
Organocatalytic Ring-Opening Polymerization of ε-Caprolactone Using bis(N-(N′-butylimidazolium)alkane Dicationic Ionic Liquids as the Metal-Free Catalysts: Polymer Synthesis, Kinetics and DFT Mechanistic Study



 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. Synthesis and Characterization of Dicationic Ionic Liquids (DILs, 2a–2d)
2.3.1. Synthesis of 1,n-bis(1H-imidazol-1-yl)alkane (1,n-bis[Bim], 6a–6d)
Synthesis of 1,4-bis(1H-imidazol-1-yl)butane (1,4-bis[Bim], 6a)
General Procedure for Synthesis of 1,n-bis[N-(N′-butylimidazolium)]alkane (6b–6d, n = 6, 8, and 10)
Synthesis of 1,8-bis(1H-imidazol-1-yl)octane (1,8-bis[Bim], 6c)
Synthesis of 1,10-bis(1H-imidazol-1-yl)decane (1,10-bis[Bim], 6d)
2.3.2. Synthesis of 1,n-bis[N-(N′-butylimidazolium)]alkane bisbromide salts (1,n-bis[Bim] [Br], 7a–7d)
General Procedure for Synthesis of 1,4-bis[N-(N′-butylimidazolium)]butane bisbromide salts (1,4-bis[Bim][Br], 7a)
Synthesis of 1,6-bis[N-(N′-butylimidazolium)]hexane bisbromide salts (1,6-bis[Bim][Br], 7b)
Synthesis of 1,8-bis[N-(N′-butylimidazolium)]octane bisbromide salts (1,8-bis[Bim][Br], 7c)
Synthesis of 1,10-bis[N-(N′-butylimidazolium)]decane bisbromide salts (1,10-bis[Bim][Br], 7d)
2.3.3. Synthesis of 1,n-bis[N-(N′-butylimidazolium)]alkane bishexafluorophosphates (1,n-bis[Bim][PF6], 2a–2d)
General Procedure for Synthesis of 1,4-bis[N-(N′-butylimidazo-lium)]butane bishexafluorophosphates (1,4-bis[Bim][PF6], 2a)
Synthesis of 1,6-bis[N-(N′-butylimidazolium)]hexane bishexafluorophosphates (1,6-bis[Bim][PF6], 2b)
Synthesis of 1,8-bis[N-(N′-butylimidazolium)]octane bishexafluorophosphates (1,8-bis[Bim][PF6], 2c)
Synthesis of 1,10-bis[N-(N′-butylimidazolium)]decane bishexafluorophosphates (1,10-bis[Bim][PF6], 2d)
2.4. Thermal Decomposition Analysis and Cytotoxicity Testing of the Synthesized Dicationic Ionic Liquids (2a–2d)
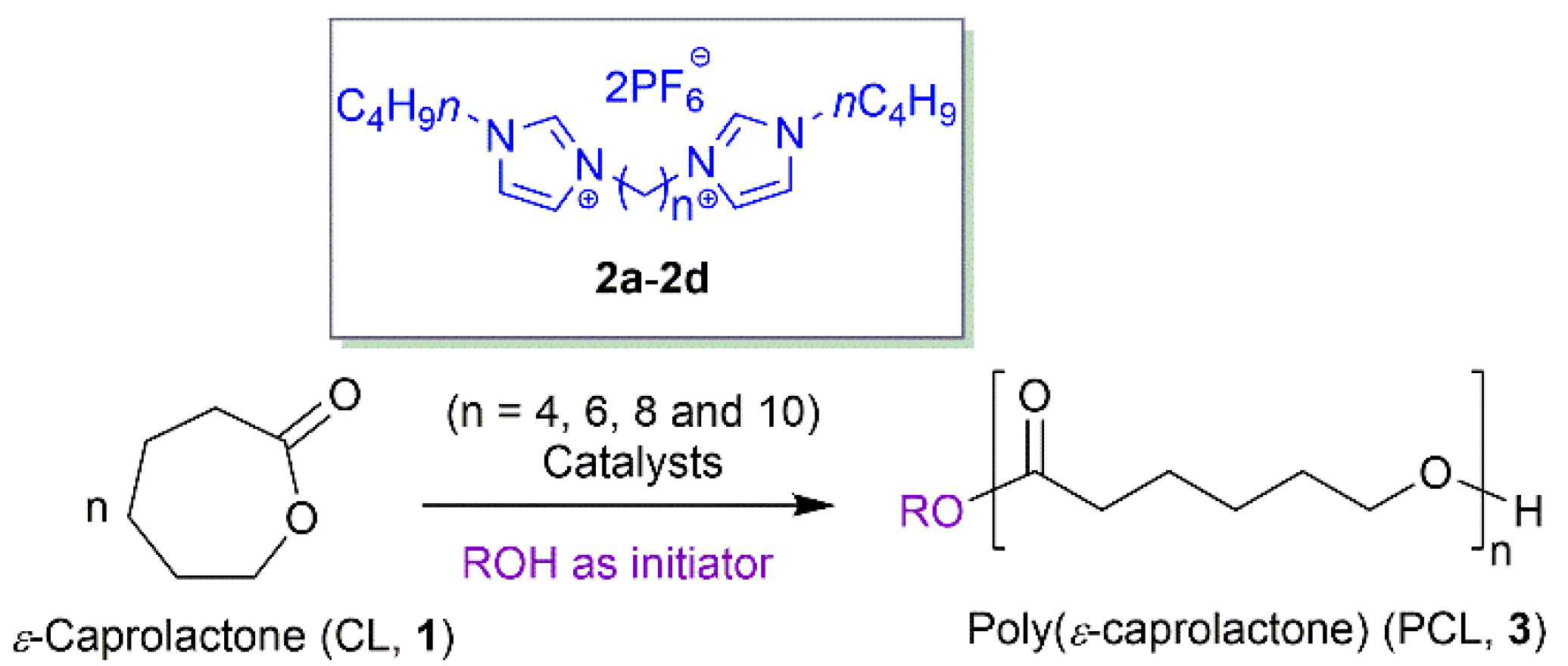
2.5. Synthesis of Poly(ε-Caprolactone) via ROP of ε-Caprolactone using the Synthesized DILs (2a–2d) and Sn(Oct)2 as Catalyst with Alcohols Initiator
2.6. Kinetic Studies of the ROP of ε-Caprolactone Catalyzed by DILs (2a–2d) Catalysts with 1-Dodecanol Initiator
2.7. Computational Study by Density Functional Theory (DFT)
3. Results and Discussion
3.1. Synthesis of Dicationic Ionic Liquids (2a–2d)
3.2. Thermal Decomposition Analysis and Cytotoxicity Testing of the Synthesized DIL Catalysts (2a–2d)


3.3. Synthesis of Poly(ε-Caprolactone) via the ROP of ε-Caprolactone using the Synthesized 1,4-bis[Bim][PF6] (2a) Catalyst with 1-Butanol Initiator
