
Degradability of Polyurethanes and Their Blends with Polylactide, Chitosan and Starch

 ,
,  ,
, 
 ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Degradability in Hydrolytic Conditions
2.2.2. Degradability in Oxidative Conditions
2.2.3. Chemical Structure
2.2.4. Molecular Weight
2.2.5. Surface Topography
- Scanning electron microscopy (SEM): The sample surfaces before and after hydrolysis were analyzed by a Hitachi S-4800 scanning electron microscope (SEM) (Hitachi High-Technologies Corporation, Tokyo, Japan). Samples were metallically covered and scanned at an accelerated voltage of 10 kV at a working distance of 8 mm. Different zones of each sample were analyzed to ensure the morphology of each sample.
- Optical Microscopy(OM) micrographs were taken using a Nikon Eclipse E600W microscope (Mettler FP 82 HT, Melvile, NY, USA). The micrographs were collected with the software analySIS docu FIVE.
2.2.6. Thermal Properties
3. Results and Discussion
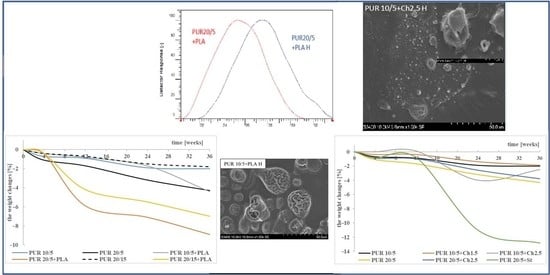
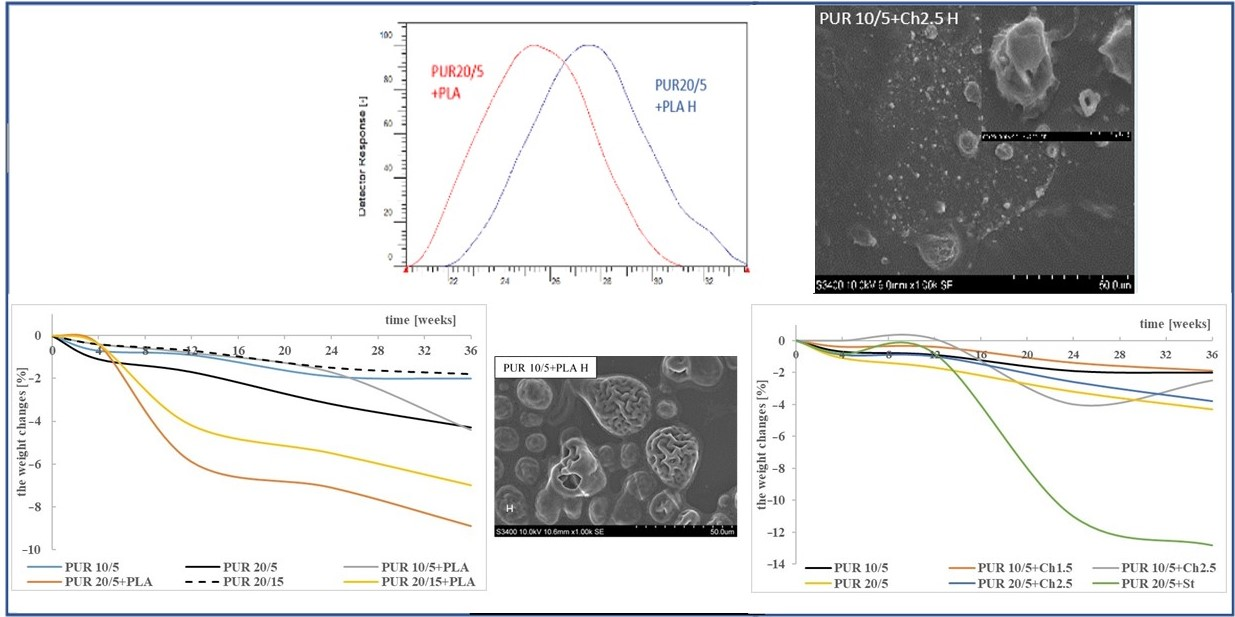
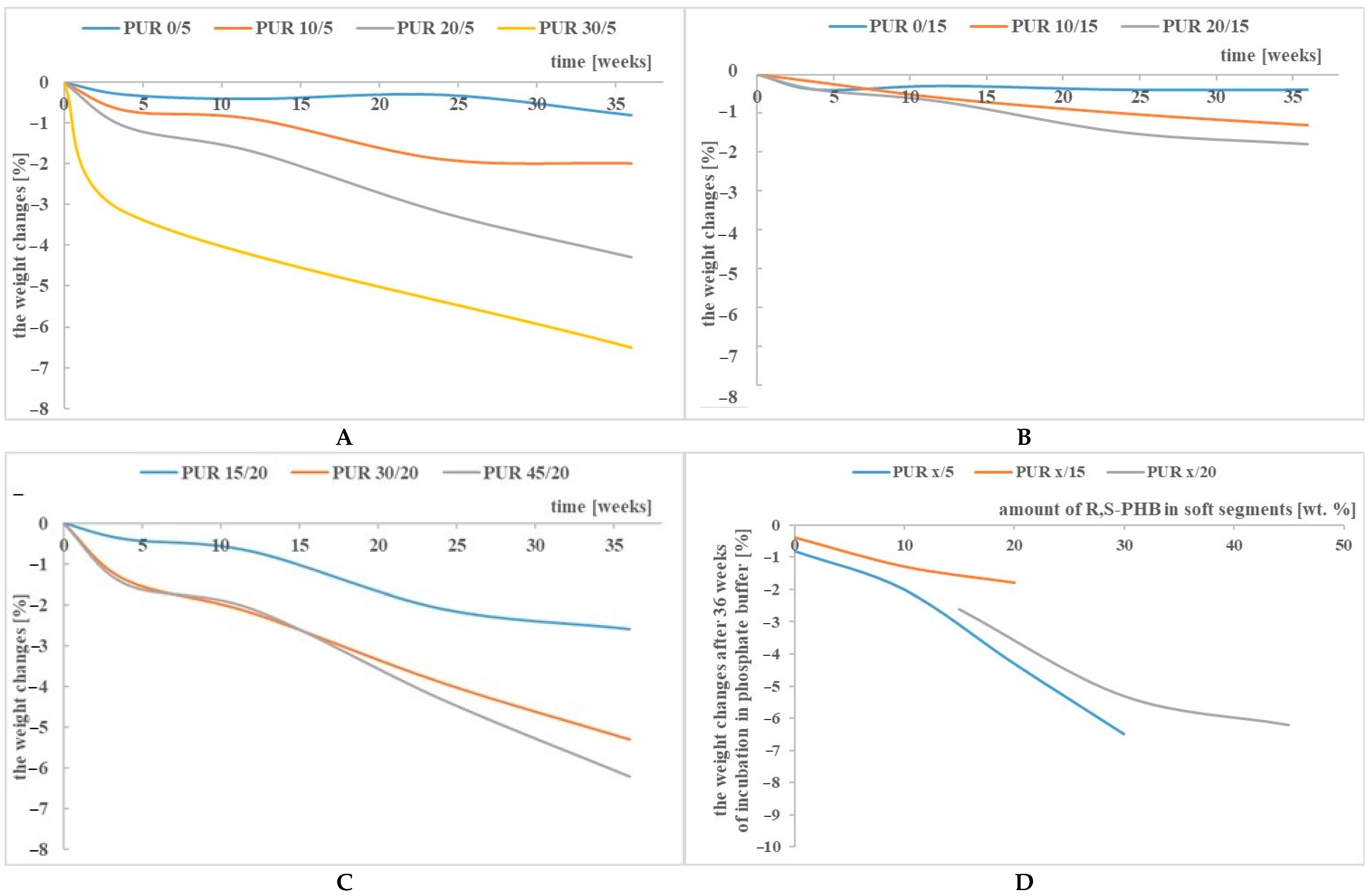
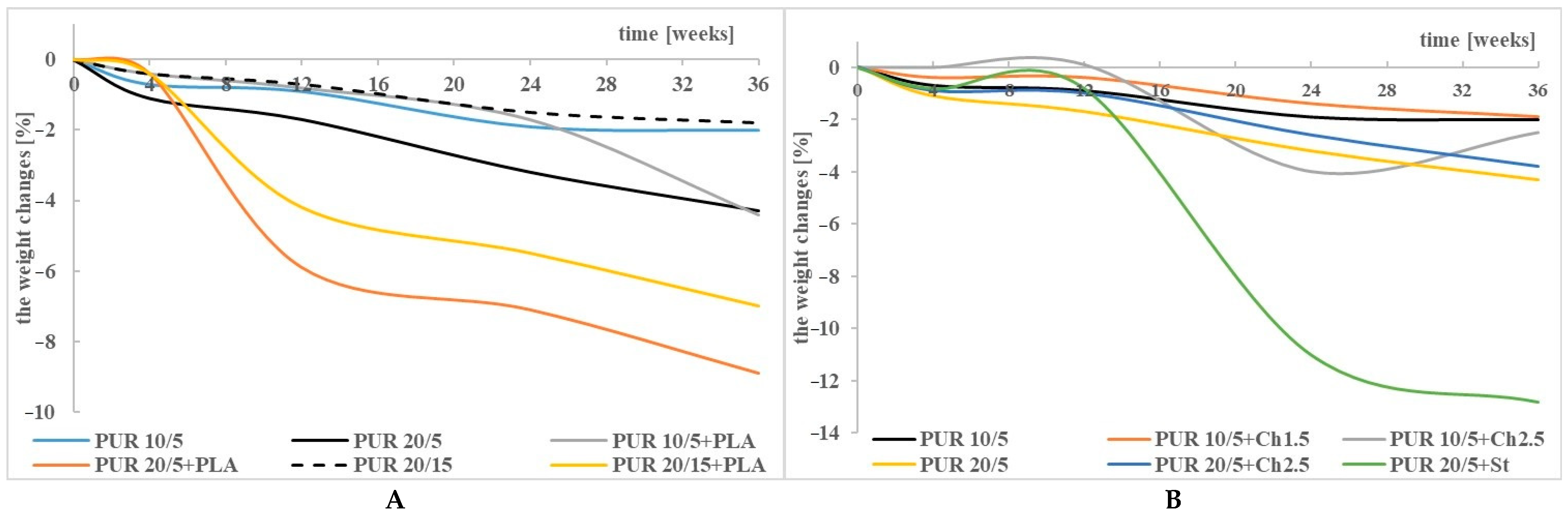
3.1. Weight Changes
3.2. Molecular Weight
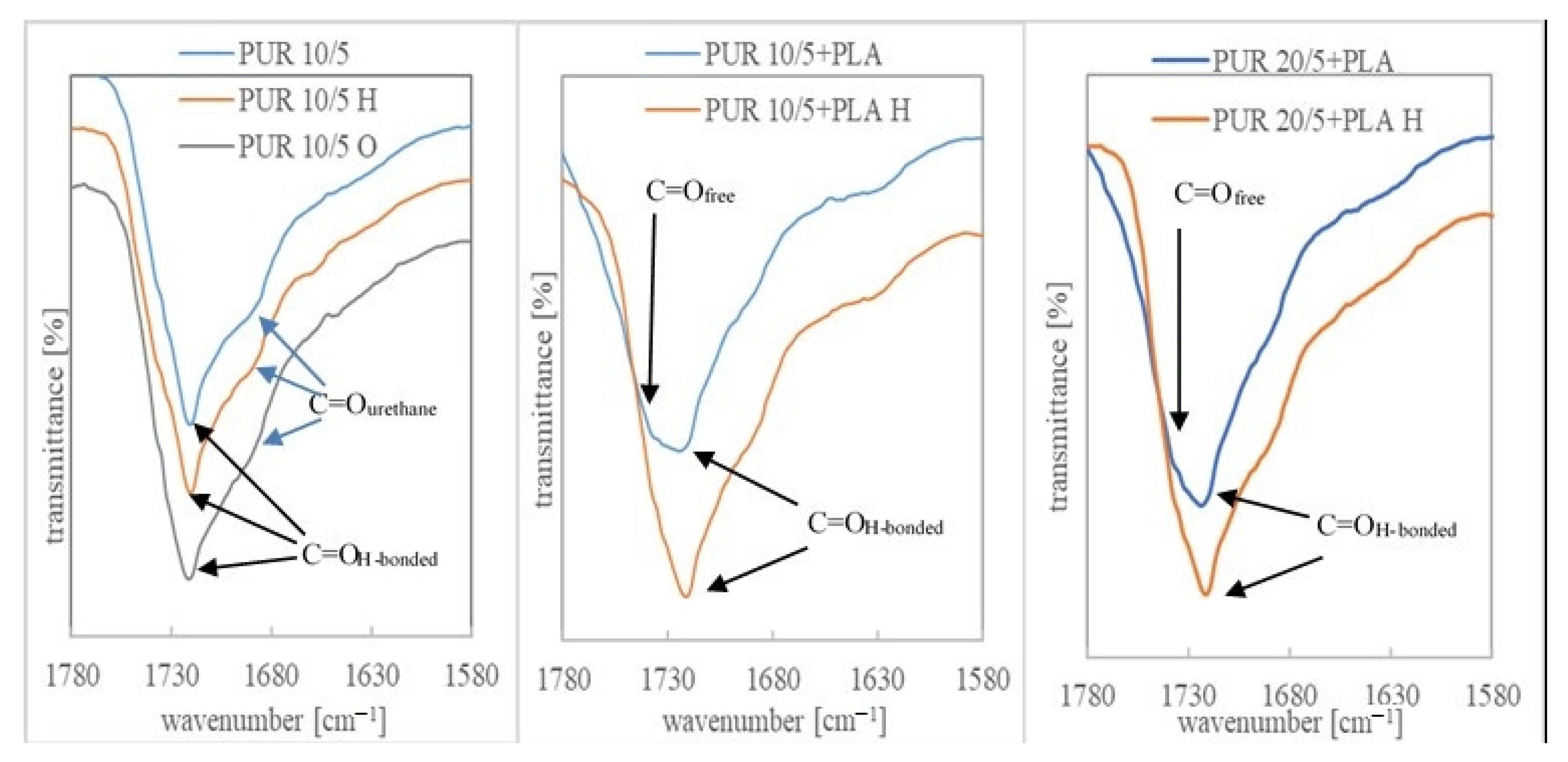
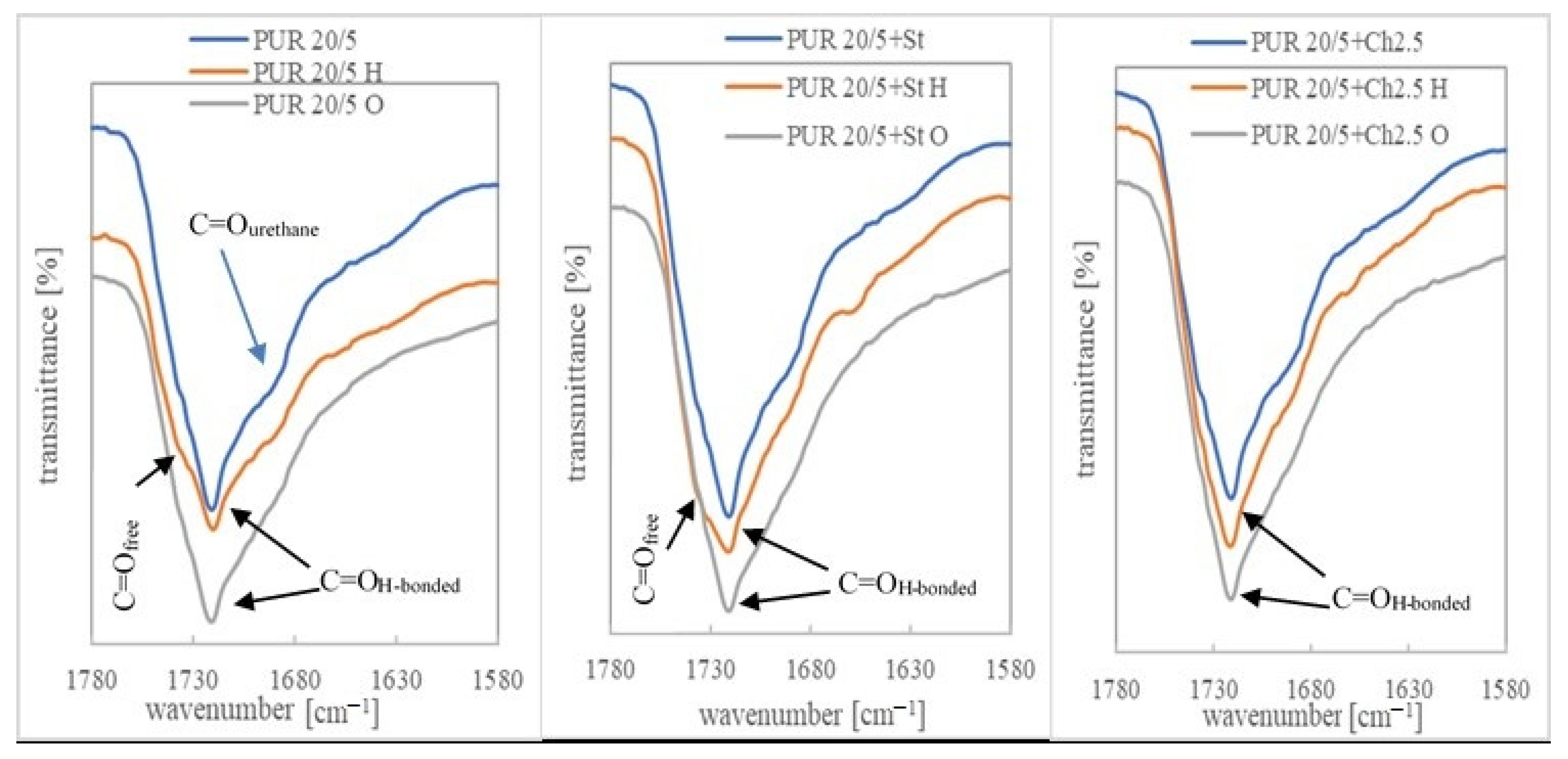
3.3. Chemical Structure
3.4. Thermal Properties
3.5. Microscopic Observation
3.6. Degradation in Oxidative Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heath, D.E.; Guelcher, S.A.; Cooper, S.L. Polyurethanes in Biomaterials Science, 4th ed.; Wagner, W., Sakiyama-Elbert, S., Zhang, G., Yaszemski, M., Eds.; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 103–107. [Google Scholar]
- Oprea, S.; