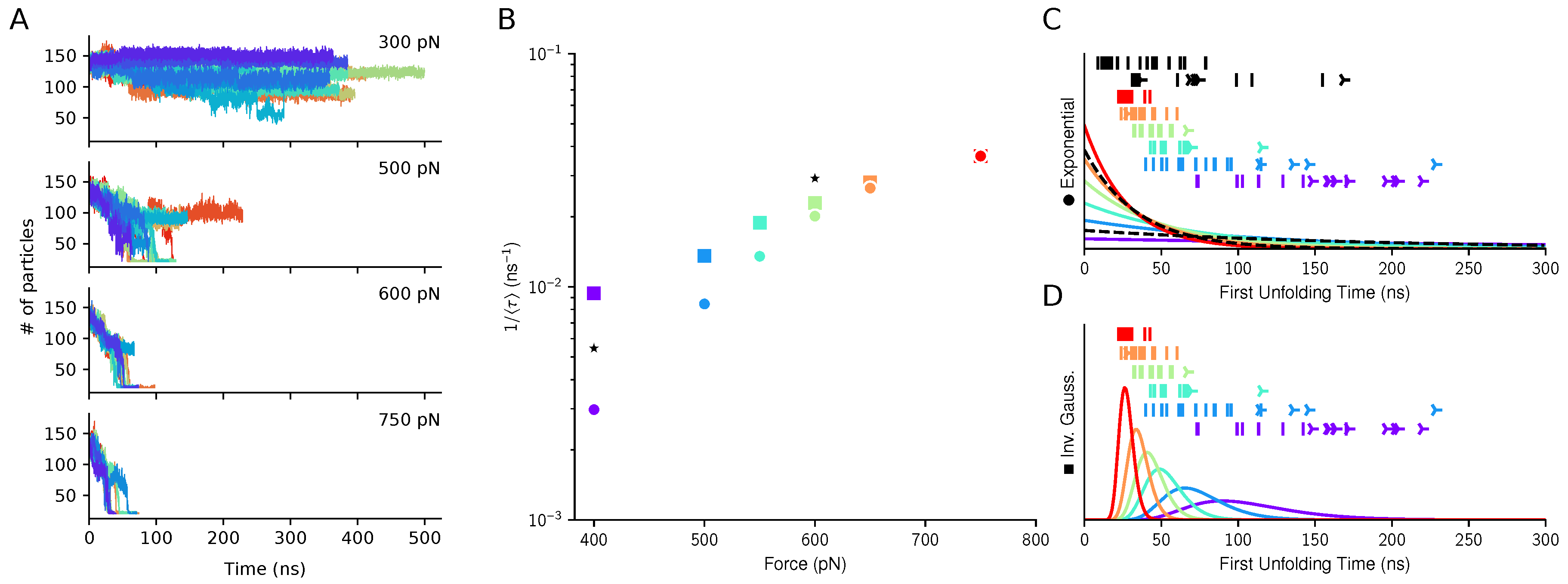
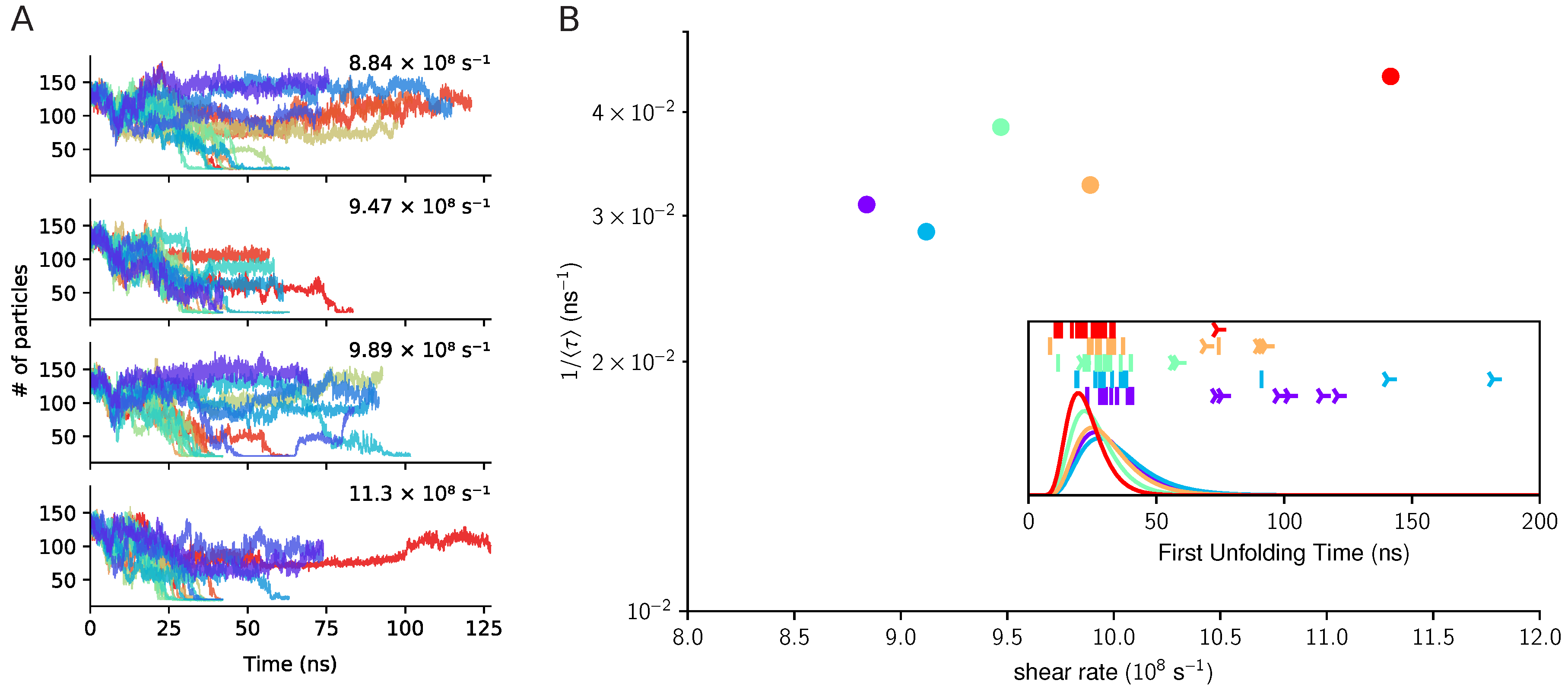
3.2. The Three Domains A1A2A3: The Unfolding of A2 in Shear Flow
3.3. The Three Domains A1A2A3: The Shear-Induced Unfolding and Mechanical Tension
3.4. The Three Domains A1A2A3: The Rotational Dynamics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Springer, T.A. von Willebrand Factor, Jedi Knight of the Bloodstream. Blood 2014, 124, 1412–1425. [Google Scholar] [CrossRef] [Green Version]
- Slayter, H.; Loscalzo, J.; Bockenstedt, P.; Handin, R.I. Native Conformation of Human von Willebrand Protein. Analysis by Electron Microscopy and Quasi-Elastic Light Scattering. J. Biol. Chem. 1985, 260, 8559–8563. [Google Scholar] [CrossRef]
- Novák, L.; Deckmyn, H.; Damjanovich, S.; Hársfalvi, J. Shear-Dependent Morphology of von Willebrand Factor Bound to Immobilized Collagen. Blood 2002, 99, 2070–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.W.; Nuschele, S.; Wixforth, A.; Gorzelanny, C.; Alexander-Katz, A.; Netz, R.R.; Schneider, M.F. Shear-Induced Unfolding Triggers Adhesion of von Willebrand Factor Fibers. Proc. Natl. Acad. Sci. USA 2007, 104, 7899–7903. [Google Scholar] [CrossRef] [Green Version]
- Rack, K.; Huck, V.; Hoore, M.; Fedosov, D.; Schneider, S.; Gompper, G. Margination and Stretching of von Willebrand Factor in the Blood Stream enable adhesion. Sci. Rep. 2017, 7, 14278. [Google Scholar] [CrossRef]
- Nesbitt, W.S.; Westein, E.; Tovar-Lopez, F.J.; Tolouei, E.; Mitchell, A.; Fu, J.; Carberry, J.; Fouras, A.; Jackson, S.P. A Shear Gradient–Dependent Platelet Aggregation Mechanism Drives Thrombus Formation. Nat. Med. 2009, 15, 665–673. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical Organization in the Hemostatic Response and its Relationship to the Platelet-Signaling Network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Nesbitt, W.S.; Westein, E. Dynamics of Platelet Thrombus Formation. J. Thromb. Haemost. 2009, 7, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Coburn, L.; Damaraju, V.; Dozic, S.; Eskin, S.; Cruz, M.; McIntire, L. Rolling under Shear Stress Shows Differences between Type 2B and 2M von Willebrand Disease. Biophys. J. 2011, 100, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Yago, T.; Lou, J.; Wu, T.; Yang, J.; Miner, J.J.; Coburn, L.; Lopez, J.A.; Cruz, M.A.; Dong, J.F.; McIntire, L.V.; et al. Platelet Glycoprotein Ibα Forms Catch Bonds with Human WT vWF but not with Type 2B von Willebrand Disease vWF. J. Clin. Investig. 2008, 118, 3195–3207. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Jiang, Y.; Yang, D.; Scheiflinger, F.; Wong, W.P.; Springer, T.A. Flow-Induced Elongation of von Willebrand Factor Precedes Tension-Dependent Activation. Nat. Commun. 2017, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Kushchenko, Y.K.; Belyaev, A.V. Effects of Hydrophobicity, Tethering and Size on Flow-Induced Activation of von Willebrand Factor Multimers. J. Theor. Biol. 2020, 485, 110050. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, S.; Sacco, M.; Basso, M.; Cristofaro, R.D. Mechanochemistry of von Willebrand Factor. Biomol. Concepts 2019, 10, 194–208. [Google Scholar] [CrossRef]
- Zhang, X.; Halvorsen, K.; Zhang, C.Z.; Wong, W.P.; Springer, T.A. Mechanoenzymatic Cleavage of the Ultralarge Vascular Protein von Willebrand Factor. Science 2009, 324, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, C.; Methia, N.; Frenette, P.S.; Rayburn, H.; Ullman-Culleré, M.; Hynes, R.O.; Wagner, D.D. A Mouse Model of Severe von Willebrand Disease: Defects in Hemostasis and Thrombosis. Proc. Natl. Acad. Sci. USA 1998, 95, 9524–9529. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; López, J.A. ADAMTS-13 Rapidly Cleaves Newly Secreted Ultralarge von Willebrand Factor Multimers on the Endothelial Surface Under Flowing Conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Zhang, C.Z.; Zhang, X.; Springer, T.A. A Mechanically Stabilized Receptor-Ligand Flex-Bond Important in the Vasculature. Nature 2010, 466, 992–995. [Google Scholar] [CrossRef] [Green Version]
- Lippok, S.; Obser, T.; Müller, J.P.; Stierle, V.K.; Benoit, M.; Budde, U.; Schneppenheim, R.; Rädler, J.O. Exponential Size Distribution of von Willebrand Factor. Biophys. J. 2013, 105, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Lippok, S.; Radtke, M.; Obser, T.; Kleemeier, L.; Schneppenheim, R.; Budde, U.; Netz, R.R.; Rädler, J.O. Shear-Induced Unfolding and Enzymatic Cleavage of Full-Length VWF Multimers. Biophys. J. 2016, 110, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Belyaev, A.V. Long Ligands Reinforce Biological Adhesion Under Shear Flow. Phys. Rev. E 2018, 97, 042407. [Google Scholar] [CrossRef] [Green Version]
- Butera, D.; Passam, F.; Ju, L.; Cook, K.M.; Woon, H.; Aponte-Santamaría, C.; Gardiner, E.; Davis, A.K.; Murphy, D.A.; Bronowska, A.; et al. Autoregulation of von Willebrand Factor Function by a Disulfide Bond Switch. Sci. Adv. 2018, 4, eaaq1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhou, Y.F.; Zhang, C.Z.; Zhang, X.; Lu, C.; Springer, T.A. Structural Specializations of A2, a Force-Sensing Domain in the Ultralarge Vascular Protein von Willebrand Factor. Proc. Natl. Acad. Sci. USA 2009, 106, 9226–9231. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.E. Biochemistry and Genetics of von Willebrand Factor. Ann. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Takeda, S.; Kokame, K.; Takagi, J.; Miyata, T. Crystal Structures of the Noncatalytic Domains of ADAMTS13 Reveal Multiple Discontinuous Exosites for von Willebrand Factor. Proc. Natl. Acad. Sci. USA 2009, 106, 19274–19279. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Choi, H.; Zhou, Z.; Nolasco, L.; Pownall, H.J.; Voorberg, J.; Moake, J.L.; Dong, J.F. Covalent Regulation of ULVWF String Formation and Elongation on Endothelial Cells Under Flow Conditions. J. Thromb. Haemost. 2008, 6, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Hovinga, J.A.K.; Lämmle, B. Role of ADAMTS13 in the Pathogenesis, Diagnosis, and Treatment of Thrombotic Thrombocytopenic Purpura. Hematology 2012, 2012, 610–616. [Google Scholar] [CrossRef]
- Chen, W.; Lou, J.; Zhu, C. Molecular Dynamics Simulated Unfolding of von Willebrand Factor A Domains by Force. Cell. Mol. Bioeng. 2009, 2, 75–86. [Google Scholar] [CrossRef]
- Zhou, M.; Dong, X.; Baldauf, C.; Chen, H.; Zhou, Y.; Springer, T.A.; Luo, X.; Zhong, C.; Gräter, F.; Ding, J. A Novel Calcium-Binding Site of von Willebrand Factor A2 Domain Regulates its Cleavage by ADAMTS13. Blood 2011, 117, 4623–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.J.; Springer, T.A. Calcium stabilizes the von Willebrand factor A2 domain by promoting refolding. Proc. Natl. Acad. Sci. USA 2012, 109, 3742–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.F.; Eng, E.T.; Nishida, N.; Lu, C.; Walz, T.; Springer, T.A. A pH-Regulated Dimeric Bouquet in the Structure of von Willebrand Factor. EMBO J. 2011, 30, 4098–4111. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, H.E.R.; Zhang, X.F.; Sanz-Hernandez, M.; Chion, A.; Shapiro, S.; Mobayen, G.; Xu, Y.; Simone, A.D.; Laffan, M.A.; McKinnon, T.A.J. Blocking von Willebrand Factor Free Thiols Inhibits Binding to Collagen Under High and Pathological Shear Stress. J. Thromb. Haemost. 2020, 19, 358–369. [Google Scholar] [CrossRef]
- Sacco, M.; Lancellotti, S.; Ferrarese, M.; Bernardi, F.; Pinotti, M.; Tardugno, M.; Candia, E.D.; Gennaro, L.D.; Basso, M.; Giusti, B.; et al. Noncanonical Type 2B von Willebrand Disease Associated with Mutations in the VWF D′D3 and D4 Domains. Blood Adv. 2020, 4, 3405–3415. [Google Scholar] [CrossRef] [PubMed]