3.4. Synthesis of Poly(ε-Caprolactone) via the ROP of ε-Caprolactone using the Synthesized DILs (2a–2d) and Sn(Oct)2 Catalyst with 1-Dodecanol Initiator
3.5. Kinetic Studies of the ROP of ε-Caprolactone Catalyzed by the Synthesized DIL Catalysts (2a–2d) with 1-Dodecanol Initiator
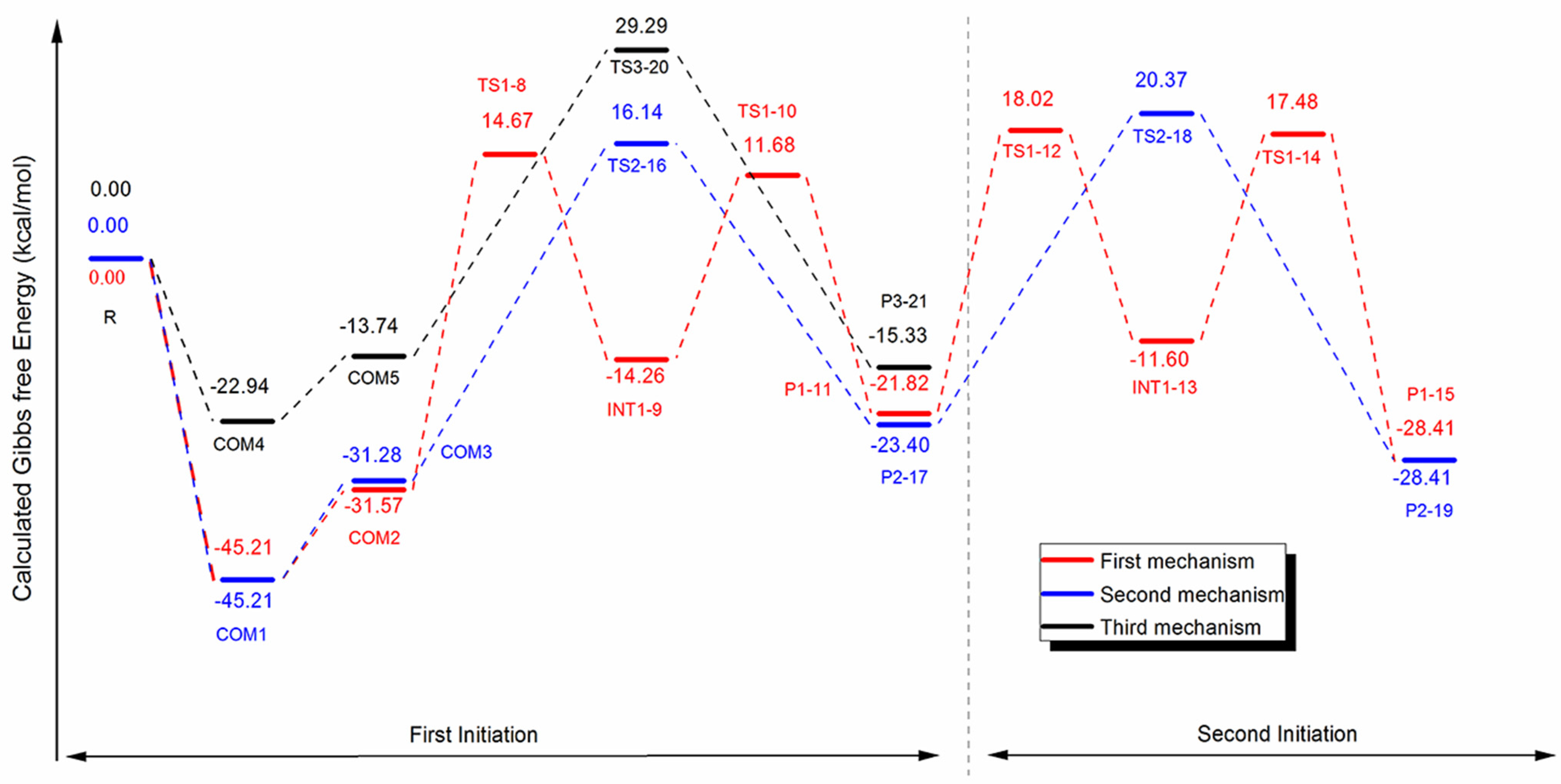
3.6. Mechanistic Study Using DFT Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Piskin, E. Biodegradable polymers as biomaterials. J. Biomater. Sci. Polym. Ed. 1995, 6, 775–795. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.S.; Daniels, A.U.; Andriano, K.P.; Heller, J. Six Bioabsorbable polymers: In vitro acute toxicity of accumulated degradation products. J. Appl. Biomater. 1994, 5, 151–157. [Google Scholar] [CrossRef]
- Kokubo, T.; Kim, H.-M.; Kawashita, M. Novel bioactive materials with different mechanical properties. Biomaterials 2003, 24, 2161–2175. [Google Scholar] [CrossRef]
- Brode, G.L.; Koleske, J.V. Lactone polymerization and polymer properties. J. Macromol. Sci. Chem. 1972, 6, 1109–1144. [Google Scholar] [CrossRef]
- Babler, J.H.; Invergo, B.J.; Sarussi, S.J. Lactone formation via oxidative cyclization of an unsaturated carboxylic acid: Application to the stereoselective synthesis of (±)-malyngolide, an antibiotic from the marine blue-green alga Lyngbya Majuscula Gomont. J. Org. Chem. 1980, 45, 4241–4243. [Google Scholar] [CrossRef]
- Carothers, W.H.; Dorough, G.L.; van Natta, F.J. Studies of polymerization and ring formation. X. The reversible polymerization of six-membered cyclic ester. J. Am. Chem. Soc. 1932, 54, 761–772. [Google Scholar] [CrossRef]
- Sosnowski, S.; Gadzinowski, M.; Slomkowski, S. Poly(L,L-lactide) microspheres by ring-opening polymerization. Macromolecules 1996, 29, 4556–4564. [Google Scholar] [CrossRef]
- Löfgren, A.; Albertsson, A.-C.; Dubois, P.; Jérôme, R. Recent advances in ring-opening polymerization of lactones and related compounds. J. Macromol. Sci. Rev. Macromol. Chem. 1995, 35, 379–418. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484–3504. [Google Scholar] [CrossRef]
- Platel, R.H.; Hodgson, L.M.; Williams, C.K. Biocompatible Initiators for lactide polymerization. Polym. Rev. 2008, 48, 11–63. [Google Scholar] [CrossRef]
- Dubois, P.; Degée, P.; Jérôme, R.; Teyssié, P. Macromolecular engineering of polylactones and polylactides. 8. Ring-opening polymerization of ε-caprolactone initiated by primary amines and trialkylaluminum. Macromolecules 1992, 25, 2614–2618. [Google Scholar] [CrossRef]
