
Non-Destructive Detection of Pentachlorophenol Residues in Historical Wooden Objects

 ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Model Wood Samples Preparation
2.2. SPME Methods
2.3. Solvent Extraction and SPE Method
2.4. HPLC-UV Analysis
2.5. GC–MS Analysis
2.6. Sampling in the Museum Depot
3. Results and Discussion
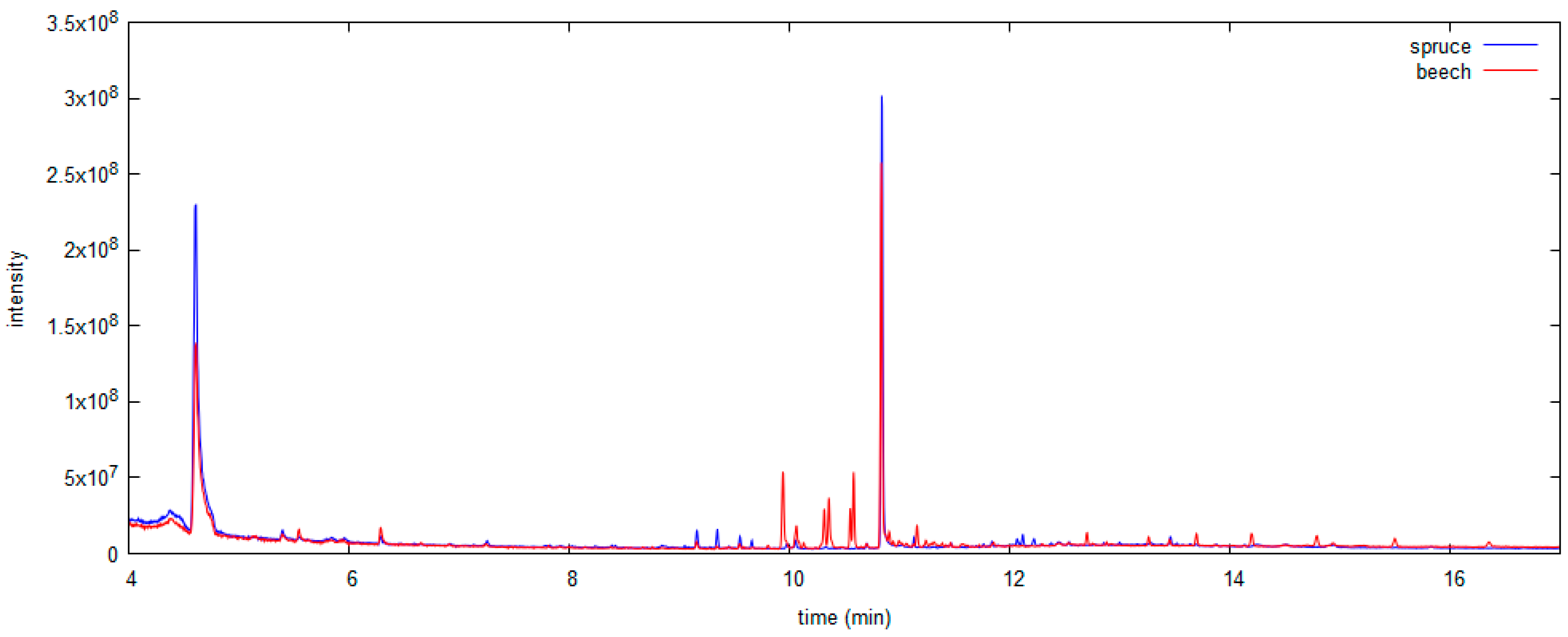
3.1. Model Wood Samples
3.2. Solvent Extraction
3.3. SPME Sampling
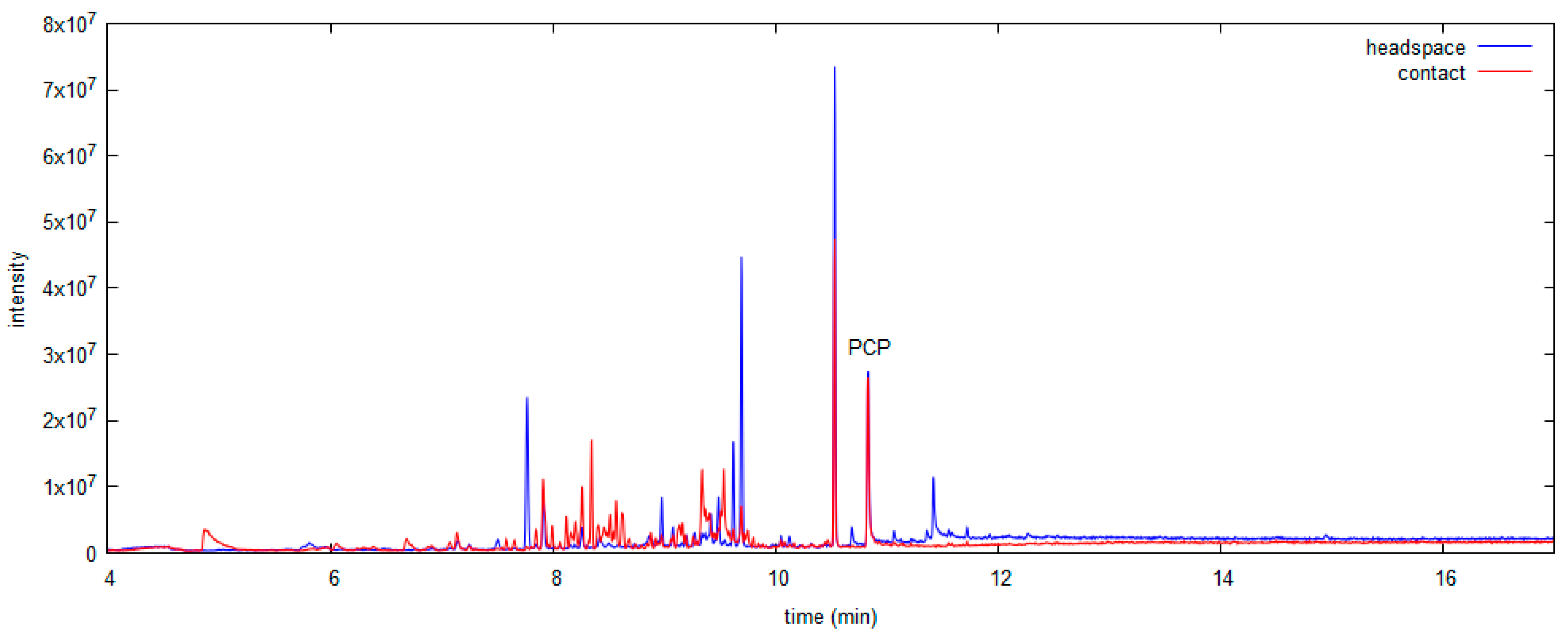
3.3.1. SPME in Headspace Mode
3.3.2. SPME in Contact Mode
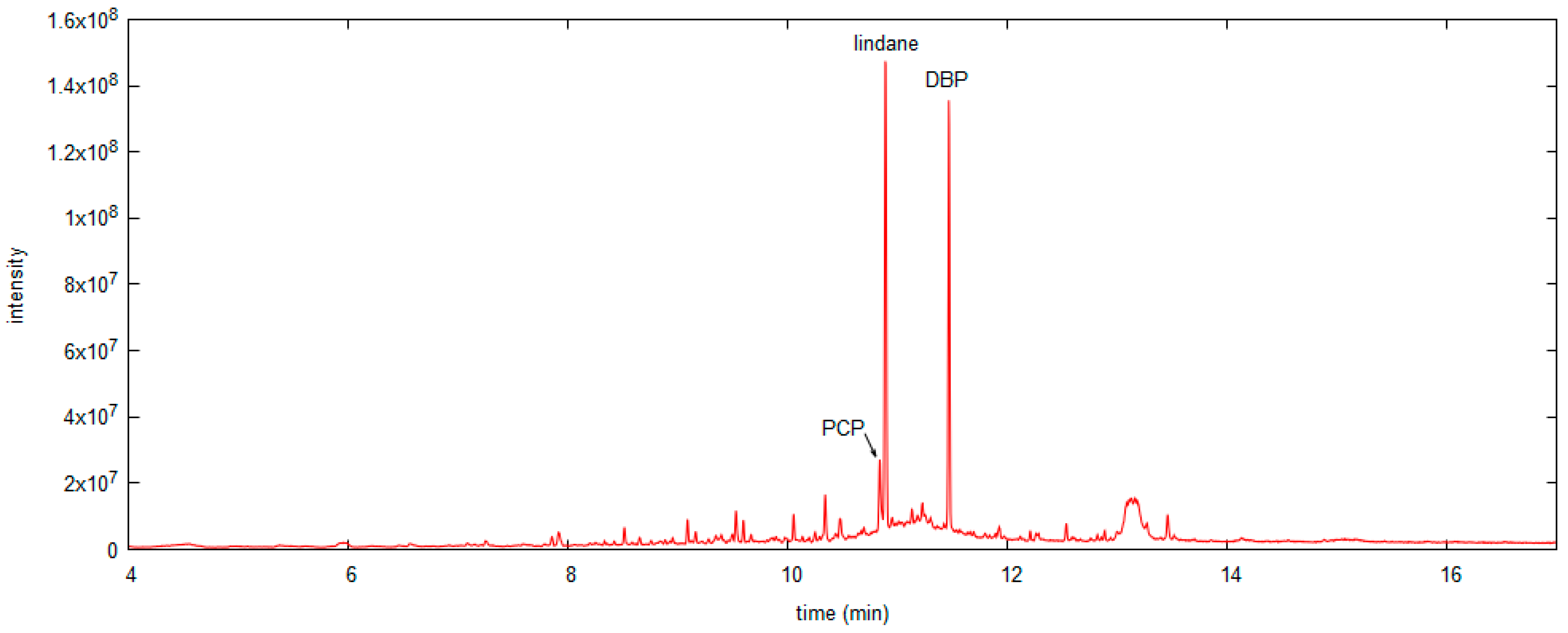
3.4. Application of the Sampling Methods to Museum Objects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Object Description | Sampling | PCP Detection | |
|---|---|---|---|
| OBJ1 | Small cabinet with doors and a drawer, open top surface. | An already detached trim collected (m = 2.709 g); contact SPME on both largest surfaces. | <LOD |
| OBJ2 | Nightstand with tall narrow legs. | An already detached piece of wood collected (m = 0.201 g); HS SPME in vial and acetone extraction. | Not detected |
| OBJ3 | Black chest of drawers, with nine drawers (Figure 6). | HS SPME sampling in the central drawer. | 40 mg/kg wood |
| OBJ4 | Dark chest, painted with still life and year 1853 (Figure 7). | HS SPME on the lid. | <LOD |
| OBJ5 | Light chest, painted with birds. | HS SPME inside the chest. | <LOD |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Unger, A.; Schniewind, A.; Unger, W. Conservation of Wood Artifacts: A Handbook; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001; ISBN 978-3-540-41580-0. [Google Scholar]
- British Standards Institution. Specification for Managing Environmental Conditions for Cultural Collections; PAS.; British Standards Institution: London, UK, 2012; ISBN 978-0-580-71315-6. [Google Scholar]
- Nguyen, H.; Lagarde, F.; Louarn, G.; Daniel, P. A New Way to Discriminate Polluted Wood by Vibrational Spectroscopies. Talanta 2017, 167, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Fierascu, R.C.; Doni, M.; Fierascu, I. Selected Aspects Regarding the Restoration/Conservation of Traditional Wood and Masonry Building Materials: A Short Overview of the Last Decade Findings. Appl. Sci. 2020, 10, 1164. [Google Scholar] [CrossRef]
- Smithsonian Museum Conservation Institute Furniture Care and Handling. Available online: https://www.si.edu/mci/downloads/taking_care/MCIFurnitureCare.pdf (accessed on 18 February 2021).
