
Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Composite Materials Based on Polyvinylpyrrolidone with Hydroxyapatite Obtained In Situ and Ex Situ
Crosslinking of Composite PVP-SA-HA Films
2.2. Thermogravimetric Analysis
2.3. The Annealing
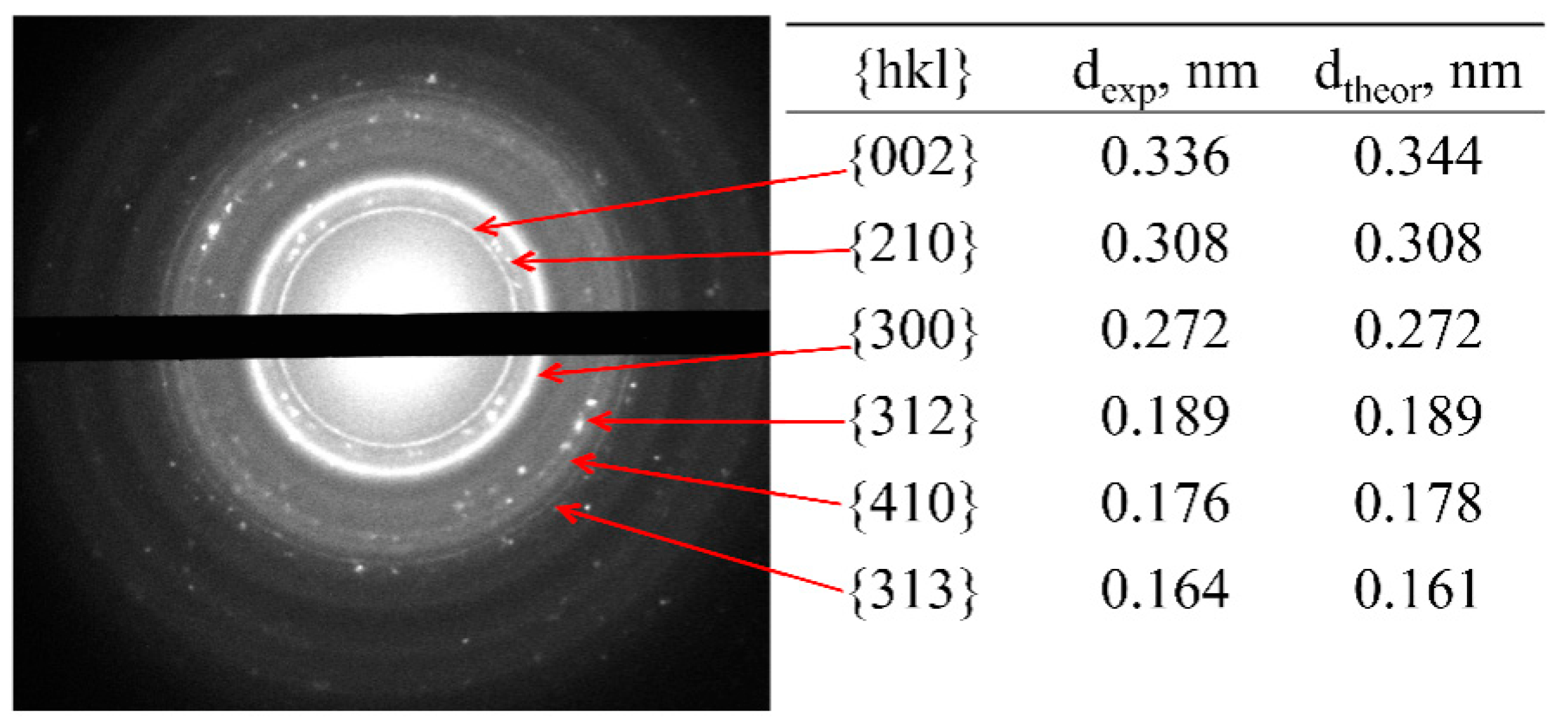
2.4. X-ray Phase Analysis
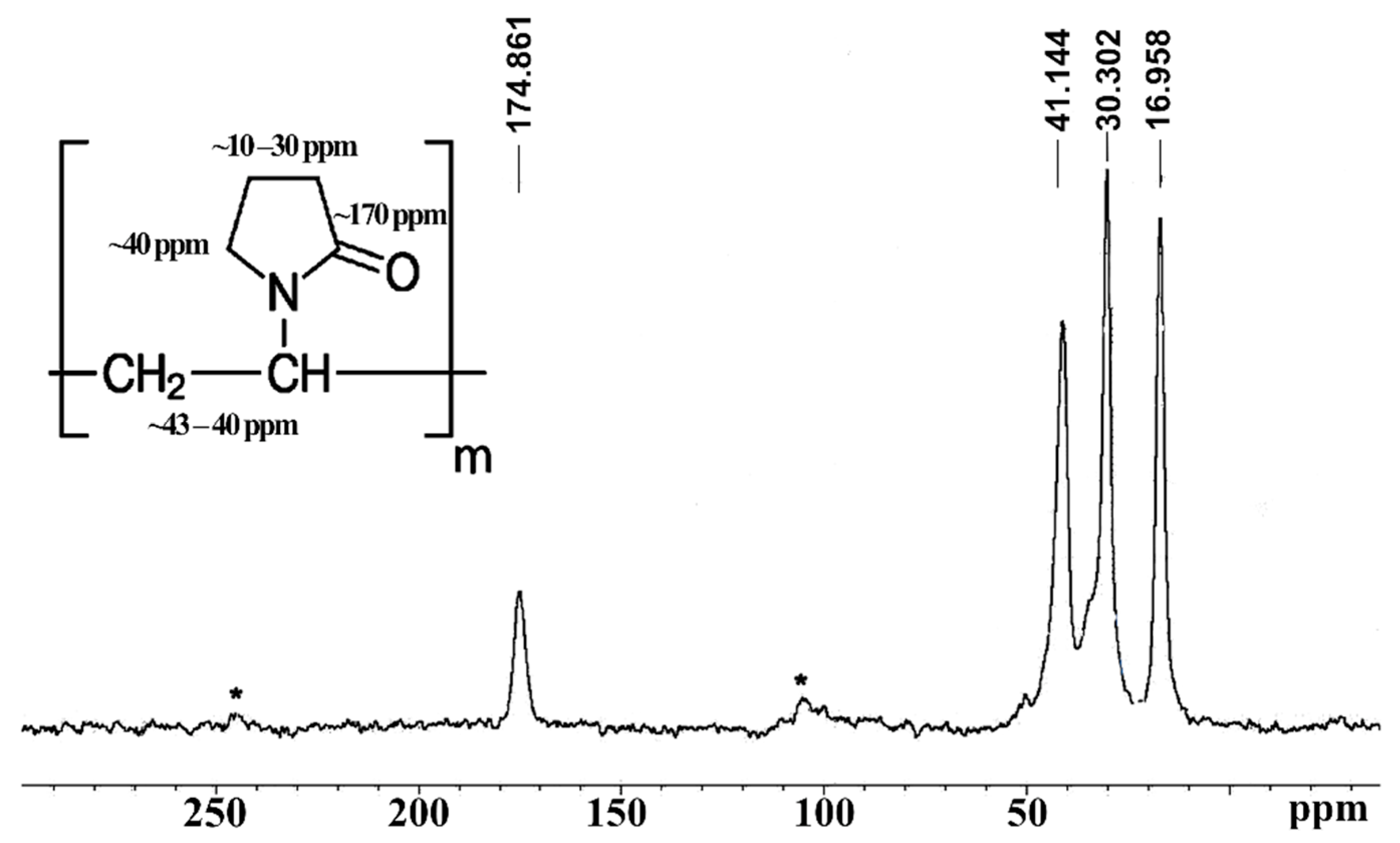
2.5. NMR Spectral Analysis
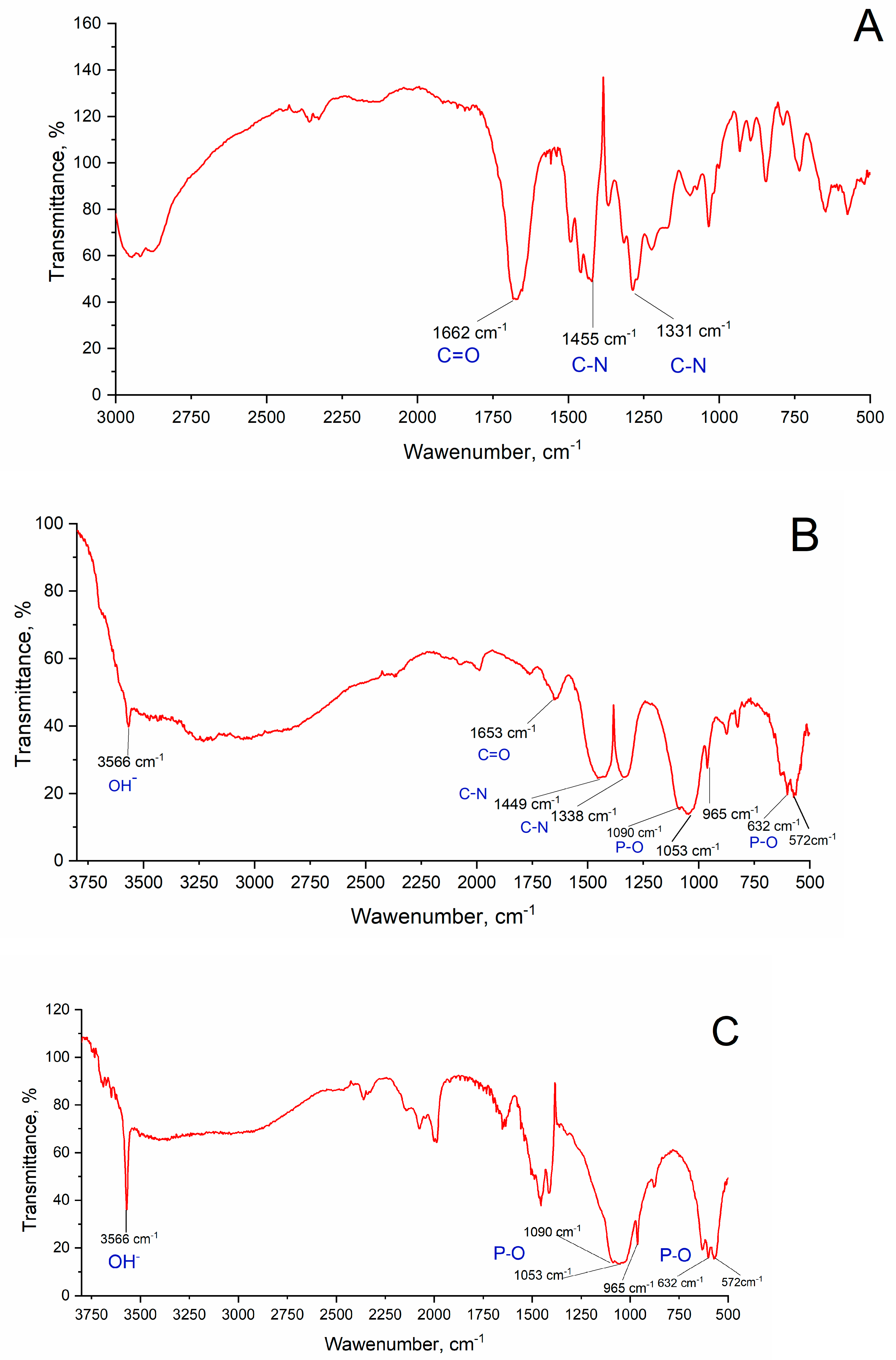
2.6. FTIR Spectral Analysis
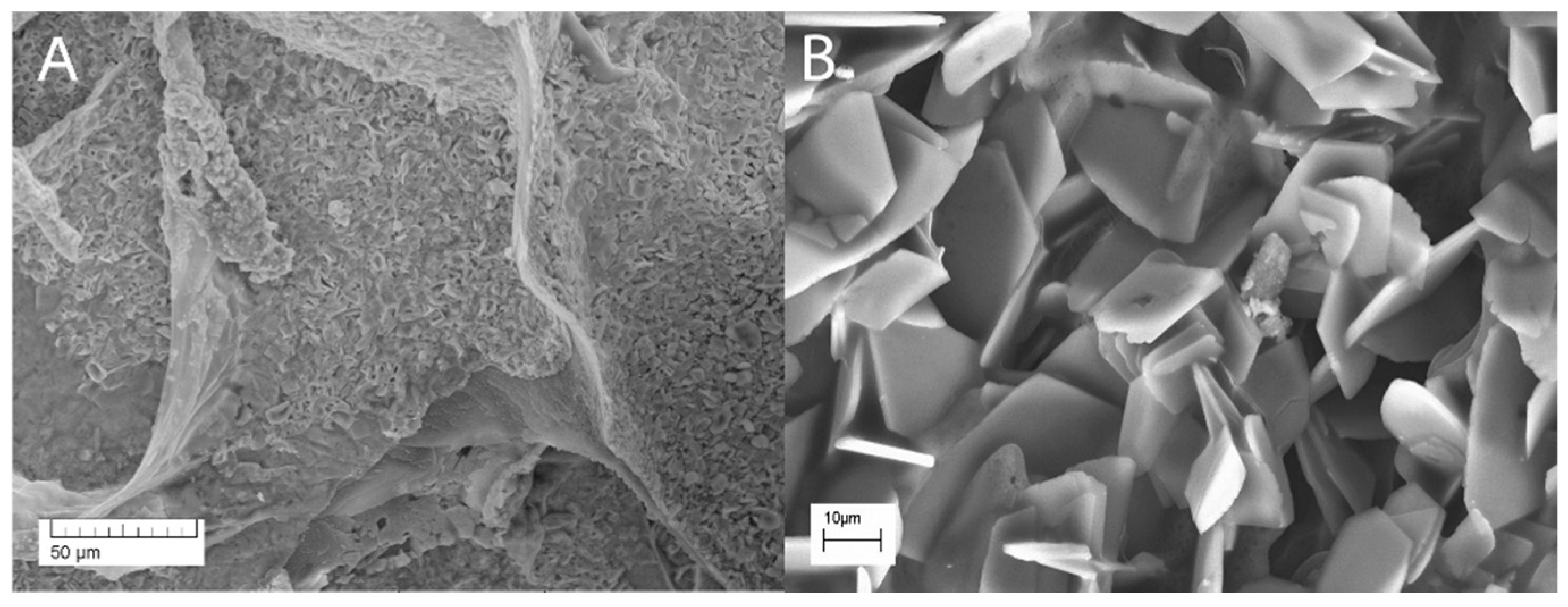
2.7. Scanning Electron Microscopy, Transmission Electron Microscopy and Specific Surface Area Analysis
2.8. Specific Surface Area Analysis
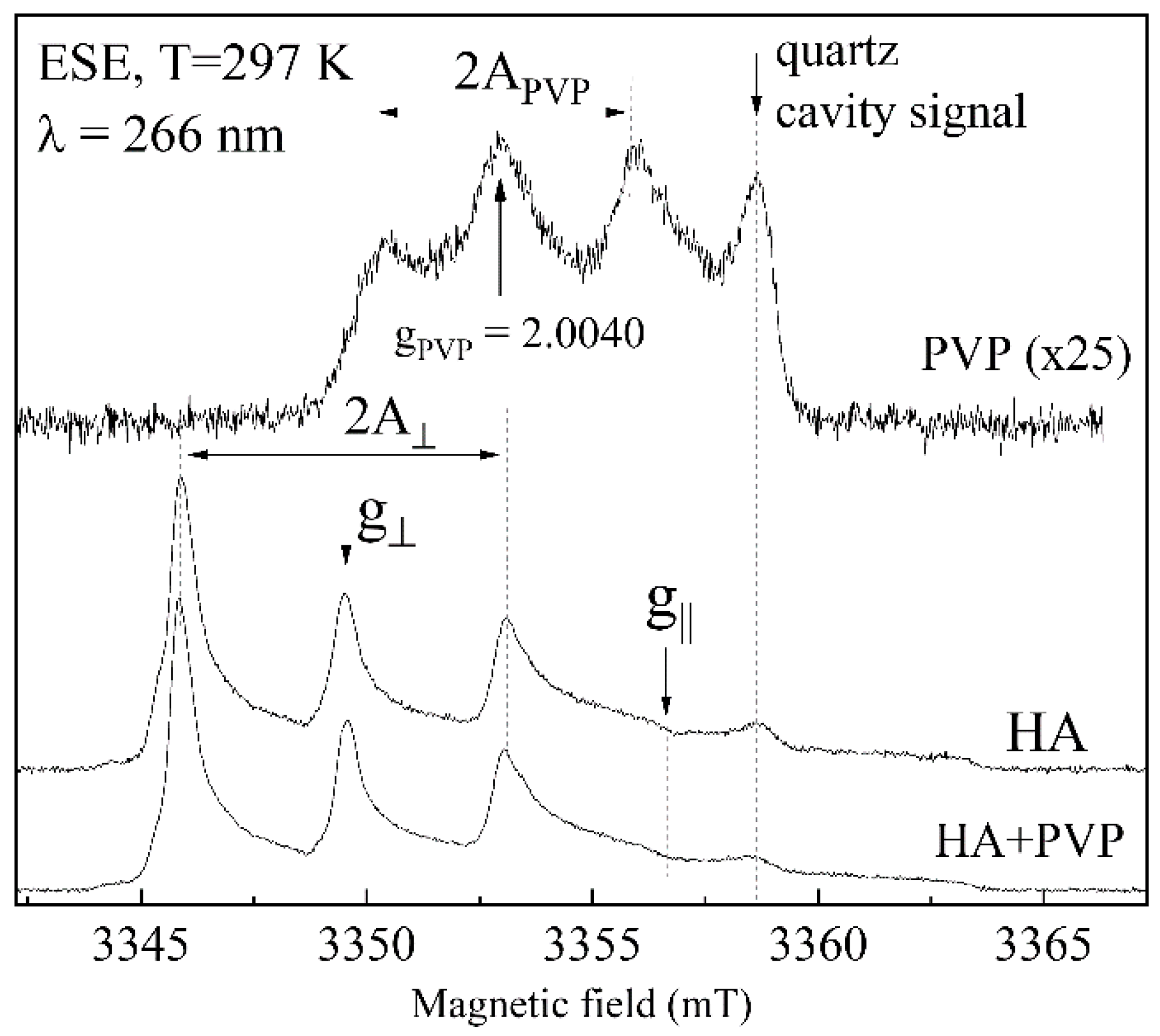
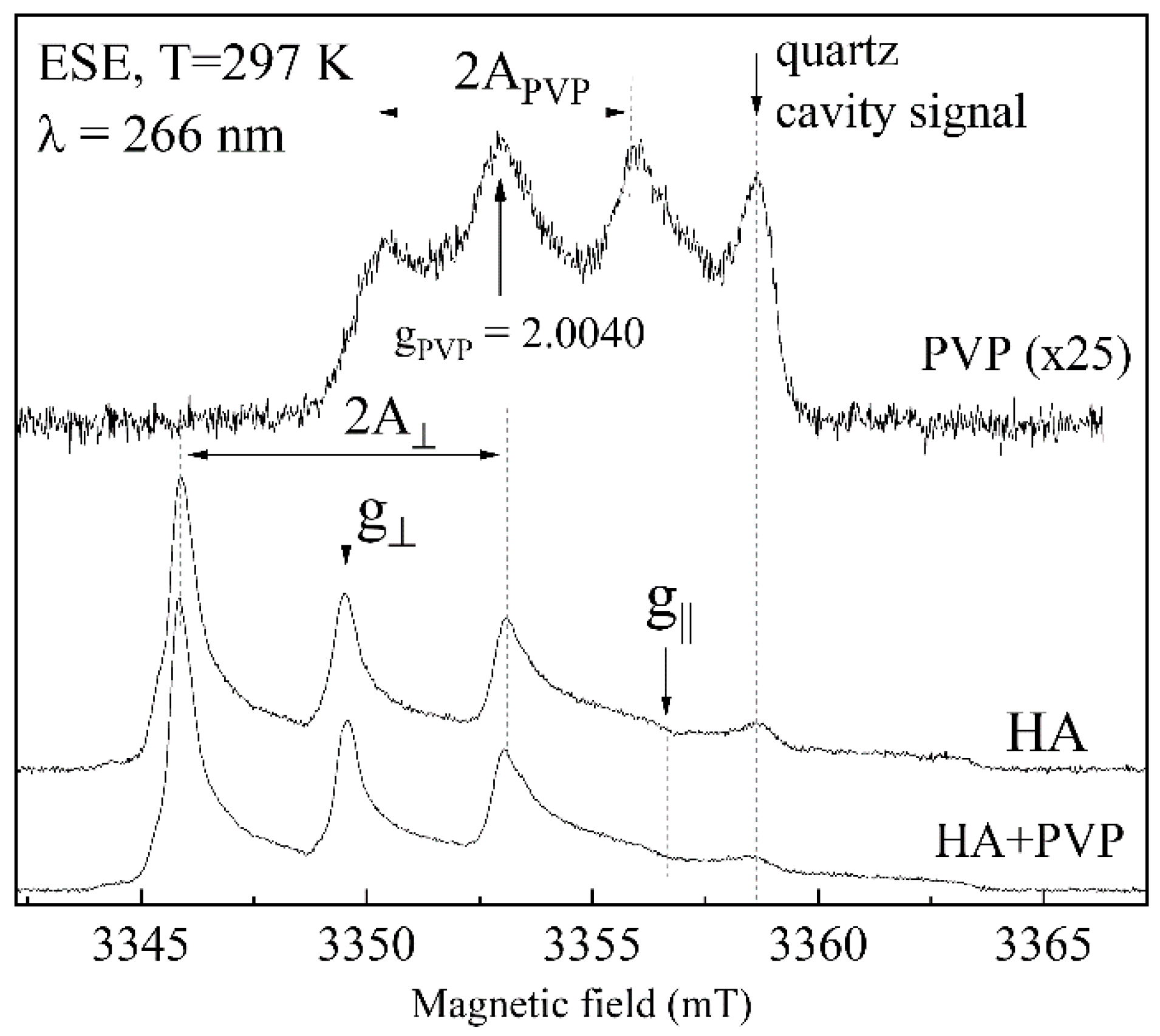
2.9. Electron Paramagnetic Resonance Investigations
2.10. Swelling Properties Investigation
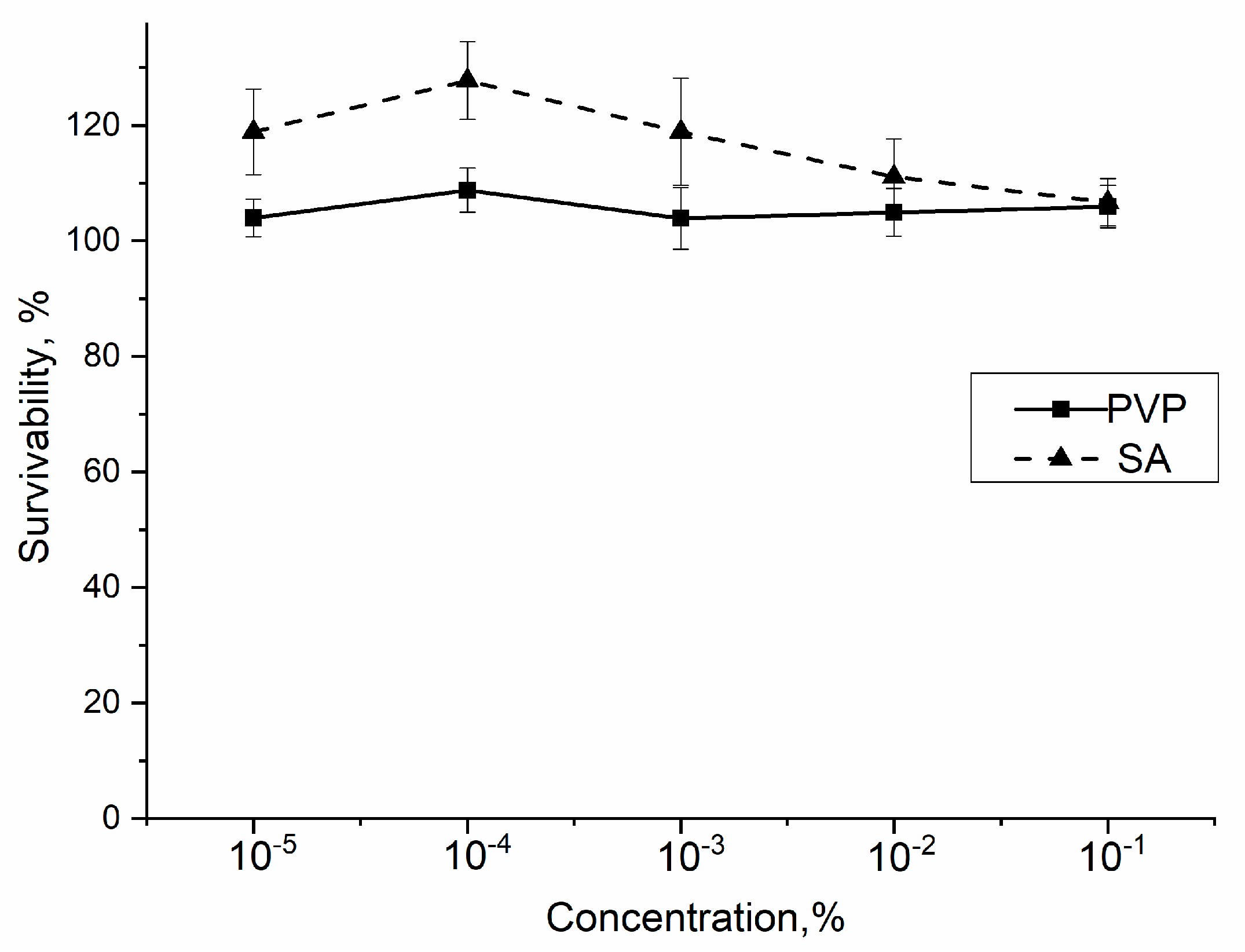
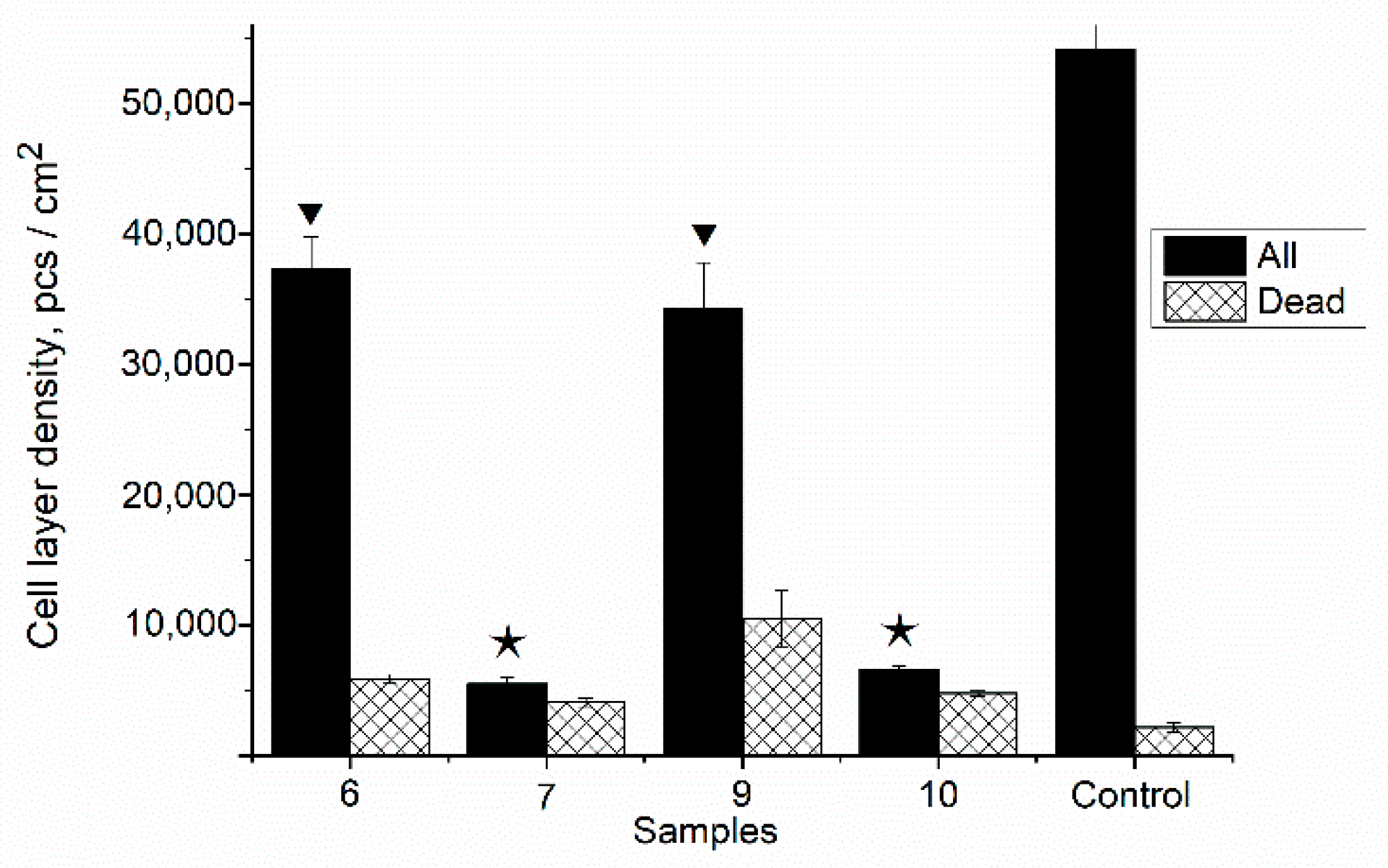
2.11. In Vitro Studies
3. Results and Discussion
3.1. TGA Investigation
3.2. X-ray Analysis
3.3. NMR Analysis
3.4. FTIR Analysis
3.5. Dimensional Analysis
3.6. EPR Analysis
3.7. Microstructural Analysis
3.8. The Crosslinking of Composite PVP-SA-HA Film
3.9. The Swelling Data
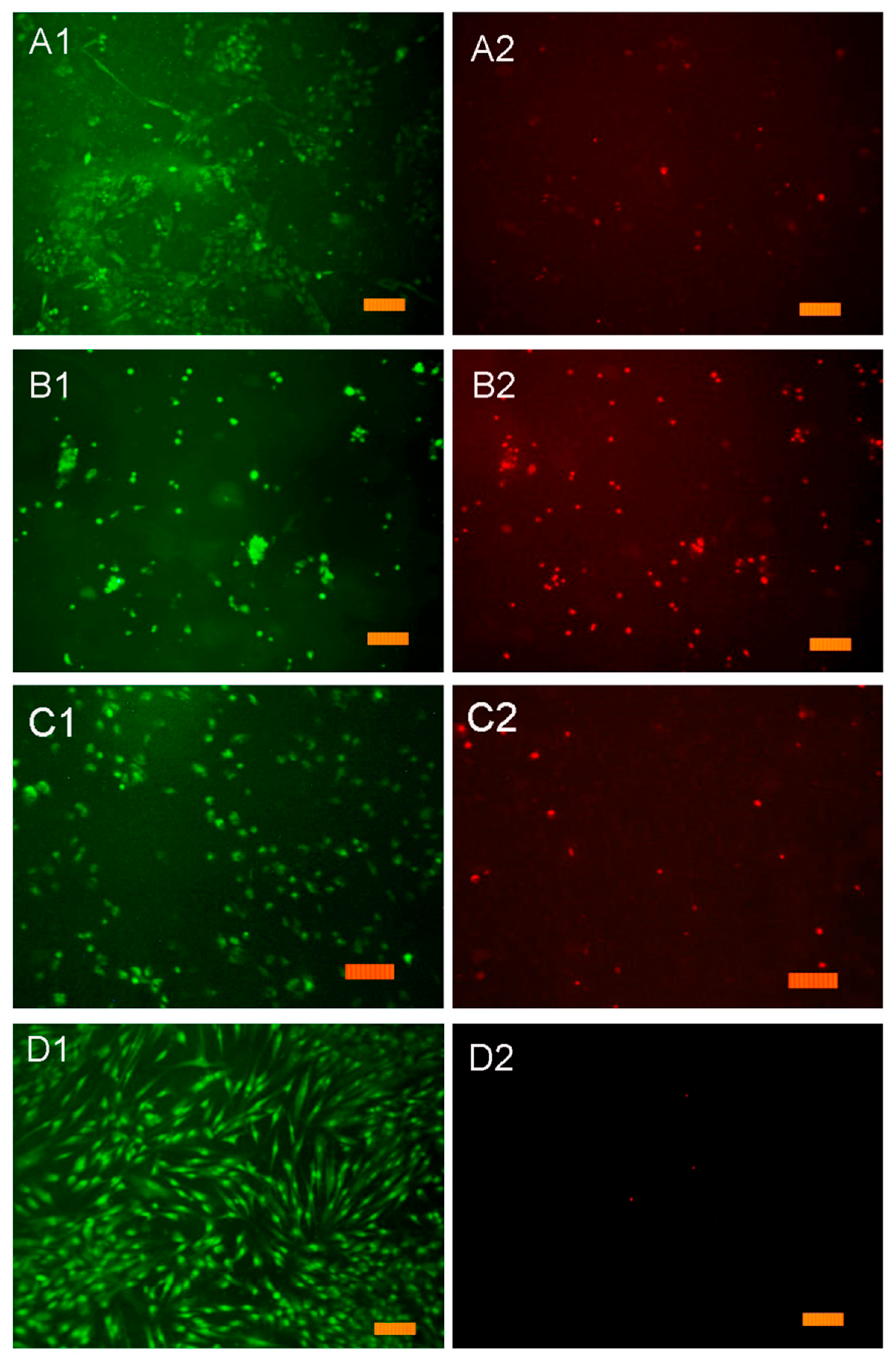
3.10. In Vitro
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hench, L.L. Bioactive Ceramics. Ann. N. Y. Acad. Sci. 1988, 523, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Schlickewei, C.W.; Laaff, G.; Andresen, A.; Klatte, T.O.; Rueger, J.M.; Ruesing, J.; Epple, M.; Lehmann, W. Bone augmentation using a new injectable bone graft substitute by combining calcium phosphate and bisphosphonate as composite—An animal model. J. Orthop. Surg. Res. 2015, 10, 116. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, A.-M. A review of calcium phosphate cements and acrylic bone cements as injectable materials for bone repair and implant fixation. J. Appl. Biomater. Funct. Mater. 2019, 17, 228080001987259. [Google Scholar] [CrossRef]
- Eliaz, N.; Metoki, N. Calcium Phosphate Bioceramics: A Review of Their History, Structure, Properties, Coating Technologies and Biomedical Applications. Materials 2017, 10, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakasam, M.; Locs, J.; Salma-Ancane, K.; Loca, D.; Largeteau, A.; Berzina-Cimdina, L. Fabrication, Properties and Applications of Dense Hydroxyapatite: A Review. J. Funct. Biomater. 2015, 6, 1099–1140. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, S.; Yamaguchi, S.; Barbani, N.; Cazzola, M.; Cristallini, C.; Miola, M.; Vernè, E.; Spriano, S. Bioactive materials: In vitro investigation of different mechanisms of hydroxyapatite precipitation. Acta Biomater. 2020, 102, 468–480. [Google Scholar] [CrossRef]
- Markel, M.D. Bone Grafts and Bone Substitutes. In Equine Fract. Repair, 1st ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2020; p. 916. ISBN 9781119108740. [Google Scholar] [CrossRef]