- Duda, A.; Florjanczyk, Z.; Hofman, A.; Slomkowski, S.; Penczek, S. Living pseudoanionic polymerization of ε-caprolactone. Poly(ε-caprolactone) free of cyclics and with controlled end groups. Macromolecules 1990, 23, 1640–1646. [Google Scholar] [CrossRef]
- Duda, A. Polymerization of ε-caprolactone initiated by aluminum isopropoxide carried out in the presence of alcohols and diols. Kinetics and mechanism. Macromolecules 1996, 29, 1399–1406. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. Polymerization of ε-caprolactone initiated by aluminum isopropoxide trimer and/or tetramer. Macromolecules 1995, 28, 5981–5992. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. On the difference of reactivities of various aggregated forms of aluminium triisopropoxide in initiating ring-opening polymerizations. Macromol. Rapid Commun. 1995, 16, 67–76. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Berl, M.; Scharnagl, N. Poly(lactones). 9. Polymerization mechanism of metal alkoxide initiated polymerizations of lactide and various lactones. Macromolecules 1988, 21, 286–293. [Google Scholar] [CrossRef]
- Martin, E.; Dubois, P.; Jérôme, R. Polymerization of ε-caprolactone initiated by y alkoxide grated onto porous silica. Macromolecules 2003, 36, 7094–7099. [Google Scholar] [CrossRef]
- Ropson, N.; Dubois, P.; Jérôme, R.; Teyssié, P. Macromolecular engineering of polylactones and polylactides. 20. Effect of monomer, solvent, and initiator on the ring-opening polymerization as initiated with aluminum alkoxides. Macromolecules 1995, 28, 7589–7598. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S.; Dubois, P.; Mecerreyes, D.; Jérôme, R. Oligomerization and copolymerization of γ-butyrolactone—A monomer known as unable to homopolymerize, 1. Copolymerization with ε-caprolactone. Macromol. Chem. Phys. 1996, 197, 1273–1283. [Google Scholar] [CrossRef]
- Benabdillah, K.M.; Coudane, J.; Boustta, M.; Engel, R.; Vert, M. Synthesis and characterization of novel degradable polyesters derived from d-gluconic and glycolic acids. Macromolecules 1999, 32, 8774–8780. [Google Scholar] [CrossRef]
- Lenoir, S.; Riva, R.; Lou, X.; Detrembleur, C.; Jerome, R.; Lecomte, P. Ring-opening polymerization of α-chloro-ε-caprolactone and chemical modification of poly(α-chloro-ε-caprolactone) by atom transfer radical processes. Macromolecules 2004, 37, 4055–4061. [Google Scholar] [CrossRef]