- Gahan, C.J. Furniture Beetles; BM(NH): London, UK, 1920; p. 23. [Google Scholar]
- Schiopu, N.; Tiruta-Barna, L. Wood preservatives. In Toxicity of Building Materials; Pacheco-Torgal, F., Jalali, S., Fucic, A., Eds.; Woodhead Publishing Series in Civil and Structural Engineering; Woodhead Publishing: Cambridge, UK, 2012; pp. 138–165. ISBN 978-0-85709-122-2. [Google Scholar]
- Ferizli, A.G.; Emekci, M. Museum Fumigation. In Proceedings of the 2008 Annual International Research Conference on Methyl Bromide Alternatives and Emissions Reductions, Orlando, FL, USA, 10–14 November 2008; pp. 72–73. [Google Scholar]
- Rushworth, I.D.; Higgitt, C.; Smith, M.; Gibson, L.T. Non-Invasive Multiresidue Screening Methods for the Determination of Pesticides in Heritage Collections. Herit. Sci. 2014, 2, 3. [Google Scholar] [CrossRef]
- Kearney, T. Chemical Contamination of Repatriated Native Californian NAGPRA Materials: Principles of Risk Assessment for Acute and Chronic Health Effects. Collect. Forum. 2001, 16, 44–53. [Google Scholar]
- Pinniger, D.B.; Child, R.E. Woodworm-a Necessary Case for Treatment? New Techniques for the Detection and Control of Furniture Beetle. In Proceedings of the Second International Conference on Urban Pests, Edinburgh, Scotland, 7–10 July 1996. [Google Scholar]
- Wörle, M.; Hubert, V.; Hildbrand, E.; Hunger, K.; Lehmann, E.; Mayer, I.; Petrak, G.; Pracher, M.; von Arx, U.; Wülfert, S. Evaluation of Decontamination Methods of Pesticide Contaminated Wooden Objects in Museum Collections: Efficiency of the Treatments and Influence on the Wooden Structure. J. Cult. Herit. 2012, 13, S209–S215. [Google Scholar] [CrossRef]
- Humar, M. Zaščita Lesa Danes—Jutri (Wood Preservation Today—Tomorrow). Les 2004, 56, 184–188. [Google Scholar]
- Morrell, J.J. Protection of Wood-Based Materials. In Handbook of Environmental Degradation of Materials, 3rd ed.; Kutz, M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 343–368. ISBN 978-0-323-52472-8. [Google Scholar]
- Agency for Toxic Substances and Disease Registry of US Department of Health and Human Services. Toxicological Profile for Pentachlorophenol; Agency for Toxic Substances and Disease Registry of US Department of Health and Human Services: Atlanta, GA, USA, 2001. [Google Scholar]
- Environmental Protection Agency. Technical Factsheet on: Pentachlorophenol; Environmental Protection Agency: Washington, DC, USA, 2005. [Google Scholar]
- Buhr, A.; Genning, C.; Salthammer, T. Trace Analysis of Pentachlorophenol (PCP) in Wood and Wood-Based Products—Comparison of Sample Preparation Procedures. Fresenius J. Anal. Chem. 2000, 367, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D. Determination of Pentachlorophenol Residue in Meat and Fish by Gas Chromatography–Electron Capture Detection and Gas Chromatography–Mass Spectrometry with Accelerated Solvent Extraction. J. Chromatogr. Sci. 2014, 52, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Schnelle-Kreis, J.; Scherb, H.; Gebefügi, I.; Kettrup, A.; Weigelt, E. Pentachlorophenol in Indoor Environments. Correlation of PCP Concentrations in Air and Settled Dust from Floors. Sci. Total Environ. 2000, 256, 125–132. [Google Scholar] [CrossRef]
- ARSO. Operativni Program Preprečevanja Onesnaževanja Vodnega Okolja z Nevarnimi Kloriranimi Ogljikovodiki Iz Razpršenih Virov Onesnaževanja; Slovenian Environment Agency: Ljubljana, Slovenia, 2005. [Google Scholar]
- Inoue, K.; Yoshida, S.; Nakayama, S.; Ito, R.; Okanouchi, N.; Nakazawa, H. Development of Stable Isotope Dilution Quantification Liquid Chromatography–Mass Spectrometry Method for Estimation of Exposure Levels of Bisphenol A, 4-Tert-Octylphenol, 4-Nonylphenol, Tetrabromobisphenol A, and Pentachlorophenol in Indoor Air. Arch. Environ. Contam. Toxicol. 2006, 51, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Schieweck, A.; Delius, W.; Siwinski, N.; Vogtenrath, W.; Genning, C.; Salthammer, T. Occurrence of Organic and Inorganic Biocides in the Museum Environment. Atmos. Environ. 2007, 41, 3266–3275. [Google Scholar] [CrossRef]