- Kalita, S.J.; Bhardwaj, A.; Bhatt, H.A. Nanocrystalline calcium phosphate ceramics in biomedical engineering. Mater. Sci. Eng. C 2007, 27, 441–449. [Google Scholar] [CrossRef]
- Sobczak-Kupiec, A.; Pluta, K.; Drabczyk, A.; Włoś, M.; Tyliszczak, B. Synthesis and characterization of ceramic-polymer composites containing bioactive synthetic hydroxyapatite for biomedical applications. Ceram. Int. 2018, 44, 13630–13638. [Google Scholar] [CrossRef]
- Park, S.-B.; Lih, E.; Park, K.-S.; Joung, Y.K.; Han, D.K. Biopolymer-based functional composites for medical applications. Prog. Polym. Sci. 2017, 68, 77–105. [Google Scholar] [CrossRef]
- Palmer, L.; Newcomb, C.J.; Kaltz, S.R.; Spoerke, E.D.; Stupp, S.I. Biomimetic Systems for Hydroxyapatite Mineralization Inspired by Bone and Enamel. Chem. Rev. 2008, 108, 4754–4783. [Google Scholar] [CrossRef] [Green Version]
- Šupová, M. Problem of hydroxyapatite dispersion in polymer matrices: A review. J. Mater. Sci. Mater. Med. 2009, 20, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regi, M.; Navarrete, D.A.A. Biomimetic Nanoceramics in Clinical Use; Royal Society of Chemistry: London, UK, 2008; ISBN 978-0-85404-142-8. [Google Scholar]
- Wahl, D.A.; Czernuszka, J.T. Collagen-Hydroxyapatite Composites for Hard Tissue Repair. Eur. Cells Mater. 2006, 11, 43–56. [Google Scholar] [CrossRef]
- Venugopal, J.; Low, S.; Choon, A.T.; Sampath Kumar, T.S.; Ramakrishna, S. Mineralization of osteoblasts with electrospun collagen/hydroxyapatite nanofibers. J. Mater. Sci. Mater. Med. 2008, 19, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Song, J.H.; Kim, H.E. Nanofiber Generation of Gelatin-Hydroxyapatite Biomimetics for Guided Tissue Regeneration. Adv. Funct. Mater. 2005, 15, 1988–1994. [Google Scholar] [CrossRef]
- Kushioka, J.; Kaito, T.; Makino, T.; Fujiwara, H.; Tsukazaki, H.; Takenaka, S.; Sakai, Y.; Yoshikawa, H. Difference in the fusion rate and bone formation between artificial bone and iliac autograft inside an inter-body fusion cage–A comparison between porous hydroxyapatite/type 1 collagen composite and autologous iliac bone. J. Orthop. Sci. 2018, 23, 622–626. [Google Scholar] [CrossRef]
- Bleek, K.; Taubert, A. New developments in polymer-controlled, bioinspired calcium phosphate mineralization from aqueous solution. Acta Biomater. 2013, 9, 6283–6321. [Google Scholar] [CrossRef] [PubMed]
- Lebourg, M.; Antón, J.S.; Ribelles, J.L.G. Characterization of calcium phosphate layers grown on polycaprolactone for tissue engineering purposes. Compos. Sci. Technol. 2010, 70, 1796–1804. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Kim, S.J.; Hwang, M.J.; Song, H.J.; Park, Y.-J. Biomimetic fabrication of calcium phosphate/chitosan nanohybrid composite in modified simulated body fluids. Express Polym. Lett. 2017, 11, 14–20. [Google Scholar] [CrossRef]