- Möller, M.; Kånge, R.; Hedrick, J.L. Sn(OTf)2 and Sc(OTf)3: Efficient and versatile catalysts for the controlled polymerization of lactones. Polym. Sci. Part A Polym. Chem. 2000, 38, 2067–2074. [Google Scholar] [CrossRef]
- Kowalski, A.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with tin(II) octoate, 1: Polymerization of ε-caprolactone. Macromol. Rapid Commun. 1998, 19, 567–572. [Google Scholar]
- Kowalski, A.; Duda, A.; Penczek, S. Mechanism of cyclic ester polymerization initiated with tin(II) octoate. 2. 1 Macromolecules fitted with tin(ii) alkoxide species observed directly in MALDI-TOF spectra. Macromolecules 2000, 33, 689–695. [Google Scholar] [CrossRef]
- Libiszowski, J.; Kowalski, A.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with covalent metal carboxylates, 5. End-group studies in the model ε-caprolactone and L,L-dilactide/tin(II) and zinc octoate/butyl alcohol systems. Macromol. Chem. Phys. 2002, 203, 1694–1701. [Google Scholar] [CrossRef]
- Dobrzynski, P. Mechanism of ε-caprolactone polymerization and ε-caprolactone/trimethylene carbonate copolymerization carried out with Zr(acac)4. Polymer 2007, 48, 2263–2279. [Google Scholar] [CrossRef]
- Abraham, G.A.; Gallardo, A.; Lozano, A.E.; Roman, J.S. ε-caprolactone/ZnCl2 complex formation: Characterization and ring-opening polymerization mechanism. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 1355–1365. [Google Scholar] [CrossRef]
- Barakat, I.; Dubois, P.; Jérôme, R.; Teyssié, P. Living polymerization and selective end functionalization of ε-caprolactone using zinc alkoxides as initiators. Macromolecules 1991, 24, 6542–6545. [Google Scholar] [CrossRef]
- Sarazin, Y.; Schormann, M.; Bochmann, M. Novel zinc and magnesium alkyl and amido cations for ring-opening polymerization reactions. Organometallics 2004, 23, 3296–3302. [Google Scholar] [CrossRef]
- Liao, L.; Liu, L.; Zhang, C.; Gong, S. Microwave-assisted ring-opening polymerization of ε-caprolactone in the presence of ionic liquid. Macromol. Rapid Commun. 2006, 27, 2060–2064. [Google Scholar] [CrossRef]
- Agarwal, S.; Mast, C.; Dehnicke, K.; Greiner, A. Rare earth metal initiated ring-opening polymerization of lactones. Macromol. Rapid Commun. 2000, 21, 195–212. [Google Scholar] [CrossRef]
- Nomura, N.; Taira, A.; Tomioka, T.; Okada, M. Catalytic approach for cationic living polymerization: Sc(OTf)3-catalyzed ring-opening polymerization of lactones. Macromolecules 2000, 33, 1497–1499. [Google Scholar] [CrossRef]
