
Temperature Dependence of Conformational Relaxation of Poly(ethylene oxide) Melts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
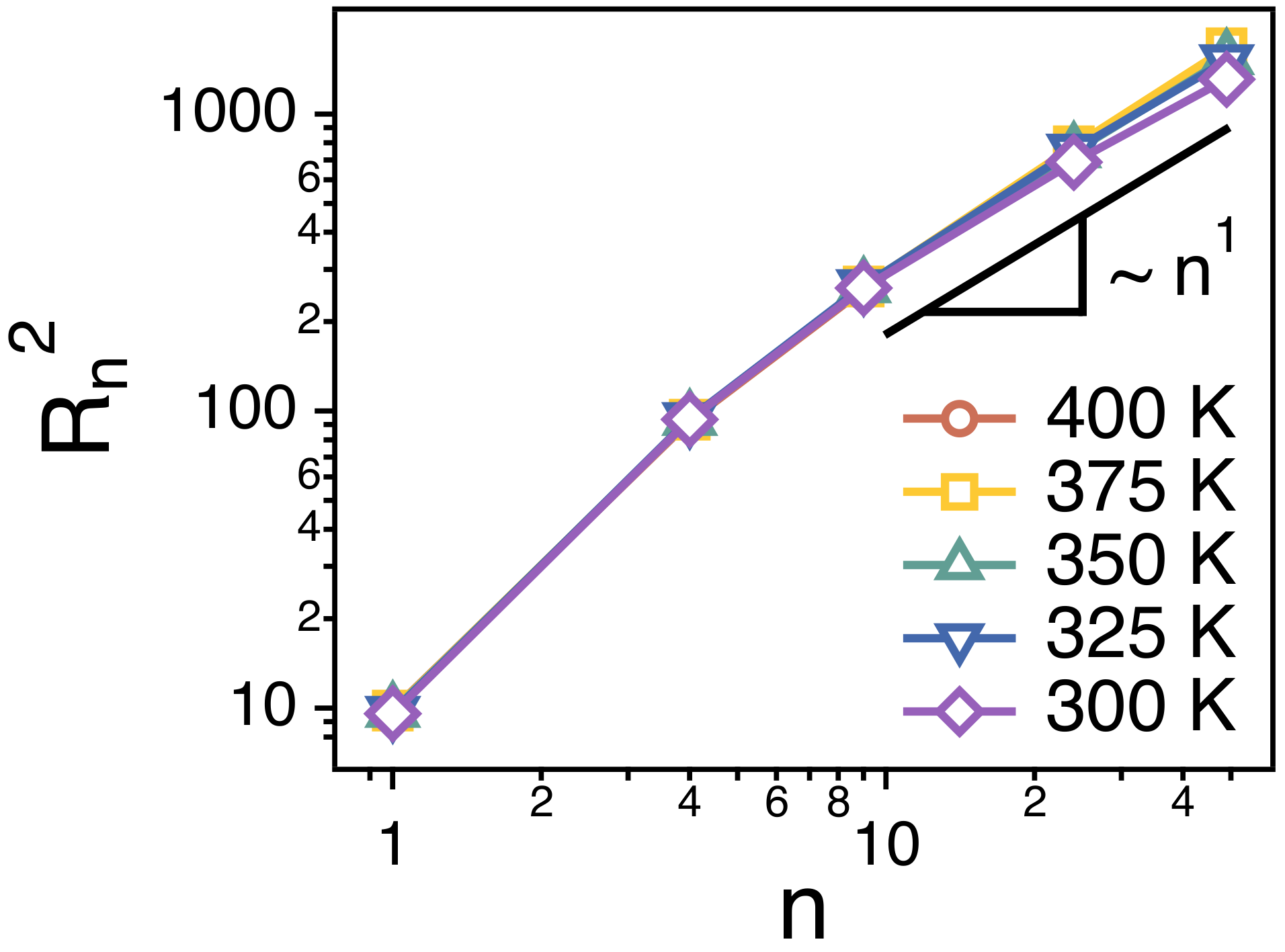
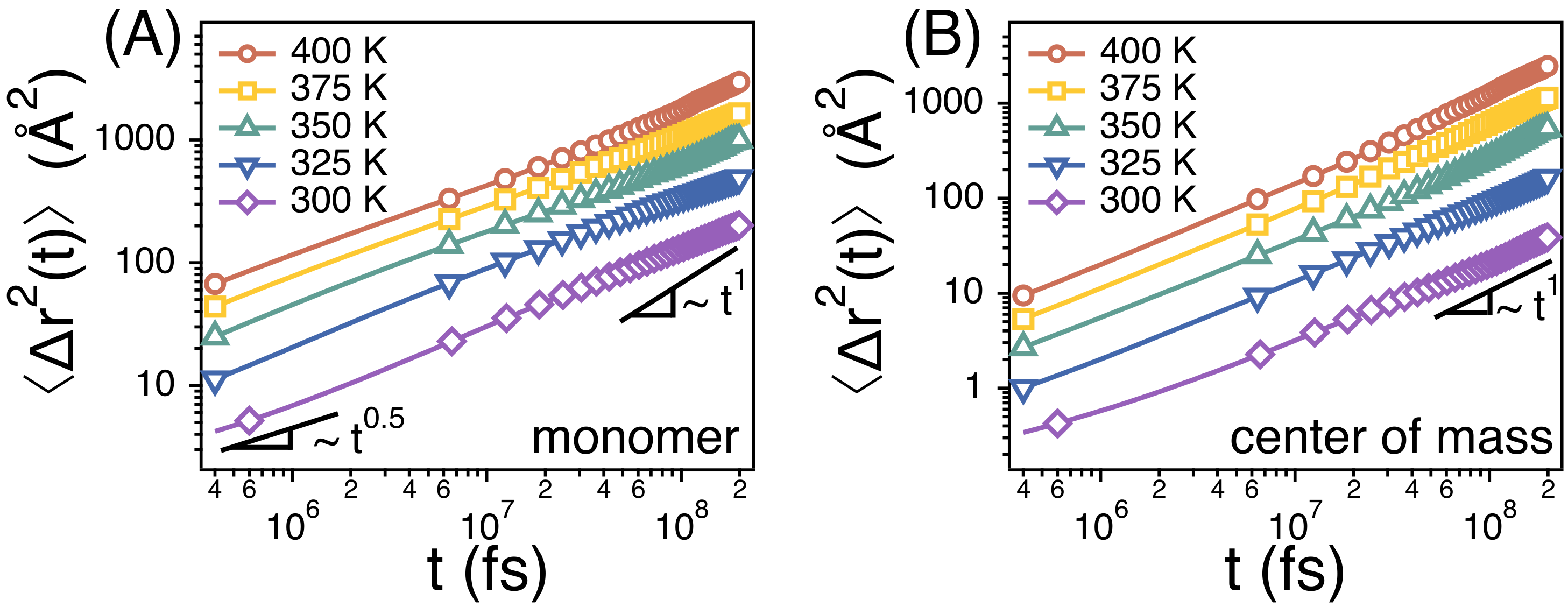
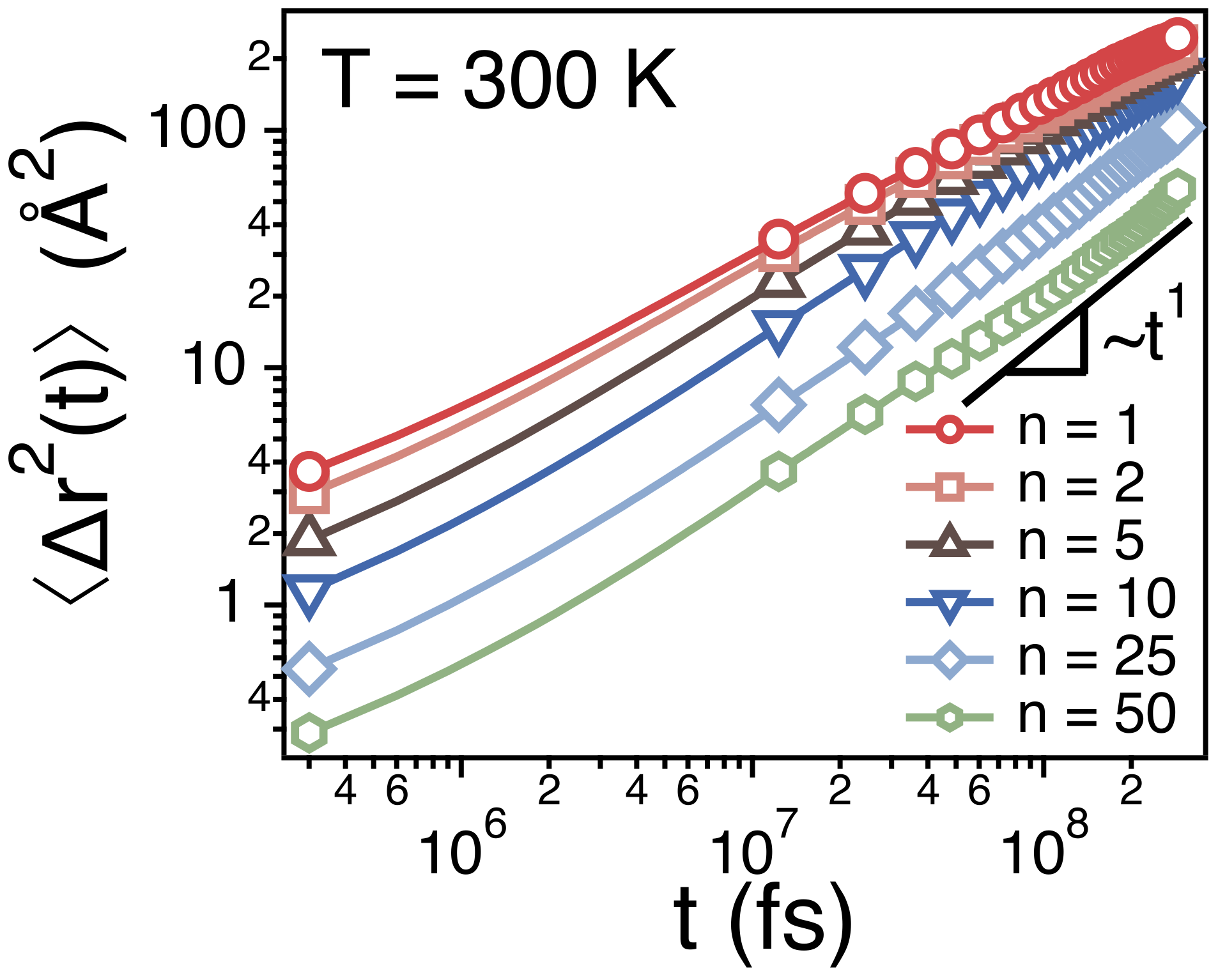
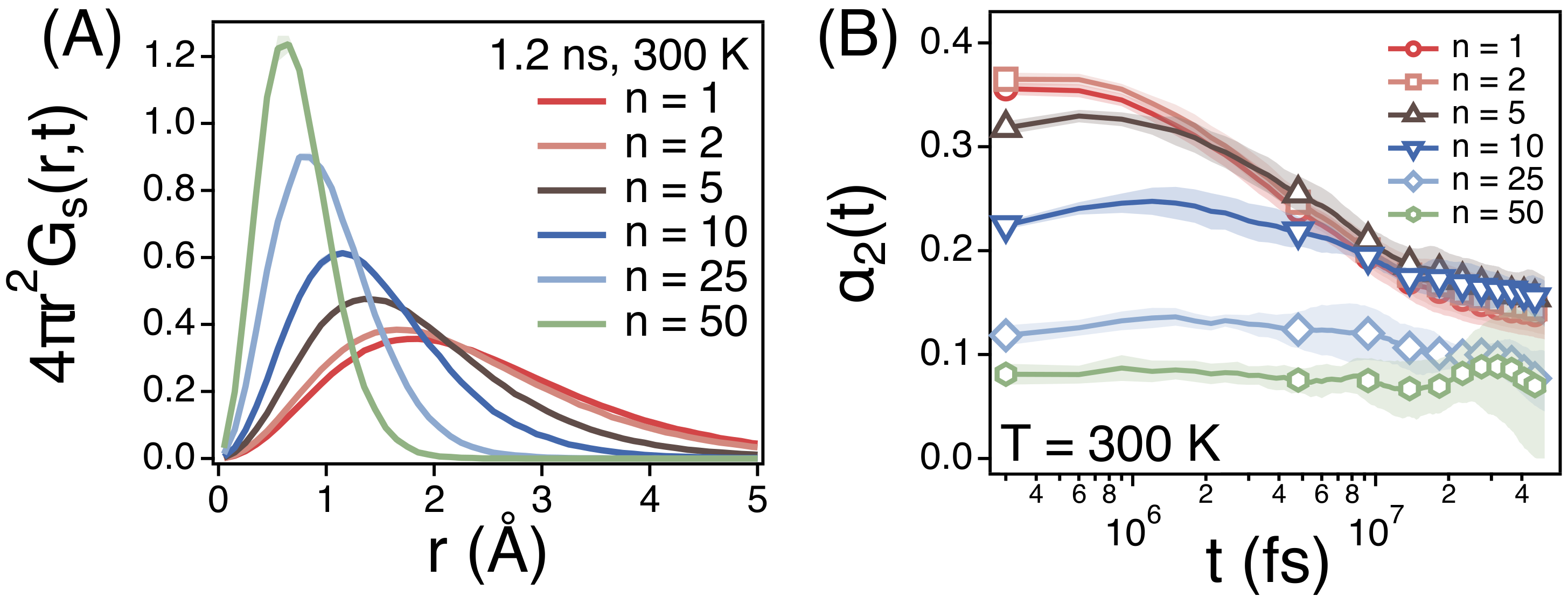
3.1. The Rouse Dynamics of PEO Melts
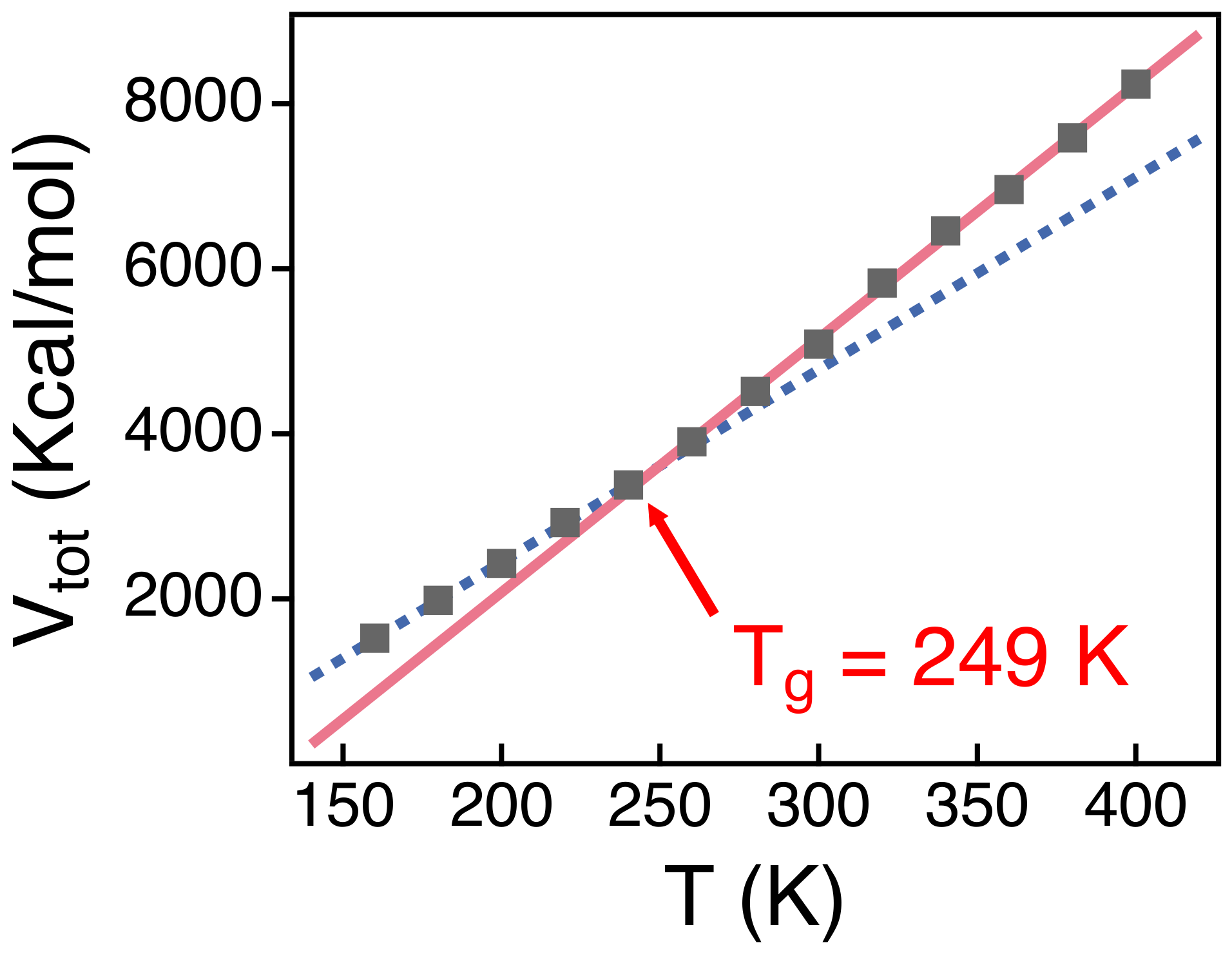
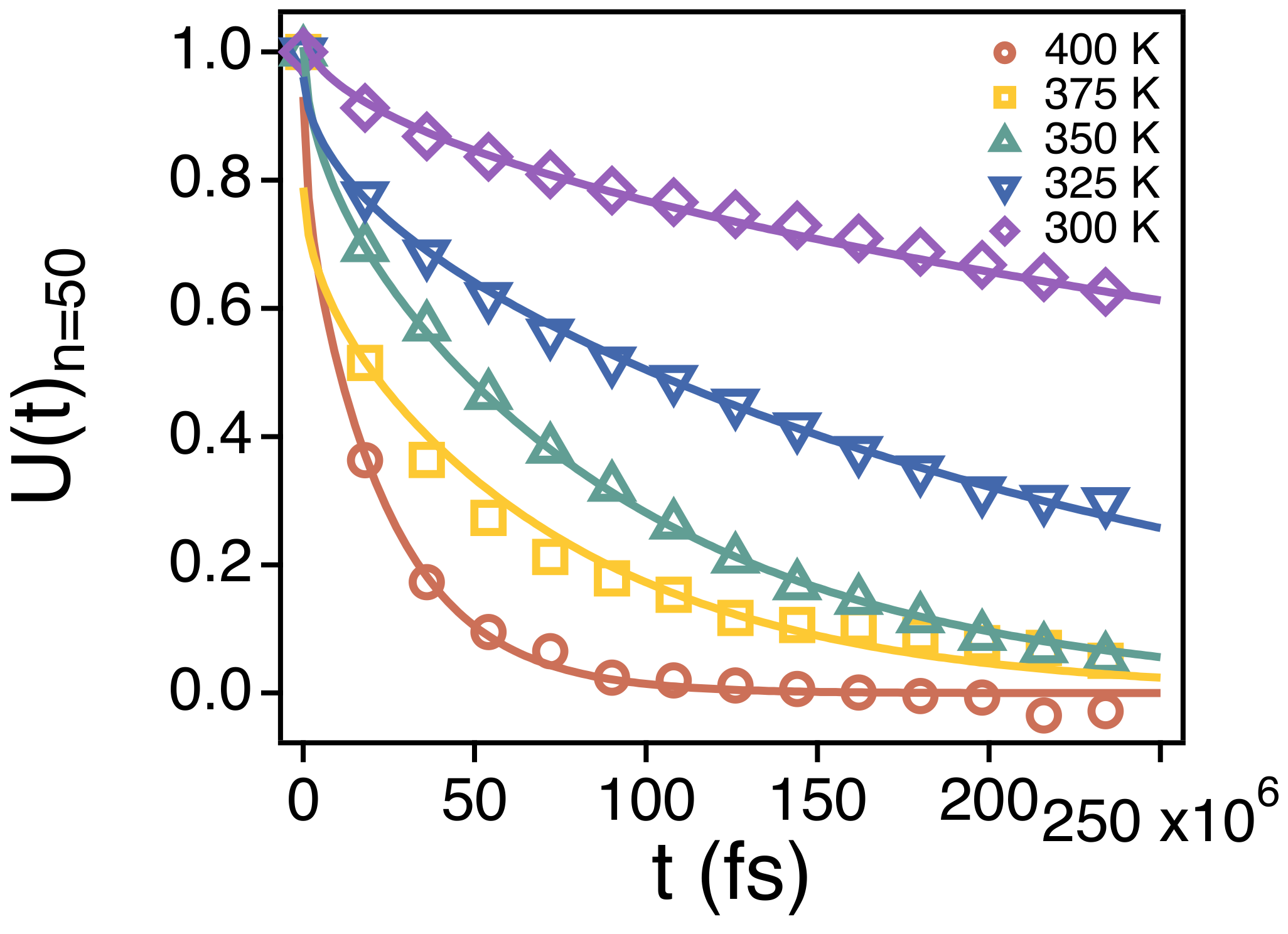
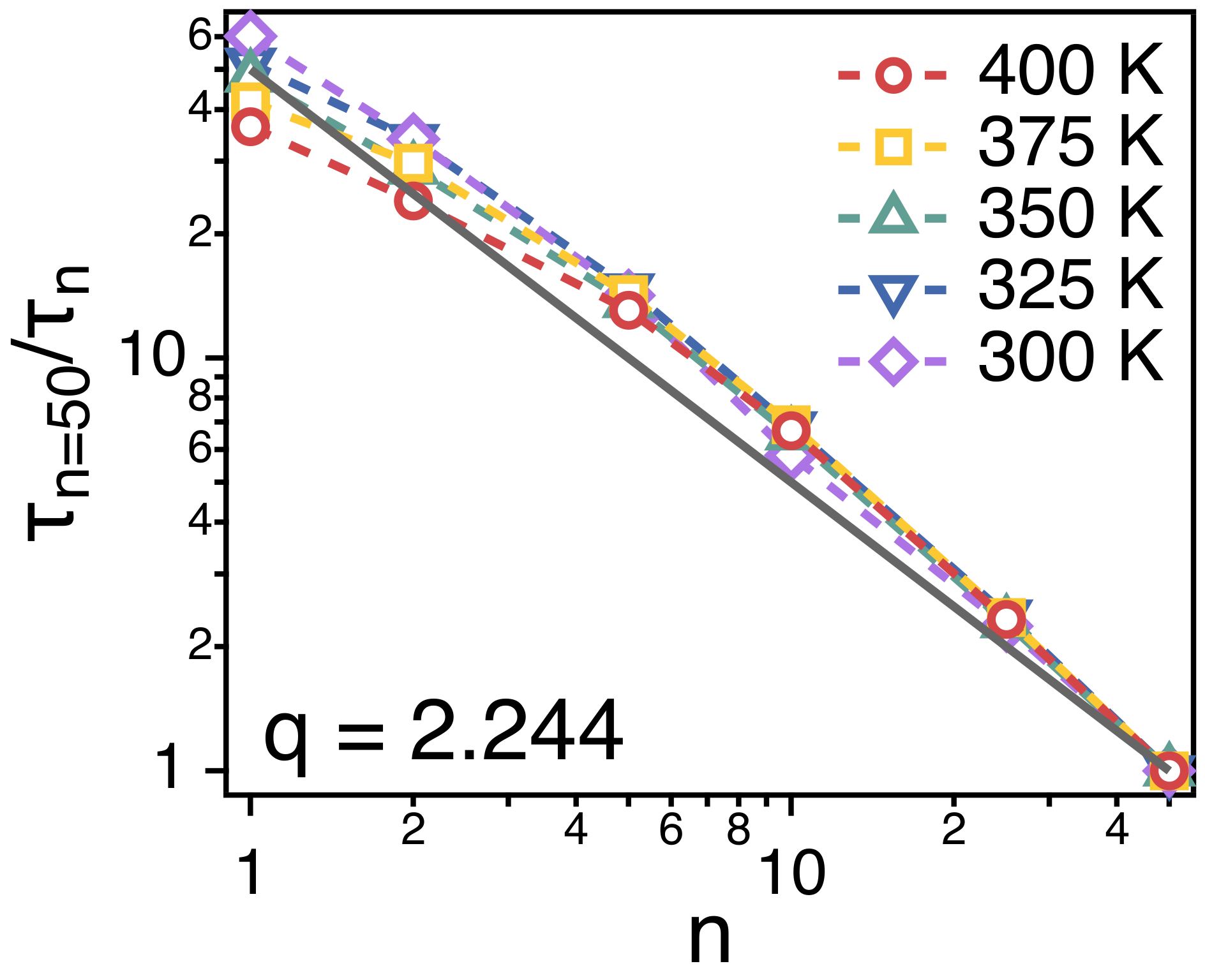
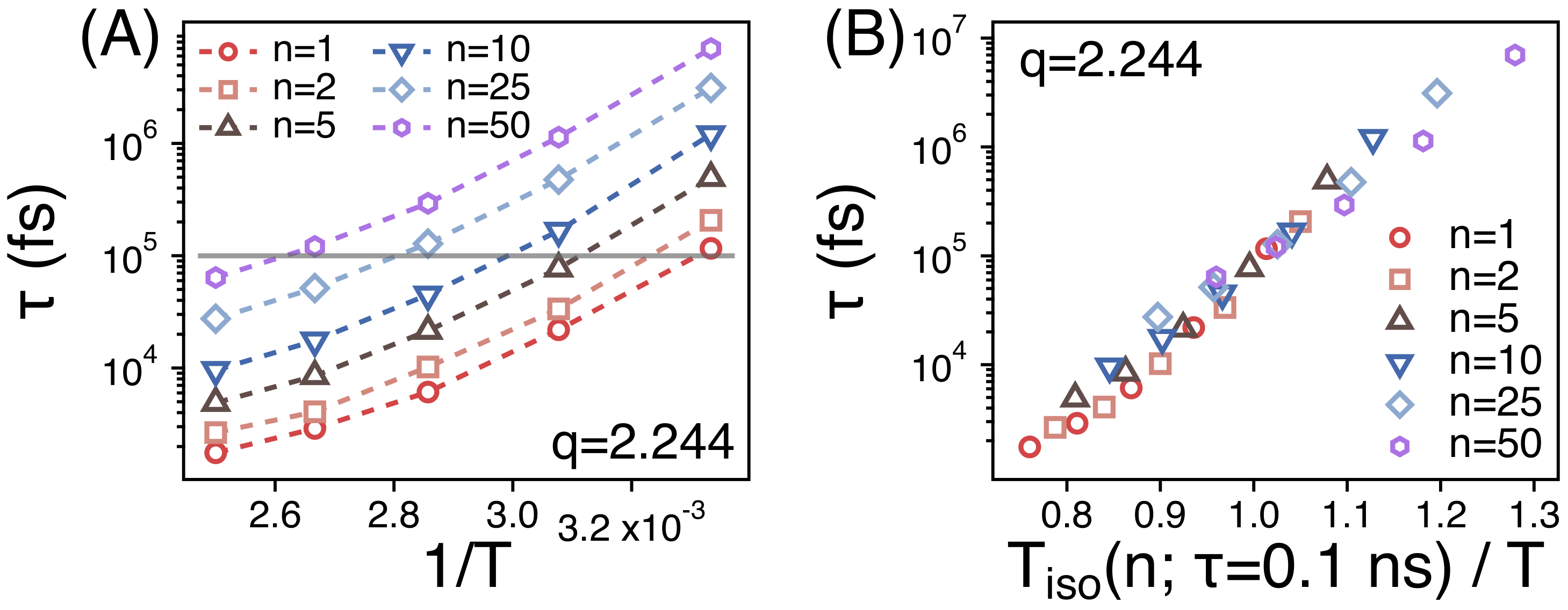
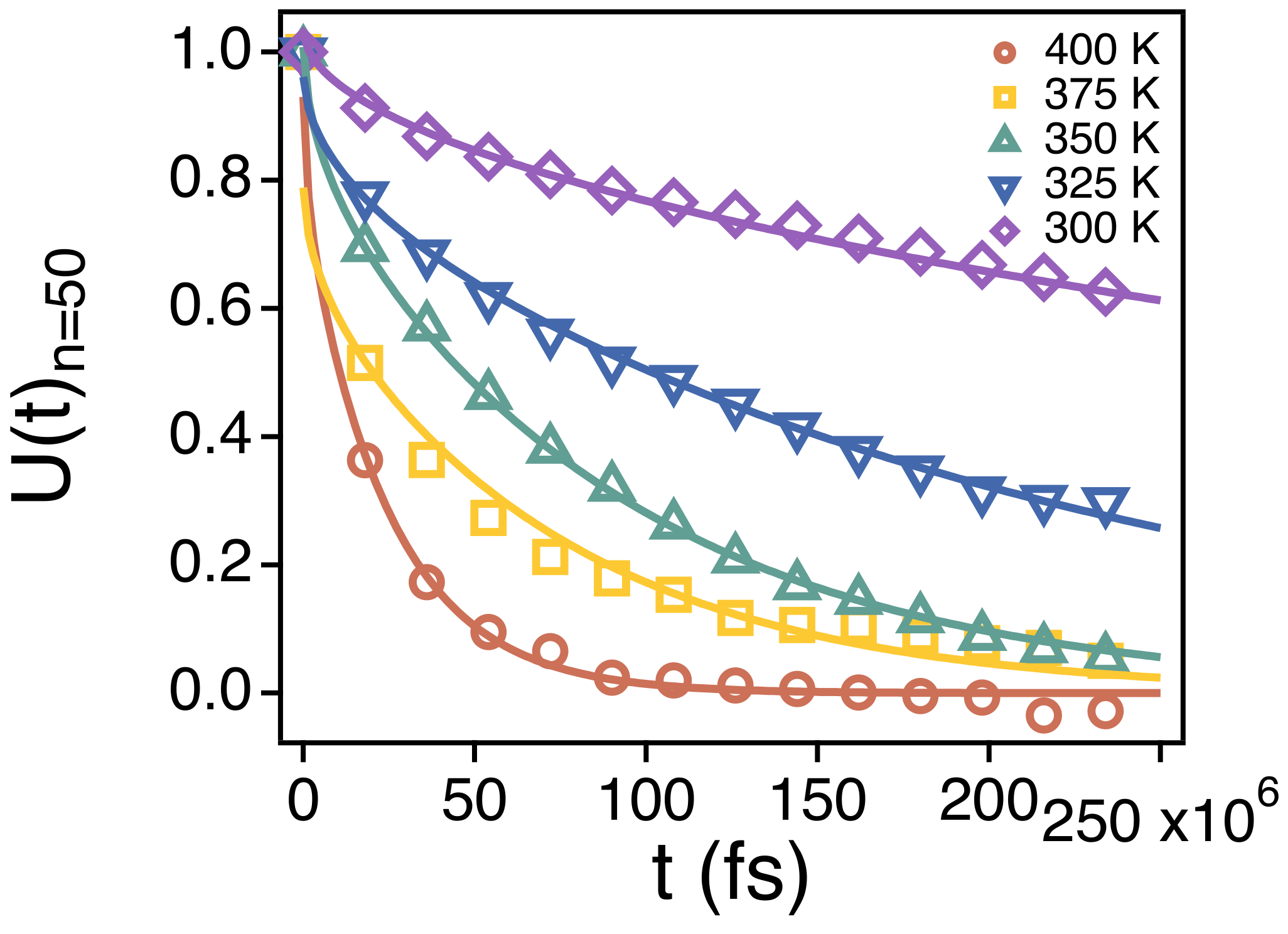
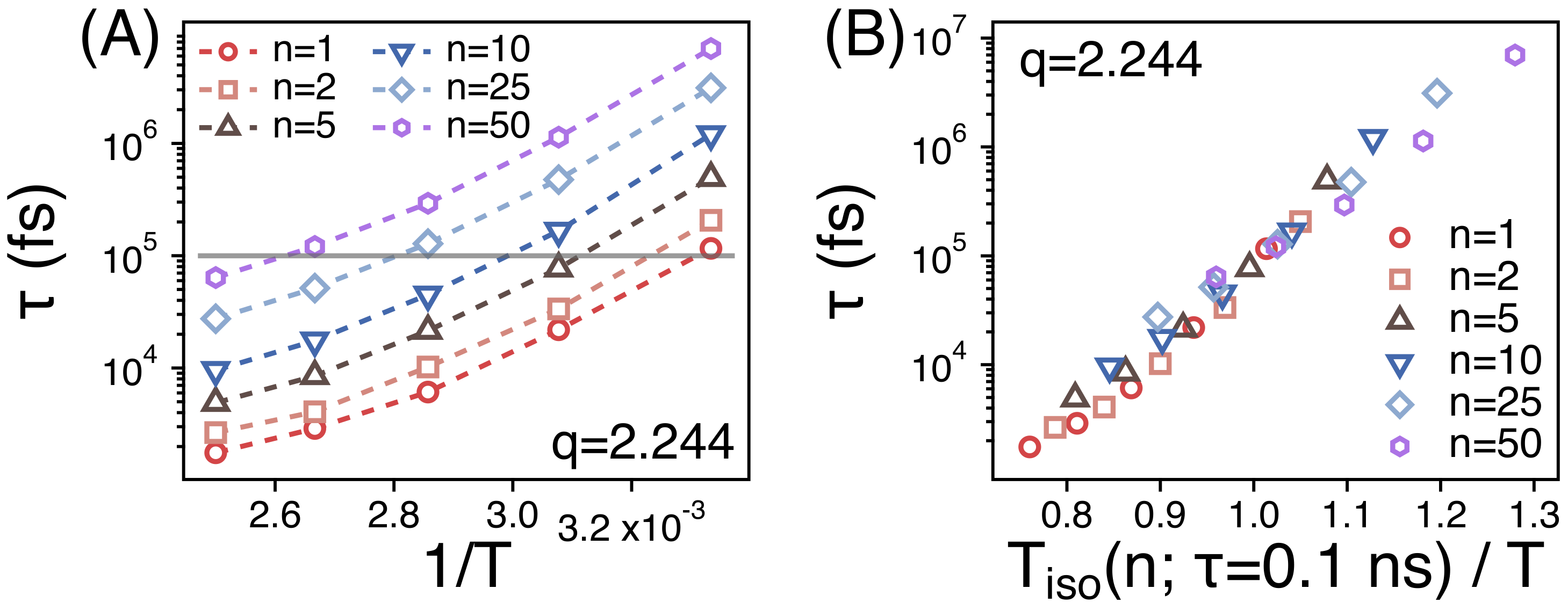
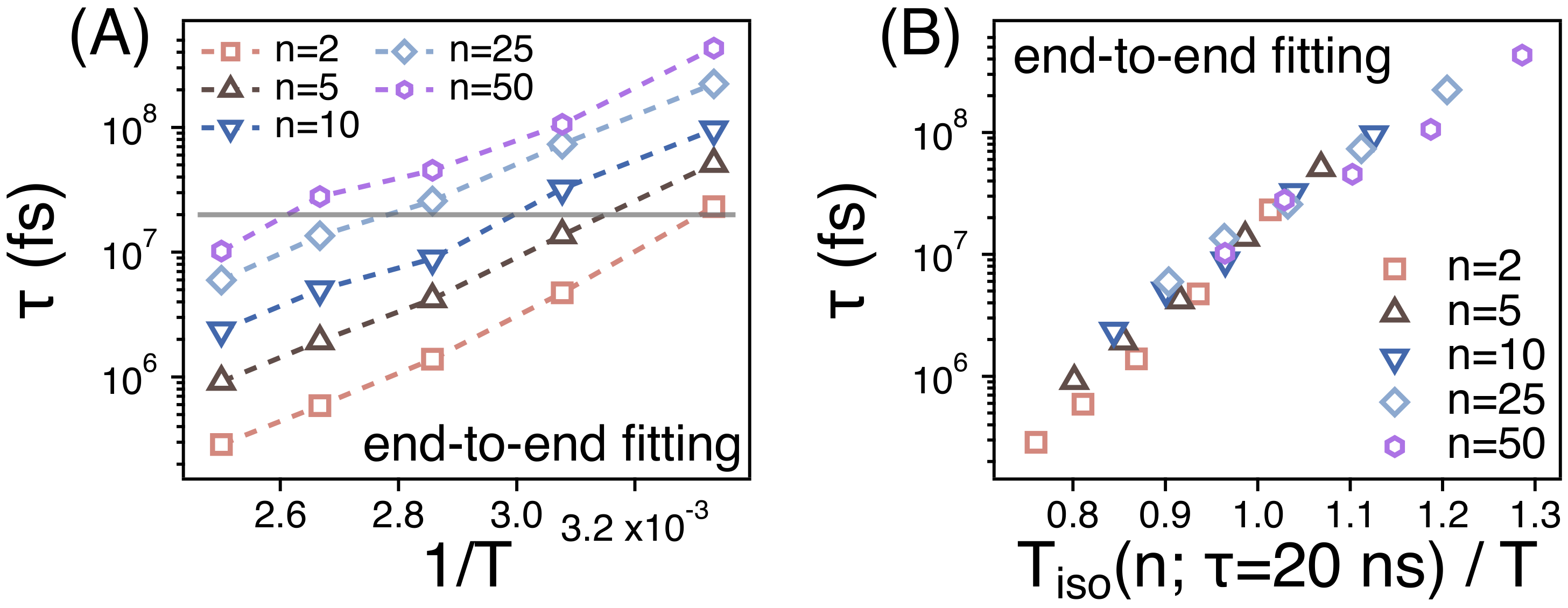
3.2. Temperature Dependence of Conformational Relaxation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palade, L.; Verney, V.; Attane, P. Time–temperature superposition and linear viscoelasticity of polybutadienes. Macromolecules 1995, 28, 7051–7057. [Google Scholar] [CrossRef]
- Van Gurp, M.; Palmen, J. Time–temperature superposition for polymeric blends. Rheol. Bull. 1998, 67, 5–8. [Google Scholar]
- Li, R. Time–temperature superposition method for glass transition temperature of plastic materials. Mater. Sci. Eng. A 2000, 278, 36–45. [Google Scholar] [CrossRef]
- Olsen, N.B.; Christensen, T.; Dyre, J.C. Time–temperature superposition in viscous liquids. Phys. Rev. Lett. 2001, 86, 1271–1274. [Google Scholar] [CrossRef] [Green Version]
- Dealy, J.; Plazek, D. Time–temperature superposition—A users guide. Rheol. Bull. 2009, 78, 16–31. [Google Scholar]
- Plazek, D.J. Temperature dependence of the viscoelastic behavior of polystyrene. J. Phys. Chem. 1965, 69, 3480–3487. [Google Scholar] [CrossRef]