- Patil, S.B.; Inamdar, S.Z.; Reddy, K.R.; Raghu, A.V.; Soni, S.K.; Kulkarni, R.V. Novel biocompatible poly(acrylamide)-grafted-dextran hydrogels: Synthesis, characterization and biomedical applications. J. Microbiol. Methods 2019, 159, 200–210. [Google Scholar] [CrossRef]
- Nasibi, S.; Khoramabadi, H.N.; Arefian, M.; Hojjati, M.; Tajzad, I.; Mokhtarzade, A.; Mazhar, M.; Jamavari, A. A review of Polyvinyl alcohol / Carboxiy methyl cellulose (PVA/CMC) composites for various applications. J. Compos. Compd. 2020, 2, 68–75. [Google Scholar] [CrossRef]
- Hasan, A.; Waibhaw, G.; Tiwari, S.; Dharmalingam, K.; Shukla, I.; Pandey, L.M. Fabrication and characterization of chitosan, polyvinylpyrrolidone, and cellulose nanowhiskers nanocomposite films for wound healing drug delivery application. J. Biomed. Mater. Res. Part A 2017, 105, 2391–2404. [Google Scholar] [CrossRef] [PubMed]
- Miculescu, F.; Maidaniuc, A.; Voicu, S.I.; Thakur, V.K.; Stan, G.E.; Ciocan, L.T. Progress in Hydroxyapatite–Starch Based Sustainable Biomaterials for Biomedical Bone Substitution Applications. ACS Sustain. Chem. Eng. 2017, 5, 8491–8512. [Google Scholar] [CrossRef] [Green Version]
- Lang, W.-Z.; Shen, J.-P.; Wei, Y.-T.; Wu, Q.-Y.; Wang, J.; Guo, Y.-J. Precipitation kinetics, morphologies, and properties of poly(vinyl butyral) hollow fiber ultrafiltration membranes with respect to polyvinylpyrrolidone molecular weight. Chem. Eng. J. 2013, 225, 25–33. [Google Scholar] [CrossRef]
- Moffitt, E.A. Blood Substitutes. Can. Anaesth Soc. J. 1975, 22, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Chirila, T.V.; Vijayasekaran, S.; Shen, W.; Lou, X.; Dalton, P.D. Biodegradation in Vitro and Retention in the Rabbit Eye of Crosslinked Poly(1-Vinyl-2-Pyrrolidinone) Hydrogel as a Vitreous Substitute. J Biomed Mater Res 1998, 39, 650–659. [Google Scholar] [CrossRef]
- Öri, F.; Dietrich, R.; Ganz, C.; Dau, M.; Wolter, D.; Kasten, A.; Gerber, T.; Frerich, B. Silicon-dioxide−polyvinylpyrrolidone as a wound dressing for skin defects in a murine model. J. Cranio-Maxillofac. Surg. 2017, 45, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E.; Rimmer, S.; MacNeil, S. Examination of the effects of poly(N-vinylpyrrolidinone) hydrogels in direct and indirect contact with cells. Biomaterials 2006, 27, 2806–2812. [Google Scholar] [CrossRef]
- Teodorescu, M.; Bercea, M.; Morariu, S. Biomaterials of PVA and PVP in medical and pharmaceutical applications: Perspectives and challenges. Biotechnol. Adv. 2019, 37, 109–131. [Google Scholar] [CrossRef]
- Ng, S.L.; Such, G.K.; Johnston, A.P.R.; Antequera-García, G.; Caruso, F. Controlled release of DNA from poly(vinylpyrrolidone) capsules using cleavable linkers. Biomaterials 2011, 32, 6277–6284. [Google Scholar] [CrossRef]