- Deering, K.; Spiegel, E.; Quaisser, C.; Nowak, D.; Schierl, R.; Bose-O’Reilly, S.; Garí, M. Monitoring of Arsenic, Mercury and Organic Pesticides in Particulate Matter, Ambient Air and Settled Dust in Natural History Collections Taking the Example of the Museum Für Naturkunde, Berlin. Environ. Monit. Assess. 2019, 191, 375. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, R.; Luan, T.; Lin, L.; Zou, S.; Yang, Q. Full Automatic Determination of Chlorophenols in Water Using Solid-Phase Microextraction/on-Fiber Derivatization and Gas Chromatography-Mass Spectrometry: Sample Preparation. J. Sep. Sci. 2012, 35, 1017–1026. [Google Scholar] [CrossRef]
- Amendola, L.; Cortese, M.; Vinatoru, D.; Sposato, S.; Insogna, S. Innovative Analytical Method for the Determination of Underivatized Tributyltin and Pentachlorophenol in Seawater by Gas Chromatography-Triple Quadrupole Mass Spectrometry. Anal. Chim. Acta 2017, 975, 70–77. [Google Scholar] [CrossRef] [PubMed]
- De Morais, P.; Stoichev, T.; Basto, M.C.P.; Carvalho, P.N.; Vasconcelos, M.T.S.D. A Headspace SPME-GC-ECD Method Suitable for Determination of Chlorophenols in Water Samples. Anal. Bioanal. Chem. 2011, 399, 2531–2538. [Google Scholar] [CrossRef]
- Liu, Y.; Wen, B.; Shan, X. Determination of Pentachlorophenol in Wastewater Irrigated Soils and Incubated Earthworms. Talanta 2006, 69, 1254–1259. [Google Scholar] [CrossRef]
- Hong, H.C.; Zhou, H.Y.; Luan, T.G.; Lan, C.Y. Residue of Pentachlorophenol in Freshwater Sediments and Human Breast Milk Collected from the Pearl River Delta, China. Environ. Int. 2005, 31, 643–649. [Google Scholar] [CrossRef]
- Reigner, B.G.; Rigod, J.F.; Tozer, T.N. Simultaneous Assay of Pentachlorophenol and Its Metabolite, Tetrachlorohydroquinone, by Gas Chromatography without Derivatization. J. Chromatogr. B Biomed. Sci. Appl. 1990, 533, 111–124. [Google Scholar] [CrossRef]
- Mardones, C.; Palma, J.; Sepúlveda, C.; Berg, A.; von Baer, D. Determination of Tribromophenol and Pentachlorophenol and Its Metabolite Pentachloroanisole InAsparagus Officinalis by Gas Chromatography/Mass Spectrometry. J. Sep. Sci. 2003, 26, 923–926. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Q.; Peng, Q.; Xuan, D.; Qu, W. Development of a Solid Phase Microextraction-Gas Chromatography–Mass Spectrometry Method for the Determination of Pentachlorophenol in Human Plasma Using Experimental Design. Chemosphere 2007, 70, 256–262. [Google Scholar] [CrossRef]
- Cline, R.E.; Hill, R.H.; Phillips, D.L.; Needham, L.L. Pentachlorophenol Measurements in Body Fluids of People in Log Homes and Workplaces. Arch. Environ. Contam. Toxicol. 1989, 18, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.L.; Durand, L.R.; Inman, R.D.; Kiigemagi, U.; Deinzer, M.L. Residues of Pentachlorophenol and Other Chlorinated Contaminants in Human Tissues: Analysis by Electron Capture Gas Chromatography and Electron Capture Negative Ion Mass Spectrometry. Arch. Environ. Contam. Toxicol. 1991, 21, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, X.; Yu, H.; Tao, X.; Zhou, Y.; Qu, W. Global Trends and Diversity in Pentachlorophenol Levels in the Environment and in Humans: A Meta-Analysis. Environ. Sci. Technol. 2011, 45, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, N.; Sadongei, A.; Boyer, L.V. Old Poisons, New Problems: A Museum Resource for Managing Contaminated Cultural Materials; Rowman Altamira: Lanham, MD, USA, 2005; ISBN 978-0-7591-0515-7. [Google Scholar]
- Tello, H. Investigations on Super Fluid Extraction (SFE) with Carbon Dioxide on Ethnological Materials and Objects Contaminated with Pesticides. Ph.D. Thesis, Fachhochschule für Technik und Wirtschaft Berlin, Berlin, Germany, 30 September 2006. [Google Scholar]
- Sirois, J.P.; Sansoucy, G. Analysis of Museum Objects for Hazardous Pesticide Residues: A Guide to Techniques. Collect. Forum. 2001, 17, 49–66. [Google Scholar]
- Gremaud, E.; Turesky, R.J. Rapid Analytical Methods To Measure Pentachlorophenol in Wood. J. Agric. Food Chem. 1997, 45, 1229–1233. [Google Scholar] [CrossRef]