- Colby, R.H. Breakdown of time–temperature superposition in miscible polymer blends. Polymer 1989, 30, 1275–1278. [Google Scholar] [CrossRef]
- Ding, Y.; Sokolov, A.P. Breakdown of time–temperature superposition principle and universality of chain dynamics in polymers. Macromolecules 2006, 39, 3322–3326. [Google Scholar] [CrossRef]
- Plazek, D.J. The temperature dependence of the viscoelastic softening and terminal dispersions of linear amorphous polymers. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 729–742. [Google Scholar] [CrossRef]
- Sokolov, A.; Hayashi, Y. Breakdown of time–temperature superposition: From experiment to the coupling model and beyond. J. Non-Cryst. Solids 2007, 353, 3838–3844. [Google Scholar] [CrossRef]
- Sokolov, A.P.; Schweizer, K.S. Resolving the mystery of the chain friction mechanism in polymer liquids. Phys. Rev. Lett. 2009, 102, 248301. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.L.; Plazek, D.J. Identification of different modes of molecular motion in polymers that cause thermorheological complexity. Rubber Chem. Technol. 1995, 68, 376–434. [Google Scholar] [CrossRef]
- Santangelo, P.G.; Ngai, K.L.; Roland, C.M. Temperature dependence of relaxation in polypropylene and poly(ethylene-co-propylene). Macromolecules 1996, 29, 3651–3653. [Google Scholar] [CrossRef]
- Plazek, D.L.; Plazek, D.J. Viscoelastic behavior of atactic polypropylene. Macromolecules 1983, 16, 1469–1475. [Google Scholar] [CrossRef]