- D’Souza, A.J.M.; Schowen, R.L.; Topp, E.M. Polyvinylpyrrolidone–drug conjugate: Synthesis and release mechanism. J. Control. Release 2004, 94, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Pornpitchanarong, C.; Rojanarata, T.; Opanasopit, P.; Ngawhirunpat, T.; Patrojanasophon, P. Synthesis of novel N-vinylpyrrolidone/acrylic acid nanoparticles as drug delivery carriers of cisplatin to cancer cells. Colloids Surf. B Biointerfaces 2020, 185, 110566. [Google Scholar] [CrossRef] [PubMed]
- Chimal-Monroy, J.; Bravo-Ruiz, T.; Furuzawa-Carballeda, G.J.; Lira, J.M.; DE LA Cruz, J.C.; Almazan, A.; Krotzsch-Gomez, F.E.; Arrellin, G.; DE Leon, L.D. Collagen-PVP Accelerates New Bone Formation of Experimentally Induced Bone Defects in Rat Skull and Promotes the Expression of Osteopontin and SPARC during Bone Repair of Rat Femora Fracturesa. Ann. N. Y. Acad. Sci. 1998, 857, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Sharma, V. Crosslinking of poly(vinylpyrrolidone)/acrylic acid with tragacanth gum for hydrogels formation for use in drug delivery applications. Carbohydr. Polym. 2017, 157, 185–195. [Google Scholar] [CrossRef]
- Buchsel, P.C.; Murphy, P.J.M. Polyvinylpyrrolidone–sodium hyaluronate gel (Gelclair®): A bioadherent oral gel for the treatment of oral mucositis and other painful oral lesions. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1449–1454. [Google Scholar] [CrossRef]
- Lavanya, K.; Chandran, S.V.; Balagangadharan, K.; Selvamurugan, N. Temperature- and pH-responsive chitosan-based injectable hydrogels for bone tissue engineering. Mater. Sci. Eng. C 2020, 111, 110862. [Google Scholar] [CrossRef] [PubMed]
- Lozinsky, V.I.; Damshkaln, L.G.; Shaskol’Skii, B.L.; Babushkina, T.A.; Kurochkin, I.N.; Kurochkin, I.I. Study of cryostructuring of polymer systems: 27. Physicochemical properties of poly(vinyl alcohol) cryogels and specific features of their macroporous morphology. Colloid J. 2007, 69, 747–764. [Google Scholar] [CrossRef]
- Wang, B.; Wan, Y.; Zheng, Y.; Lee, X.; Liu, T.; Yu, Z.; Huang, J.; Ok, Y.S.; Chen, J.; Gao, B. Alginate-based composites for environmental applications: A critical review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 318–356. [Google Scholar] [CrossRef]
- Augst, A.D.; Kong, H.J.; Mooney, D.J. Alginate Hydrogels as Biomaterials. Macromol. Biosci. 2006, 6, 623–633. [Google Scholar] [CrossRef]
- Eiselt, P.; Yeh, J.; Latvala, R.K.; Shea, L.D.; Mooney, D.J. Porous carriers for biomedical applications based on alginate hydrogels. Biomaterials 2000, 21, 1921–1927. [Google Scholar] [CrossRef]