- Portoni, F.; Grau-Bové, J.; Strlič, M. Application of a Non-Invasive, Non-Destructive Technique to Quantify Naphthalene Emission Rates from Museum Objects. Herit. Sci. 2019, 7, 58. [Google Scholar] [CrossRef]
- Kearney, M.; Parkin, I.; Townsend, J.H.; Hidalgo, M.; Curran, K. Characterisation of VOCs Surrounding Naum Gabo’s Construction in Space ‘Two Cones’, (Tate) by in Situ SPME GC-MS Monitoring. Stud. Conserv. 2018, 63, 369–371. [Google Scholar] [CrossRef]
- Ormsby, M.; Johnson, J.S.; Heald, S.; Chang, L.; Bosworth, J. Investigation of Solid Phase Microextraction Sampling for Organic Pesticide Residues on Museum Collections. Collect. Forum. 2006, 20, 1–12. [Google Scholar]
- Alvarez-Martin, A.; George, J.; Kaplan, E.; Osmond, L.; Bright, L.; Newsome, G.A.; Kaczkowski, R.; Vanmeert, F.; Kavich, G.; Heald, S. Identifying VOCs in Exhibition Cases and Efflorescence on Museum Objects Exhibited at Smithsonian’s National Museum of the American Indian-New York. Herit. Sci. 2020, 8, 115. [Google Scholar] [CrossRef]
- Alvarez-Martin, A.; McHugh, K.; Martin, C.; Kavich, G.; Kaczkowski, R. Understanding Air-Tight Case Environments at the National Museum of the American Indian (Smithsonian Institution) by SPME-GC-MS Analysis. J. Cult. Herit. 2020, 44, 38–46. [Google Scholar] [CrossRef]
- Goewie, C.E.; Berkhof, R.J.; Maris, F.A.; Treskes, M.; Brinkman, U.T. Determination of Pentachlorophenol in Wood Samples Using Liquid Chromatography With UV Absorbance, Amperometric and Electron-Capture Detection. Int. J. Environ. Anal. Chem. 1986, 26, 305–318. [Google Scholar] [CrossRef]
- Pohlandt, K.; Bockelmann, C.; Marutzky, R. Concentrations of Pentachlorophenol and Lindane in Various Assortments of Wood. Chemosphere 1995, 31, 4025–4031. [Google Scholar] [CrossRef]
- Bartelt, G.; Buge, H.G.; Görner, W.; Win, T. Determination of Chlorine and Pentachlorophenol in Wood. Fresenius J. Anal. Chem. 1998, 360, 433–434. [Google Scholar] [CrossRef]
- Becker, R.; Buge, H.-G.; Win, T. Determination of Pentachlorophenol (PCP) in Waste Wood—Method Comparison by a Collaborative Trial. Chemosphere 2002, 47, 1001–1006. [Google Scholar] [CrossRef]
- Besner, A.; Gilbert, R.; Tetreault, P.; Lepine, L.; Archambault, J.-F. Determination of Pentachlorophenol and Its Hydrocarbon Solvent in Wood, Soil, and Water by Gas Chromatography and FT-LR Spectroscopy in a Single-Sample Treatment. Anal. Chem. 1995, 67, 442–446. [Google Scholar] [CrossRef]
- European Committee for Standardization. Durability of Wood and Wood-Based Products—Quantitative Determination of Pentachlorophenol in Wood—Gas Chromatographic Method; PD CEN/TR 14823:2003; European Committee for Standardization (CEN): Brussels, Belgium, 2003. [Google Scholar]
- Koyano, S.; Ueno, D.; Yamamoto, T.; Kajiwara, N. Concentrations of POPs Based Wood Preservatives in Waste Timber from Demolished Buildings and Its Recycled Products in Japan. Waste Manag. 2019, 85, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Covaci, A.; Kawaki, P.; Indekeu, C.; Schepens, P.; Neels, H. Highly Chlorinated Toxic Contaminants in Pesticide-Treated Wooden Art Objects. Arch. Environ. Occup. Health 2006, 61, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Mauruschat, D.; Schumann, A.; Meinlschmidt, P.; Gunschera, J.; Salthammer, T. Application of Gas Chromatography—Field Asymmetric Ion Mobility Spectrometry (GC-FAIMS) for the Detection of Organic Preservatives in Wood. Int. J. Ion Mobil. Spec. 2014, 17, 1–9. [Google Scholar] [CrossRef]
- Mayer, I.; Hunger, K. Destructive and Non-Destructive Methods for the Evaluation of Chlorinated Pesticides Concentration and Emissions from Wooden Art Objects. In Proceedings of the International Conference on Wooden Cultural Heritage: Evaluation of Deterioration and Management of Change, Hamburg, Germany, 7–10 October 2009. [Google Scholar]
- PubChem Pentachlorophenol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/992 (accessed on 14 December 2020).