- Plazek, D.J.; Bero, C.A.; Neumeister, S.; Floudas, G.; Fytas, G.; Ngai, K.L. Viscoelastic properties of amorphous polymers 3: Low molecular weight poly(methylphenylsiloxane). Colloid Polym. Sci. 1994, 272, 1430–1438. [Google Scholar] [CrossRef]
- Ilan, B.; Loring, R.F. Local vitrification model for melt dynamics. Macromolecules 1999, 32, 949–951. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003; ISBN 978-019-852-059-7. [Google Scholar]
- Gao, Q.; Jian, Z. Fragility and Vogel-Fulcher-Tammann parameters near glass transition temperature. Mater. Chem. Phys. 2020, 252, 123252. [Google Scholar] [CrossRef]
- Frick, B.; Richter, D. The microscopic basis of the glass transition in polymers from neutron scattering studies. Science 1995, 267, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Bormuth, A.; Henritzi, P.; Vogel, M. Chain-length dependence of the segmental relaxation in polymer melts: Molecular dynamics simulation studies on poly(propylene oxide). Macromolecules 2010, 43, 8985–8992. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Koukoulas, T.; Mavrantzas, V.G.; Pasquino, R.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Wischnewski, A.; Monkenbusch, M.; Richter, D. Microscopic Structure, Conformation, and Dynamics of Ring and Linear Poly(ethylene oxide) Melts from Detailed Atomistic Molecular Dynamics Simulations: Dependence on Chain Length and Direct Comparison with Experimental Data. Macromolecules 2017, 50, 2565–2584. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Alatas, P.V.; Peristeras, L.D.; Mavrantzas, V.G. Scaling laws for the conformation and viscosity of ring polymers in the crossover region around Me from detailed molecular dynamics simulations. ACS Macro Lett. 2018, 7, 916–920. [Google Scholar] [CrossRef]