- Löf, A.; Müller, J.P.; Benoit, M.; Brehm, M.A. Biophysical Approaches Promote Advances in the Understanding of von Willebrand Factor Processing and Function. Adv. Biol. Regul. 2017, 63, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.P.; Mielke, S.; Löf, A.; Obser, T.; Beer, C.; Bruetzel, L.K.; Pippig, D.A.; Vanderlinden, W.; Lipfert, J.; Schneppenheim, R.; et al. Force Sensing by the Vascular Protein von Willebrand Factor is Tuned by a Strong Intermonomer Interaction. Proc. Natl. Acad. Sci. USA 2016, 113, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aponte-Santamaria, C.; Huck, V.; Posch, S.; Bronowska, A.K.; Grässle, S.; Brehm, M.A.; Obser, T.; Schneppenheim, R.; Hinterdorfer, P.; Schneider, S.W.; et al. Force-Sensitive Autoinhibition of the von Willebrand Factor Is Mediated by Interdomain Interactions. Biophys. J. 2015, 108, 2312–2321. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, C.; Schneppenheim, R.; Stacklies, W.; Obser, T.; Pieconka, A.; Schneppenheim, S.; Budde, U.; Zhou, J.; Grater, F. Shear-induced unfolding activates von Willebrand factor A2 domain for proteolysis. J. Thromb. Haemost. 2009, 7, 2096–2105. [Google Scholar] [CrossRef] [Green Version]
- Posch, S.; Obser, T.; König, G.; Schneppenheim, R.; Tampé, R.; Hinterdorfer, P. Interaction of von Willebrand Factor Domains with Collagen Investigated by Single Molecule Force Spectroscopy. J. Chem. Phys. 2018, 148, 123310. [Google Scholar] [CrossRef]
- Wu, T.; Lin, J.; Cruz, M.A.; Dong, J.F.; Zhu, C. Force-Induced Cleavage of Single VWF A1A2A3 tridomains by ADAMTS-13. Blood 2010, 115, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, M.; Lippok, S.; Rädler, J.O.; Netz, R.R. Internal Tension in a Collapsed Polymer under Shear Flow and the Connection to Enzymatic Cleavage of von Willebrand factor. Eur. Phys. J. E 2016, 39, 32. [Google Scholar] [CrossRef] [Green Version]
- Alexander-Katz, A.; Schneider, M.F.; Schneider, S.W.; Wixforth, A.; Netz, R.R. Shear-Flow-Induced Unfolding of Polymeric Globules. Phys. Rev. Lett. 2006, 97, 138101. [Google Scholar] [CrossRef]
- Belyaev, A.V. Intradimer Forces and their Implication for Conformations of von Willebrand Factor Multimers. Biophys. J. 2021, 120, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Okhota, S.; Melnikov, I.; Avtaeva, Y.; Kozlov, S.; Gabbasov, Z. Shear Stress-Induced Activation of von Willebrand Factor and Cardiovascular Pathology. Int. J. Mol. Sci. 2020, 21, 7804. [Google Scholar] [CrossRef]