- Nomura, N.; Taira, A.; Nakase, A.; Tomioka, T.; Okada, M. Ring-opening polymerization of lactones by rare-earth metal triflates and by their reusable system in ionic liquids. Tetrahedron 2007, 63, 8478–8484. [Google Scholar] [CrossRef]
- Deng, X.M.; Zhu, Z.; Xiong, C.; Zhang, L. Ring-opening polymerization of ε-caprolactone initiated by rare earth complex catalysts. J. Appl. Polym. Sci. 1997, 64, 1295–1299. [Google Scholar] [CrossRef]
- ASTM. ASTM F1925–17, Standard Specification for Semi-Crystalline Poly(Lactide) Polymer and Copolymer Resins for Surgical Implants; ASTM International: West Conshohocken, PA, USA, 2017. [Google Scholar]
- Roslan, N.A.; Hasnan, M.H.C.; Abdullah, N.; Abdullah, S.B.; Abidin, S.Z. A preliminary study: Esterification of free fatty acids (FFA) in artificially modified feedstock using ionic liquids as catalysts. Bull. Chem. React. Eng. Catal. 2016, 11, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Amendola, S.C.; Sharp-Goldman, S.L.; Janjua, M.S.; Spencer, N.; Kelly, M.T.; Petillo, P.J.; Binder, M. A safe, portable, hydrogen gas generator using aqueous borohydride solution and Ru catalyst. Int. J. Hydrogen Energy 2000, 25, 969–975. [Google Scholar] [CrossRef]
- Vafaeezadeh, M.; Hashemi, M.M. Efficient fatty acid esterification using silica supported brønsted acidic ionic liquid catalyst: Experimental study and DFT modeling. Chem. Eng. J. 2014, 250, 35–41. [Google Scholar] [CrossRef]
- Shi, F.; Gu, Y.; Zhang, Q.; Deng, Y. Development of ionic liquids as green reaction media and catalysts. Catal. Surv. Asia. 2004, 8, 179–186. [Google Scholar] [CrossRef]
- Li, Y.; Hu, S.; Cheng, J.; Lou, W. Acidic ionic liquid-catalyzed esterification of oleic acid for biodiesel synthesis. Chin. J. Catal. 2014, 35, 396–406. [Google Scholar] [CrossRef]
- Zhang, L.; Xian, M.; He, Y.; Li, L.; Yang, J.; Yu, S.; Xu, X. A BrØnsted acidic ionic liquid as an efficient and environmentally benign catalyst for biodiesel synthesis from free fatty acids and alcohols. Bioresour. Technol. 2009, 100, 4368–4373. [Google Scholar] [CrossRef] [PubMed]
- Galiński, M.; Lewandowski, A.; Stępniak, I. Ionic liquids as electrolytes. Electrochim. Acta 2006, 51, 5567–5580. [Google Scholar] [CrossRef]
- Hough, W.L.; Smiglak, M.; Rodríguez, H.; Swatloski, R.P.; Spear, S.K.; Daly, D.T.; Pernak, J.; Grisel, J.E.; Carliss, R.D.; Soutullo, M.D.; et al. The third evolution of ionic liquids: Active pharmaceutical ingredients. New J. Chem. 2007, 31, 1429–1436. [Google Scholar] [CrossRef]