- Huang, L.; Nagapudi, K.; Apkarian, R.P.; Chaikof, E.L. Engineered collagen–PEO nanofibers and fabrics. J. Biomater. Sci. Polym. Ed. 2001, 12, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.K.; Reilly, M.J.; Jones, D.N.; Robinson, P.M.; Bhatia, S.R. The effect of pharmaceuticals on the nanoscale structure of PEO–PPO–PEO micelles. Colloids Surf. B 2008, 61, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Pesko, D.M.; Savoie, B.M.; Timachova, K.; Hasan, A.L.; Smith, M.C.; Miller, T.F.; Coates, G.W.; Balsara, N.P. Optimizing ion transport in polyether-based electrolytes for lithium batteries. Macromolecules 2018, 51, 2847–2858. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.W.; Salomon, M. Improvement of ionic conductivity in plasticized PEO–based solid polymer electrolytes. J. Electrochem. Soc. 1993, 140, 3409–3412. [Google Scholar] [CrossRef]
- Long, L.; Wang, S.; Xiao, M.; Meng, Y. Polymer electrolytes for lithium polymer batteries. J. Mater. Chem. A 2016, 4, 10038–10069. [Google Scholar] [CrossRef]
- Li, H.; Xu, Z.; Yang, J.; Wang, J.; Hirano, S.I. Polymer electrolytes for rechargeable lithium metal batteries. Sustain. Energy Fuels 2020, 4, 5469–5487. [Google Scholar] [CrossRef]
- Brodeck, M.; Alvarez, F.; Arbe, A.; Juranyi, F.; Unruh, T.; Holderer, O.; Colmenero, J.; Richter, D. Study of the dynamics of poly(ethylene oxide) by combining molecular dynamic simulations and neutron scattering experiments. J. Chem. Phys. 2009, 130, 094908. [Google Scholar] [CrossRef]