- Kreeger, P.K.; Fernandes, N.N.; Woodruff, T.K.; Shea, L.D. Regulation of Mouse Follicle Development by Follicle-Stimulating Hormone in a Three-Dimensional In Vitro Culture System Is Dependent on Follicle Stage and Dose1. Biol. Reprod. 2005, 73, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Pangas, S.A.; Saudye, H.; Shea, L.D.; Woodruff, T.K. Novel Approach for the Three-Dimensional Culture of Granulosa Cell–Oocyte Complexes. Tissue Eng. 2003, 9, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, E.R.; Xu, M.; Woodruff, T.; Shea, L.D. Physical properties of alginate hydrogels and their effects on in vitro follicle development. Biomaterials 2007, 28, 4439–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Woodruff, T.K.; Shea, L.D. Bioengineering and the ovarian follicle. Hematop. Growth Factors Oncol. 2007, 138, 75–82. [Google Scholar] [CrossRef]
- Gao, X.; Yu, Z.; Liu, B.; Yang, J.; Yang, X.; Yu, Y. A smart drug delivery system responsive to pH/enzyme stimuli based on hydrophobic modified sodium alginate. Eur. Polym. J. 2020, 133, 109779. [Google Scholar] [CrossRef]
- García-Astrain, C.; Avérous, L. Synthesis and evaluation of functional alginate hydrogels based on click chemistry for drug delivery applications. Carbohydr. Polym. 2018, 190, 271–280. [Google Scholar] [CrossRef]
- Pawar, V.; Topkar, H.; Srivastava, R. Chitosan nanoparticles and povidone iodine containing alginate gel for prevention and treatment of orthopedic implant associated infections. Int. J. Biol. Macromol. 2018, 115, 1131–1141. [Google Scholar] [CrossRef]
- Teleky, B.-E.; Vodnar, D.C. Recent Advances in Biotechnological Itaconic Acid Production, and Application for a Sustainable Approach. Polymers 2021, 13, 3574. [Google Scholar] [CrossRef]
- Hong, X.; Ding, H.; Li, J.; Xue, Y.; Sun, L.; Ding, F. Poly(acrylamide-co-acrylic acid)/chitosan semi-interpenetrating hydrogel for pressure sensor and controlled drug release. Polym. Adv. Technol. 2021, 8, 3050–3058. [Google Scholar] [CrossRef]
- Dhand, A.P.; Galarraga, J.H.; Burdick, J.A. Enhancing Biopolymer Hydrogel Functionality through Interpenetrating Networks. Trends Biotechnol. 2021, 39, 519–538. [Google Scholar] [CrossRef]
- Werner, S.; Grose, R. Regulation of Wound Healing by Growth Factors and Cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Matricardi, P.; Di Meo, C.; Coviello, T.; Hennink, W.E.; Alhaique, F. Interpenetrating Polymer Networks polysaccharide hydrogels for drug delivery and tissue engineering. Adv. Drug Deliv. Rev. 2013, 65, 1172–1187. [Google Scholar] [CrossRef]
- Priya, S.G.; Gupta, A.; Jain, E.; Sarkar, J.; Damania, A.; Jagdale, P.R.; Chaudhari, B.P.; Gupta, K.C.; Kumar, A. Bilayer Cryogel Wound Dressing and Skin Regeneration Grafts for the Treatment of Acute Skin Wounds. ACS Appl. Mater. Interfaces 2016, 8, 15145–15159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J. A Mild and Efficient Biomimetic Synthesis of Rodlike Hydroxyapatite Particles with a High Aspect Ratio Using Polyvinylpyrrolidone As Capping Agent. Cryst. Growth Des. 2008, 8, 2101–2107. [Google Scholar] [CrossRef]