- Li, Z.; Lin, J.; Sulchek, T.; Cruz, M.A.; Wu, J.; Dong, J.F.; Zhu, C. Domain-Specific Mechanical Modulation of VWF–ADAMTS13 Interaction. Mol. Biol. Cell. 2019, 30, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Bernaschi, M.; Melchionna, S.; Succi, S. Mesoscopic simulations at the physics-chemistry-biology interface. Rev. Mod. Phys. 2019, 91, 025004. [Google Scholar] [CrossRef] [Green Version]
- Sterpone, F.; Melchionna, S.; Tuffery, P.; Pasquali, S.; Mousseau, N.; Cragnolini, T.; Chebaro, Y.; St-Pierre, J.F.; Kalimeri, M.; Barducci, A.; et al. The OPEP Protein Model: From Single Molecules, Amyloid Formation, Crowding and Hydrodynamics to DNA/RNA Systems. Chem. Soc. Rev. 2014, 43, 4871–4893. [Google Scholar] [CrossRef] [Green Version]
- Languin-Cattoen, O.; Melchionna, S.; Derreumaux, P.; Stirnemann, G.; Sterpone, F. Three Weaknesses for Three Perturbations: Comparing Protein Unfolding under Shear, Force, and Thermal Stresses. J. Phys. Chem. B 2018, 122, 11922–11930. [Google Scholar] [CrossRef]
- Emsley, J.; Cruz, M.; Handin, R.; Liddington, R. Crystal Structure of the von Willebrand Factor A1 Domain and Implications for the Binding of Platelet Glycoprotien IB. J. Biol. Chem. 1998, 273, 10396. [Google Scholar] [CrossRef] [Green Version]
- Bienkowska, J.; Cruz, M.; Atiemo, A.; Handin, R.; Liddington, R. The von Willebrand Factor A3 Domain Does not Contain a Metal Ion-dependent Adhesion Site Motif. J. Biol. Chem. 1997, 272, 25162–25167. [Google Scholar] [CrossRef] [Green Version]
- Chebaro, Y.; Pasquali, S.; Derreumaux, P. The Coarse-Grained OPEP Force Field for Non-Amyloid and Amyloid Proteins. J. Phys. Chem. B 2012, 116, 8741–8752. [Google Scholar] [CrossRef] [PubMed]
- Sterpone, F.; Nguyen, P.; Kalimeri, M.; Derreumaux, P. Importance of the Ion-Pair Interactions in the OPEP Coarse-Grained Force Field: Parametrization and Validation. J. Chem. Theory Comput. 2013, 9, 4574–4584. [Google Scholar] [CrossRef] [Green Version]
- Kalimeri, M.; Derreumaux, P.; Sterpone, F. Are coarse-grained models apt to detect protein thermal stability? The case of OPEP force field. J. Non-Cryst. Solids 2015, 407, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Ahlrichs, P.; Dünweg, B. Simulation of a Single Polymer Chain in Solution by Combining Lattice Boltzmann and Molecular Dynamics. J. Chem. Phys. 1999, 111, 8225–8239. [Google Scholar] [CrossRef] [Green Version]
- Ahlrichs, P.; Dünweg, B. Lattice-Boltzmann Simulation of Polymer-Solvent Systems. Int. J. Mod. Phys. C 1998, 9, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Dunweg, B.; Ladd, A.J.C. Lattice Boltzmann Simulations of Soft Matter Systems. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1–78. [Google Scholar]