- Annis, B.K.; Borodin, O.; Smith, G.D.; Benmore, C.J.; Soper, A.K.; Londono, J.D. The structure of a poly(ethylene oxide) melt from neutron scattering and molecular dynamics simulations. J. Chem. Phys. 2001, 115, 10998–11003. [Google Scholar] [CrossRef]
- Wu, C. Glass transition in single poly(ethylene oxide) chain: A molecular dynamics simulation study. J. Polym. Sci. B Polym. Phys. 2017, 55, 178–188. [Google Scholar] [CrossRef]
- Zackrisson, M.; Stradner, A.; Schurtenberger, P.; Bergenholtz, J. Structure, dynamics, and rheology of concentrated dispersions of poly(ethylene glycol)-grafted colloids. Phys. Rev. E 2006, 73, 011408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic molecular dynamics simulation of polydisperse linear polyethylene melts. Macromolecules 1998, 31, 7934–7943. [Google Scholar] [CrossRef]
- van Zon, A.; Mos, B.; Verkerk, P.; de Leeuw, S. On the dynamics of PEO-NaI polymer electrolytes. Electrochim. Acta 2001, 46, 1717–1721. [Google Scholar] [CrossRef]
- Chen, C.; Depa, P.; Sakai, V.G.; Maranas, J.K.; Lynn, J.W.; Peral, I.; Copley, J.R.D. A comparison of united atom, explicit atom, and coarse-grained simulation models for poly(ethylene oxide). J. Chem. Phys. 2006, 124, 234901. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, D.N. Hierarchical modeling of amorphous polymers. Comput. Phys. Commun. 2005, 169, 82–88. [Google Scholar] [CrossRef]
- Smith, G.D.; Jaffe, R.L.; Yoon, D.Y. Force field for simulations of 1,2-dimethoxyethane and poly(oxyethylene) based upon ab initio electronic structure calculations on model molecules. J. Phys. Chem. 1993, 97, 12752–12759. [Google Scholar] [CrossRef]
- Wick, C.D.; Theodorou, D.N. Connectivity-altering monte carlo simulations of the end group effects on volumetric properties for poly(ethylene oxide). Macromolecules 2004, 37, 7026–7033. [Google Scholar] [CrossRef]
- Borodin, O.; Douglas, R.; Smith, G.D.; Trouw, F.; Petrucci, S. MD simulations and experimental study of structure, dynamics, and thermodynamics of poly(ethylene oxide) and its oligomers. J. Phys. Chem. B 2003, 107, 6813–6823. [Google Scholar] [CrossRef]
- Wu, C. Simulated glass transition of poly(ethylene oxide) bulk and film: A comparative study. J. Phys. Chem. B 2011, 115, 11044–11052. [Google Scholar] [CrossRef] [PubMed]
- Mazo, M.A.; Manevitch, L.I.; Gusarova, E.B.; Shamaev, M.Y.; Berlin, A.A.; Balabaev, N.K.; Rutledge, G.C. Molecular dynamics simulation of thermomechanical properties of montmorillonite crystal. 3. montmorillonite crystals with PEO oligomer intercalates. J. Phys. Chem. B 2008, 112, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Chrissopoulou, K.; Afratis, A.; Anastasiadis, S.H.; Elmahdy, M.M.; Floudas, G.; Frick, B. Structure and dynamics in PEO nanocomposites. Eur. Phys. J. Spec. Top. 2007, 141, 267–271. [Google Scholar] [CrossRef]
- Halley, J.W.; Duan, Y.; Nielsen, B.; Redfern, P.C.; Curtiss, L.A. Simulation of polyethylene oxide: Improved structure using better models for hydrogen and flexible walls. J. Chem. Phys. 2001, 115, 3957–3966. [Google Scholar] [CrossRef]
- Prasitnok, K.; Wilson, M.R. A coarse-grained model for polyethylene glycol in bulk water and at a water/air interface. Phys. Chem. Chem. Phys. 2013, 15, 17093–17104. [Google Scholar] [CrossRef] [Green Version]