- Tian, T.; Hu, X.; Guan, P.; Tang, Y. Investigation of surface and solubility properties of N-vinylimidazolium tetrahalogenidoferrate(III) magnetic ionic liquids using density functional theory. J. Chem. Eng. Data 2016, 61, 721–730. [Google Scholar] [CrossRef]
- Kore, R.; Srivastava, S. A simple, eco-friendly and recyclable bi-functional acidic ionic liquid catalysts for beckmann rearrangement. J. Mol. Catal. A Chem. 2013, 376, 90–97. [Google Scholar] [CrossRef]
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis, 2nd ed.; Wasserscheid, P., Welton, T., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2002; Chapter 10; pp. 698–704. [Google Scholar]
- Dong, F.-X.; Zhang, L.; Tong, X.-Z.; Chen, H.-B.; Wang, X.-L.; Wang, Y.-Z. Ionic liquid coated lipase: Green synthesis of high molecular weight poly(1,4-dioxan-2-one). J. Mol. Catal. B Enzym. 2012, 77, 46–52. [Google Scholar] [CrossRef]
- Xu, Q.; Kennedy, J.; Liu, L. An ionic liquid as reaction media in the ring-opening graft polymerization of ε-caprolactone onto starch granules. Carbohydr. Polym. 2008, 72, 113–121. [Google Scholar] [CrossRef]
- Kadokawa, J.; Iwasaki, Y.; Tagaya, H. Ring-opening polymerization of ethylene carbonate catalyzed with ionic liquids: Imidazolium chloroaluminate and chlorostannate melts. Macromol. Rapid Commun. 2002, 23, 757–760. [Google Scholar] [CrossRef]
- Zhou, J.; Cheng, L.; Wu, D. Ring-opening polymerization of ethylene carbonate using ionic liquids as catalysts. e-Polymers 2011, 11. [Google Scholar] [CrossRef]
- Kaoukabi, A.; Guillen, F.; Qayouh, H.; Bouyahya, A.; Balieu, S.; Belachemi, L.; Gouhier, G.; Lahcini, M. The use of ionic liquids as an organocatalyst for controlled ring-opening polymerization of ε-caprolactone. Ind. Crops Prod. 2015, 72, 16–23. [Google Scholar] [CrossRef]
- Kammakakam, I.; Nam, Y.S.; Kim, T.-H. Ionic group-mediated crosslinked polyimide membranes for enhanced CO2 separation. RSC Adv. 2015, 5, 69907–69914. [Google Scholar] [CrossRef]
- Howarth, J.; Hanlon, K.; Fayne, D.; McCormac, P. Moisture stable dialkylimidazolium salts as heterogeneous and homogeneous lewis acids in the diels–alder reaction. Tetrahedron Lett. 1997, 38, 3097–3100. [Google Scholar] [CrossRef]
- Wang, G.; Xu, X.; Sun, Y.; Zhuang, L.; Yao, C. Relationship between structure and biodegradability of gemini imidazolium surface active ionic liquids. J. Mol. Liq. 2019, 278, 145–155. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Mitu, S.; Roman, G.; Patricia, R.E.; Crandall, I.E.; Szarek, W.A. The antimalarial activity of bivalent imidazolium salts. Bioorg. Med. Chem. 2011, 19, 6525–6542. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Wang, Y.; Hatakeyama, M.; Ogata, K.; Wakabayashi, M.; Jin, F.; Nakamura, S. Activation of CO2 by ionic liquid EMIM–BF4 in the electrochemical system: A theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 23521–23531. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [CL]0/[nBuOH]0/ [1,n-bis[Bim][PF6]]0 | Conv. a (%) | Mn,theob (g mol−1) | Mn,NMRc (g mol−1) | Mn,GPCd (g mol−1) | Mw,GPCd (g mol−1) | Ðd |
|---|---|---|---|---|---|---|---|
| 1 | 200/1.0/0.25 | 99 | 22,670 | 6583 | 6747 | 9582 | 1.42 |
| 2 | 200/1.0/0.50 | 98 | 22,440 | 7399 | 3645 | 4234 | 1.16 |
| 3 | 200/1.0/0.75 | 99 | 22,670 | 9381 | 5304 | 8688 | 1.63 |
| 4 | 200/1.0/1.00 | 98 | 22,440 | 8986 | 3569 | 4175 | 1.16 |
| 5 | 300/1.0/0.25 | 98 | 33,630 | 8921 | 6634 | 8392 | 1.26 |
| 6 | 300/1.0/0.50 | 98 | 33,630 | 7657 | 8384 | 11,342 | 1.35 |
| 7 | 300/1.0/0.75 | 99 | 33,970 | 6483 | 8891 | 14,985 | 1.68 |
| 8 | 300/1.0/1.00 | 99 | 33,970 | 8102 | 9899 | 15,789 | 1.59 |
| 9 | 400/1.0/0.25 | 98 | 44,743 | 14,393 | 12,272 | 23,081 | 1.63 |
| 10 | 400/1.0/0.50 | 98 | 44,743 | 13,252 | 12,317 | 20,130 | 1.88 |
| 11 | 400/1.0/0.75 | 98 | 44,743 | 14,450 | 9786 | 18,019 | 1.84 |
| 12 | 400/1.0/1.00 | 97 | 44,287 | 15,911 | 10,329 | 19,433 | 1.88 |
| 13 | 500/1.0/0.25 | 98 | 56,000 | 14,455 | 9563 | 16,505 | 1.72 |
| 14 | 500/1.0/0.50 | 98 | 56,000 | 9142 | 9053 | 16,159 | 1.78 |
| 15 | 500/1.0/0.75 | 97 | 55,430 | 11,420 | 10,322 | 19,112 | 1.85 |
| 16 | 500/1.0/1.00 | 98 | 56,000 | 9657 | 8503 | 15,445 | 1.81 |
| 17 | 600/1.0/0.25 | 97 | 66,500 | 10,290 | 10,535 | 16,203 | 1.53 |
| 18 | 600/1.0/0.50 | 99 | 67,870 | 13,481 | 11,826 | 21,339 | 1.80 |
| 19 | 600/1.0/0.75 | 98 | 67,180 | 10,020 | 9970 | 18,010 | 1.80 |
| 20 | 600/1.0/1.00 | 98 | 67,180 | 10,629 | 9151 | 17,076 | 1.86 |
| 21 | 700/1.0/0.25 | 98 | 78,370 | 9214 | 8163 | 12,773 | 1.56 |
| 22 | 700/1.0/0.50 | 99 | 79,170 | 10,835 | 11,468 | 20,774 | 1.81 |
| 23 | 700/1.0/0.75 | 98 | 78,370 | 12,120 | 9358 | 16,877 | 1.80 |
| 24 | 700/1.0/1.00 | 97 | 77,570 | 12,896 | 10,098 | 17,631 | 1.74 |
| Entry | Catalyst | [CL]0/[nC12H25OH]0/ [1,n-bis[Bim][PF6]]0 | Temp. (°C) | Conv. a (%) | Mn,theob (g mol−1) | Mn,NMRc (g mol−1) | Mn,GPCd (g mol−1) | Mw,GPCd (g mol−1) | Ðd |
|---|---|---|---|---|---|---|---|---|---|
| 25 | 2a | 400/1.0/0.50 | 150 | 98 | 44,744 | 12,265 | 12,588 | 23,180 | 1.84 |
| 26 | 2b | 400/1.0/0.50 | 150 | 98 | 44,744 | 16,642 | 13,618 | 24,468 | 1.79 |