- Costa, M.J.; Marques, A.M.; Pastrana, L.M.; Teixeira, J.A.; Sillankorva, S.M.; Cerqueira, M.A. Physicochemical properties of alginate-based films: Effect of ionic crosslinking and mannuronic and guluronic acid ratio. Food Hydrocoll. 2018, 81, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Hilbig, J.; Hartlieb, K.; Gibis, M.; Herrmann, K.; Weiss, J. Rheological and mechanical properties of alginate gels and films containing different chelators. Food Hydrocoll. 2020, 101, 105487. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Grabovenko, F.I.; Fomin, A.S.; Barinov, S.M.; Murzakhanov, F.F.; Ahmed, A.I.; Mamin, G.V. Mineral-polymer composites based on hydroxyapatite with polyvynylpyrrolidon for medicine. Dokl. Chem. 2019, 487, 270–274. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Formin, A.S.; Barinov, S.M.; Trofimchuk, E.S. Method for Producing Porous Materials from Sodium Alginate and Polyvinylpyrrolidone Containing Calcium Phosphates. Patent RU N 2705084, 1 November 2019. [Google Scholar]
- Fadeeva, I.V.; Lazoryak, B.I.; Davidova, G.A.; Murzakhanov, F.F.; Gabbasov, B.F.; Petrakova, N.V.; Fosca, M.; Barinov, S.M.; Vadalà, G.; Uskoković, V.; et al. Antibacterial and cell-friendly copper-substituted tricalcium phosphate ceramics for biomedical implant applications. Mater. Sci. Eng. C 2021, 129, 112410. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, Low-Technology MIC Determination with Clinical Mycobacterium tuberculosis Isolates by Using the Microplate Alamar Blue Assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Guesmi, Y.; Agougui, H.; Jabli, M.; Alsharabasy, A.M. Bioactive composites of hydroxyapatite/polyvinylpyrrolidone for bone regeneration applications. Chem. Eng. Commun. 2018, 206, 279–288. [Google Scholar] [CrossRef]
- Langroudi, M.M.; Saravani, M.G.; Nouri, A. Surfactant-Assisted Synthesis of Polyvinylpyrrolidone-hydroxyapatite Composites as a Bone Filler. J. Appl. Biomater. Funct. Mater. 2017, 15, e334–e340. [Google Scholar] [CrossRef] [Green Version]
- Nicoara, A.I.; Stoica, A.E.; Ene, D.-I.; Vasile, B.S.; Holban, A.M.; Neacsu, I.A. In Situ and Ex Situ Designed Hydroxyapatite: Bacterial Cellulose Materials with Biomedical Applications. Materials 2020, 13, 4793. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds: Tables of Spectral Data, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 978-3-540-93810-1. [Google Scholar]
- Sofronia, A.M.; Baies, R.; Anghel, E.M.; Marinescu, C.A.; Tanasescu, S. Thermal and structural characterization of synthetic and natural nanocrystalline hydroxyapatite. Mater. Sci. Eng. C 2014, 43, 153–163. [Google Scholar] [CrossRef]