- Genix, A.C.; Arbe, A.; Alvarez, F.; Colmenero, J.; Willner, L.; Richter, D. Dynamics of poly(ethylene oxide) in a blend with poly(methyl methacrylate): A quasielastic neutron scattering and molecular dynamics simulations study. Phys. Rev. E 2005, 72, 031808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, A.; Heuer, A. Cation Transport in Polymer Electrolytes: A Microscopic Approach. Phys. Rev. Lett. 2007, 98, 227802. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Jiang, J. Molecular dynamics and dissipative particle dynamics simulations for the miscibility of poly(ethylene oxide)/poly(vinyl chloride) blends. Polymer 2010, 51, 291–299. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Yang, H.; Ze-Sheng, L.; Qian, H.J.; Yang, Y.B.; Zhang, X.B.; Sun, C.C. Molecular dynamics simulation studies of binary blend miscibility of poly(3-hydroxybutyrate) and poly(ethylene oxide). Polymer 2004, 45, 453–457. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Kwon, T.; Sung, B.J. Confinement effects on the mechanical heterogeneity of polymer fiber glasses. Phys. Rev. E 2020, 102, 052501. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedz, K.; Wischnewski, A.; Pyckhout-Hintzen, W.; Allgaier, J.; Richter, D.; Faraone, A. Chain dynamics and viscoelastic properties of poly(ethylene oxide). Macromolecules 2008, 41, 4866–4872. [Google Scholar] [CrossRef]
- Faucher, J.A.; Koleske, J.V.; Santee, E.R., Jr.; Stratta, J.J.; Wilson, C.W., III. Glass transitions of ethylene oxide polymers. J. Appl. Phys. 1966, 37, 3962–3964. [Google Scholar] [CrossRef]