- Sterpone, F.; Derreumaux, P.; Melchionna, S. Protein Simulations in Fluids: Coupling the OPEP Coarse-Grained Force Field with Hydrodynamics. J. Chem. Theory Comput. 2015, 11, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiricotto, M.; Sterpone, F.; Derreumaux, P.; Melchionna, S. Multiscale Simulation of Molecular Processes in Cellular Environments. Philos. Trans. A 2016, 374, 20160225. [Google Scholar] [CrossRef]
- Chiriccotto, M.; Melchionna, S.; Derreumaux, P.; Sterpone, F. Multiscale Aggregation of the Amyloid AB16–22 Peptide: From Disordered Coagulation and Lateral Branching to Amorphous Prefibrils. J. Phys. Chem. Lett. 2019, 10, 1594–1599. [Google Scholar] [CrossRef]
- Chiricotto, M.; Melchionna, S.; Derreumaux, P.; Sterpone, F. Hydrodynamic Effects on Beta-Amyloid (16-22) Peptide Aggregation. J. Chem. Phys. 2016, 145, 035102. [Google Scholar] [CrossRef]
- Timr, S.; Gnutt, D.; Ebbinghaus, S.; Sterpone, F. The Unfolding Journey of Superoxide Dismutase 1 Barrels under Crowding: Atomistic Simulations Shed Light on Intermediate States and Their Interactions with Crowders. J. Phys. Chem. Lett. 2020, 11, 4206–4212. [Google Scholar] [CrossRef]
- Brander, A.; Timr, S.; Melchionna, S.; Derreumaux, P.; Baaden, M.; Sterpone, F. Modelling lipid systems in fluid with Lattice Boltzmann Molecular Dynamics simulations and hydrodynamics. Sci. Rep. 2019, 9, 16450. [Google Scholar] [CrossRef]
- Sterpone, F.; Melchionna, S.; Derreumaux, P. Molecular Mechanism of Protein Unfolding Under Shear: A Lattice Boltzmann Molecular Dynamics Study. J. Phys. Chem. B 2018, 122, 1573–1579. [Google Scholar] [CrossRef]
- Benzi, R.; Succi, S.; Vergassola, M. The lattice Boltzmann equation: Theory and applications. Phys. Rep. 1992, 222, 145–197. [Google Scholar] [CrossRef]
- Bernaschi, M.; Melchionna, S.; Succi, S.; Fyta, M.; Kaxiras, E.; Sircar, J. MUPHY: A parallel MUlti PHYsics/scale code for high performance bio-fluidic simulations. Comput. Phys. Comm. 2009, 180, 1495–1502. [Google Scholar] [CrossRef]
- Robustellia, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Bell, G.I. Models for the Specific Adhesion of Cells to Cells. Science 1978, 200, 618–627. [Google Scholar] [CrossRef]
- Satija, R.; Berezhkovskii, A.M.; Makarov, D.E. Broad Distributions of Transition-Path Times are Fingerprints of Multidimensionality of the Underlying Free Energy Landscapes. Proc. Natl. Acad. Sci. USA 2020, 117, 27116–27123. [Google Scholar] [CrossRef]
- Luccioli, S.; Imparato, A.; Mitternacht, S.; Irbäck, A.; Torcini, A. Unfolding Times for Proteins in a Force Clamp. Phys. Rev. E 2010, 81, 010902. [Google Scholar] [CrossRef] [Green Version]