- Sander, R. Compilation of Henry’s Law Constants (Version 4.0) for Water as Solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Mauruschat, D.; Plinke, B.; Aderhold, J.; Gunschera, J.; Meinlschmidt, P.; Salthammer, T. Application of Near-Infrared Spectroscopy for the Fast Detection and Sorting of Wood–Plastic Composites and Waste Wood Treated with Wood Preservatives. Wood Sci. Technol. 2016, 50, 313–331. [Google Scholar] [CrossRef]
- Kearney, M.; Townsend, J.H.; Parkin, I.P.; Hidalgo, M.; Curran, K. Factors Affecting the Practicality of Solid-Phase Microextraction VOC Analysis of Artworks Featuring Polymeric Materials in Open Environments. Microchem. J. 2020, 155, 104711. [Google Scholar] [CrossRef]
- Faraca, G.; Boldrin, A.; Astrup, T. Resource Quality of Wood Waste: The Importance of Physical and Chemical Impurities in Wood Waste for Recycling. Waste Manag. 2019, 87, 135–147. [Google Scholar] [CrossRef] [PubMed]









| Sampling Mode | Effect on Object/Sample | Sampling Time | * Linearity (γ − PCP Content in Wood (mg/g), Ar − Peak Area, R2 − Correlation Coefficient) | ** Repeatability (%RSD) | *** LOD (mg PCP/kg Wood) |
|---|---|---|---|---|---|
| Headspace SPME | Non-destructive | 40 min | Ar = 8.31 × 108 γ − 9.98 × 106 R2 = 0.9524 | 9 | 20 |
| Contact SPME | Non-destructive | 30 min | Ar = 1.41 × 109 γ − 1.15 × 107 R2 = 0.8993 | 16 | 30 |
| Extraction time | Extraction recovery (%) | ||||
| Acetone extraction | Destructive | 24 h | 84 | 11 | 10 |
| Acetone/H2SO4 extraction | Destructive | 24 h | 66 | 5 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraševec, I.; Nemeček, N.; Lozar Štamcar, M.; Kralj Cigić, I.; Prosen, H. Non-Destructive Detection of Pentachlorophenol Residues in Historical Wooden Objects. Polymers 2021, 13, 1052. https://doi.org/10.3390/polym13071052
Kraševec I, Nemeček N, Lozar Štamcar M, Kralj Cigić I, Prosen H. Non-Destructive Detection of Pentachlorophenol Residues in Historical Wooden Objects. Polymers. 2021; 13(7):1052. https://doi.org/10.3390/polym13071052
Chicago/Turabian StyleKraševec, Ida, Nataša Nemeček, Maja Lozar Štamcar, Irena Kralj Cigić, and Helena Prosen. 2021. "Non-Destructive Detection of Pentachlorophenol Residues in Historical Wooden Objects" Polymers 13, no. 7: 1052. https://doi.org/10.3390/polym13071052
APA StyleKraševec, I., Nemeček, N., Lozar Štamcar, M., Kralj Cigić, I., & Prosen, H. (2021). Non-Destructive Detection of Pentachlorophenol Residues in Historical Wooden Objects. Polymers, 13(7), 1052. https://doi.org/10.3390/polym13071052