- Jung, J.; Kwon, T.; Oh, Y.; Lee, Y.R.; Sung, B.J. Spatial dependence of non-gaussian diffusion of nanoparticles in free-standing thin polymer films. J. Phys. Chem. B 2019, 123, 9250–9259. [Google Scholar] [CrossRef]
- Song, E.S.; Oh, Y.; Sung, B.J. Interdomain exchange and the flip-flop of cholesterol in ternary component lipid membranes and their effects on heterogeneous cholesterol diffusion. Phys. Rev. E 2021, 104, 044402. [Google Scholar] [CrossRef]
- Oh, Y.; Song, E.S.; Sung, B.J. The effects of the lipid type on the spatial arrangement and dynamics of cholesterol in binary component lipid membranes. J. Chem. Phys. 2021, 154, 135101. [Google Scholar] [CrossRef]
- Kwon, T.; Sung, B.J. Effects of nanoparticles on the stability of polymer fibers. Phys. Rev. E 2018, 98, 042503. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.S.; Kwon, T.; Park, C.B.; Sung, B.J. Temperature Dependence of Conformational Relaxation of Poly(ethylene oxide) Melts. Polymers 2021, 13, 4049. https://doi.org/10.3390/polym13224049
Kim HS, Kwon T, Park CB, Sung BJ. Temperature Dependence of Conformational Relaxation of Poly(ethylene oxide) Melts. Polymers. 2021; 13(22):4049. https://doi.org/10.3390/polym13224049
Chicago/Turabian StyleKim, Hye Sol, Taejin Kwon, Chung Bin Park, and Bong June Sung. 2021. "Temperature Dependence of Conformational Relaxation of Poly(ethylene oxide) Melts" Polymers 13, no. 22: 4049. https://doi.org/10.3390/polym13224049
APA StyleKim, H. S., Kwon, T., Park, C. B., & Sung, B. J. (2021). Temperature Dependence of Conformational Relaxation of Poly(ethylene oxide) Melts. Polymers, 13(22), 4049. https://doi.org/10.3390/polym13224049