| 27 | 2c | 400/1.0/0.50 | 150 | 99 | 45,200 | 17,847 | 10,916 | 15,820 | 1.44 |
| 28 | 2d | 400/1.0/0.50 | 150 | 98 | 44,744 | 15,415 | 14,236 | 27,697 | 1.94 |
| 29 | Sn(Oct)2 | 400/1.0/1.00 | 150 | 98 | 44,744 | 12,385 | 14,391 | 24,455 | 1.69 |
| 30 | 2a | 400/1.0/0.50 | 160 | 97 | 44,280 | 14,359 | 13,908 | 26,337 | 1.89 |
| 31 | 2b | 400/1.0/0.50 | 160 | 96 | 43,830 | 20,666 | 13,440 | 25,553 | 1.90 |
| 32 | 2c | 400/1.0/0.50 | 160 | 98 | 44,744 | 16,437 | 15,665 | 30,867 | 1.97 |
| 33 | 2d | 400/1.0/0.50 | 160 | 98 | 44,744 | 15,237 | 14,100 | 28,149 | 1.99 |
| 34 | Sn(Oct)2 | 400/1.0/1.00 | 160 | 98 | 44,744 | 11,991 | 17,355 | 30,247 | 1.74 |
| 35 | 2a | 400/1.0/0.50 | 170 | 97 | 44,280 | 12,636 | 12,558 | 24,124 | 1.92 |
| 36 | 2b | 400/1.0/0.50 | 170 | 98 | 44,744 | 16,163 | 16,788 | 32,227 | 1.92 |
| 37 | 2c | 400/1.0/0.50 | 170 | 98 | 44,744 | 16,089 | 14,336 | 28,008 | 1.95 |
| 38 | 2d | 400/1.0/0.50 | 170 | 97 | 44,280 | 15,233 | 16,769 | 31,622 | 1.89 |
| 39 | Sn(Oct)2 | 400/1.0/1.00 | 170 | 98 | 44,744 | 10,290 | 14,418 | 25,711 | 1.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheechana, N.; Benchaphanthawee, W.; Akkravijitkul, N.; Rithchumpon, P.; Junpirom, T.; Limwanich, W.; Punyodom, W.; Kungwan, N.; Ngaojampa, C.; Thavornyutikarn, P.; et al. Organocatalytic Ring-Opening Polymerization of ε-Caprolactone Using bis(N-(N′-butylimidazolium)alkane Dicationic Ionic Liquids as the Metal-Free Catalysts: Polymer Synthesis, Kinetics and DFT Mechanistic Study. Polymers 2021, 13, 4290. https://doi.org/10.3390/polym13244290
Cheechana N, Benchaphanthawee W, Akkravijitkul N, Rithchumpon P, Junpirom T, Limwanich W, Punyodom W, Kungwan N, Ngaojampa C, Thavornyutikarn P, et al. Organocatalytic Ring-Opening Polymerization of ε-Caprolactone Using bis(N-(N′-butylimidazolium)alkane Dicationic Ionic Liquids as the Metal-Free Catalysts: Polymer Synthesis, Kinetics and DFT Mechanistic Study. Polymers. 2021; 13(24):4290. https://doi.org/10.3390/polym13244290
Chicago/Turabian StyleCheechana, Nathaporn, Wachara Benchaphanthawee, Natthapol Akkravijitkul, Puracheth Rithchumpon, Thiti Junpirom, Wanich Limwanich, Winita Punyodom, Nawee Kungwan, Chanisorn Ngaojampa, Praput Thavornyutikarn, and et al. 2021. "Organocatalytic Ring-Opening Polymerization of ε-Caprolactone Using bis(N-(N′-butylimidazolium)alkane Dicationic Ionic Liquids as the Metal-Free Catalysts: Polymer Synthesis, Kinetics and DFT Mechanistic Study" Polymers 13, no. 24: 4290. https://doi.org/10.3390/polym13244290