- Muia, J.; Zhu, J.; Greco, S.C.; Vanhoorelbeke, K.; Gupta, G.; Westfield, L.A.; Sadler, J.E. Phylogenetic and Functional Analysis of ADAMTS13 Identifies Highly Conserved Domains Essential for Allosteric Regulation. Blood 2019, 133, 1899–1908. [Google Scholar] [CrossRef]
- Takeda, S. ADAM and ADAMTS Family Proteins and Snake Venom Metalloproteinases: A Structural Overview. Toxins 2016, 8, 155. [Google Scholar] [CrossRef] [Green Version]
- de Groot, R.; Bardhan, A.; Ramroop, N.; Lane, D.A.; Crawley, J.T.B. Essential role of the disintegrin-like domain in ADAMTS13 function. Blood 2009, 113, 5609–5616. [Google Scholar] [CrossRef] [Green Version]
- Petri, A.; Kim, H.J.; Xu, Y.; de Groot, R.; Li, C.; Vandenbulcke, A.; Vanhoorelbeke, K.; Emsley, J.; Crawley, J.T.B. Crystal Structure and Substrate-Induced Activation of ADAMTS13. Nat. Commun. 2019, 10, 3781. [Google Scholar] [CrossRef] [Green Version]
- Belyaev, A.V. Catching Platelets from the Bloodflow: The Role of the Conformation of von Willebrand Factor. Mat. Mod. Nat. Phenom. 2018, 13, 44. [Google Scholar] [CrossRef]
- Jaspe, J.; Hagen, S.J. Do Protein Molecules Unfold in a Simple Shear Flow? Biophys. J. 2006, 91, 3415–3424. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; de Groot, R.; Crawley, J.T.B.; Lane, D.A. Mechanism of von Willebrand factor scissile bond cleavage by a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13). Proc. Natl. Acad. Sci. USA 2011, 108, 11602–11607. [Google Scholar] [CrossRef] [Green Version]
- Voevodin, V.; Antonov, A.; Nikitenko, D.; Shvets, P.; Sobolev, S.; Sidorov, I.; Stefanov, K.; Voevodin, V.; Zhumatiy, S. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Languin-Cattoën, O.; Laborie, E.; Yurkova, D.O.; Melchionna, S.; Derreumaux, P.; Belyaev, A.V.; Sterpone, F. Exposure of Von Willebrand Factor Cleavage Site in A1A2A3-Fragment under Extreme Hydrodynamic Shear. Polymers 2021, 13, 3912. https://doi.org/10.3390/polym13223912
Languin-Cattoën O, Laborie E, Yurkova DO, Melchionna S, Derreumaux P, Belyaev AV, Sterpone F. Exposure of Von Willebrand Factor Cleavage Site in A1A2A3-Fragment under Extreme Hydrodynamic Shear. Polymers. 2021; 13(22):3912. https://doi.org/10.3390/polym13223912
Chicago/Turabian StyleLanguin-Cattoën, Olivier, Emeline Laborie, Daria O. Yurkova, Simone Melchionna, Philippe Derreumaux, Aleksey V. Belyaev, and Fabio Sterpone. 2021. "Exposure of Von Willebrand Factor Cleavage Site in A1A2A3-Fragment under Extreme Hydrodynamic Shear" Polymers 13, no. 22: 3912. https://doi.org/10.3390/polym13223912