- Nathanael, A.J.; Seo, Y.H.; Oh, T.H. PVP Assisted Synthesis of Hydroxyapatite Nanorods with Tunable Aspect Ratio and Bioactivity. J. Nanomater. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Fomin, A.S.; Barinov, S.M.; Ievlev, V.M.; Fadeeva, I.V.; Komlev, V.S.; Belonogov, E.K.; Turaeva, T.L. Nanosized hydroxyapatite synthesized by precipitation in a gelatin solution. Dokl. Chem. Nauka/Interperiodica 2006, 411, 219–222. [Google Scholar] [CrossRef]
- Gabbasov, B.; Gafurov, M.; Starshova, A.; Shurtakova, D.; Murzakhanov, F.; Mamin, G.; Orlinskii, S. Conventional, pulsed and high-field electron paramagnetic resonance for studying metal impurities in calcium phosphates of biogenic and synthetic origins. J. Magn. Magn. Mater. 2019, 470, 109–117. [Google Scholar] [CrossRef]
- Rau, J.V.; Fadeeva, I.V.; Fomin, A.S.; Barbaro, K.; Galvano, E.; Ryzhov, A.P.; Murzakhanov, F.; Gafurov, M.; Orlinskii, S.; Antoniac, I.V.; et al. Sic Parvis Magna: Manganese-Substituted Tricalcium Phosphate and Its Biophysical Properties. ACS Biomater. Sci. Eng. 2019, 5, 6632–6644. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Gafurov, M.R.; Kiiaeva, I.A.; Orlinskii, S.B.; Kuznetsova, L.M.; Filippov, Y.Y.; Fomin, A.S.; Davydova, G.A.; Selezneva, I.I.; Barinov, S.M. Tricalcium Phosphate Ceramics Doped with Silver, Copper, Zinc, and Iron (III) Ions in Concentrations of Less Than 0.5 wt.% for Bone Tissue Regeneration. BioNanoScience 2017, 7, 434–438. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Kalita, V.I.; Komlev, D.I.; Radiuk, A.A.; Fomin, A.S.; Davidova, G.A.; Fursova, N.K.; Murzakhanov, F.F.; Gafurov, M.R.; Fosca, M.; et al. In Vitro Properties of Manganese-Substituted Tricalcium Phosphate Coatings for Titanium Biomedical Implants Deposited by Arc Plasma. Materials 2020, 13, 4411. [Google Scholar] [CrossRef]
- Gafurov, M.; Biktagirov, T.; Mamin, G.; Orlinskii, S. A DFT, X- and W-band EPR and ENDOR Study of Nitrogen-Centered Species in (Nano)Hydroxyapatite. Appl. Magn. Reson. 2014, 45, 1189–1203. [Google Scholar] [CrossRef]
- Biktagirov, T.; Gafurov, M.; Mamin, G.; Klimashina, E.; Putlayev, V.; Orlinskii, S. Combination of EPR Measurements and DFT Calculations to Study Nitrate Impurities in the Carbonated Nanohydroxyapatite. J. Phys. Chem. A 2014, 118, 1519–1526. [Google Scholar] [CrossRef]
- Frunze, N.; Berlin, A. Sensitized formation of radicals in polymers under UV-irradiation. Polym. Sci. USSR 1969, 11, 1638–1647. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Trofimchuk, E.S.; Dedushenko, S.K.; Fomin, A.S.; Davydova, G.A.; Selezneva, I.I.; Perfiliev, Y.D.; Barinov, S.M. Methylcellulose films partially crosslinked by iron compounds for medical applications. Mater. Today Commun. 2019, 18, 54–59. [Google Scholar] [CrossRef]
- Becker, T.A.; Kipke, D.R.; Brandon, T. Calcium alginate gel: A biocompatible and mechanically stable polymer for endovascular embolization. J. Biomed. Mater. Res. 2001, 54, 76–86. [Google Scholar] [CrossRef]
- Ho, S.S.; Murphy, K.C.; Binder, B.Y.; Vissers, C.B.; Leach, J.K. Increased Survival and Function of Mesenchymal Stem Cell Spheroids Entrapped in Instructive Alginate Hydrogels. Stem Cells Transl. Med. 2016, 5, 773–781. [Google Scholar] [CrossRef]
- Guo, X.; Huang, S.; Sun, J.; Wang, F. Comparison of the Cytotoxicities and Wound Healing Effects of Hyaluronan, Carbomer, and Alginate on Skin Cells In Vitro. Adv. Ski. Wound Care 2015, 28, 410–414. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Sample Designation | Sample Composition | Polymer Concentration in Polymer-HA * Mixture, wt % | HA/Polymer Ratio (Mass) |
|---|---|---|---|---|
| 1 | PVP *-HA | PVP, HA in situ | 2.76 | 0.028 |
| 2 | PVP-HA | PVP, HA in situ | 5.52 | 0.014 |
| 3 | PVP-HA | PVP, HA in situ | 11.00 | 0.007 |
| 4 | PVP-HA | PVP, HA ex situ | 11.00 | 0.007 |
| 5 | PVP-HA | PVP, HA ex situ | 11.00 | 0.028 |
| Films for Swelling and In Vitro Investigations | ||||
| 6 | PVP-SA * | PVP:SA = 1:1 | 5.52 | - |
| 7 | PVP-SA-HA | PVP:SA = 1:1, HA in situ | 5.52 | 0.014 |
| 8 | PVP-SA-HA | PVP:SA = 1:1, HA ex situ | 5.52 | 0.014 |
| 9 | PVP-SA | PVP:SA = 1:2 | 5.52 | - |
| 10 | PVP-SA-HA | PVP:SA = 1:2, HA in situ | 5.52 | 0.014 |
| 11 | PVP-SA-HA | PVP:SA = 1:2, HA ex situ | 5.52 | 0.014 |
| Sample | Polymer Concentration in PVP *-HA * Mixture, wt % | PVP/HA Ratio, wt % | Specific Surface Area (BET) *, m2/g | CSR *, nm | Particles Size from TEM Data, nm |
|---|---|---|---|---|---|
| 1 | 2.76 | 0.028 | 16.7 | 17.7 | 30 |
| 2 | 5.52 | 0.014 | 19 | 19.6 | 33 |
| 3 | 11.00 | 0.07 | 37 | 17.9 | 45 |
| HA | 0 | - | 18 | 31.7 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadeeva, I.V.; Trofimchuk, E.S.; Forysenkova, A.A.; Ahmed, A.I.; Gnezdilov, O.I.; Davydova, G.A.; Kozlova, S.G.; Antoniac, A.; Rau, J.V. Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications. Polymers 2021, 13, 3989. https://doi.org/10.3390/polym13223989
Fadeeva IV, Trofimchuk ES, Forysenkova AA, Ahmed AI, Gnezdilov OI, Davydova GA, Kozlova SG, Antoniac A, Rau JV. Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications. Polymers. 2021; 13(22):3989. https://doi.org/10.3390/polym13223989
Chicago/Turabian StyleFadeeva, Inna V., Elena S. Trofimchuk, Anna A. Forysenkova, Abdulrahman I. Ahmed, Oleg I. Gnezdilov, Galina A. Davydova, Svetlana G. Kozlova, Aurora Antoniac, and Julietta V. Rau. 2021. "Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications" Polymers 13, no. 22: 3989. https://doi.org/10.3390/polym13223989
APA StyleFadeeva, I. V., Trofimchuk, E. S., Forysenkova, A. A., Ahmed, A. I., Gnezdilov, O. I., Davydova, G. A., Kozlova, S. G., Antoniac, A., & Rau, J. V. (2021). Composite Polyvinylpyrrolidone–Sodium Alginate—Hydroxyapatite Hydrogel Films for Bone Repair and Wound Dressings Applications. Polymers, 13(22), 3989. https://doi.org/10.3390/polym13223989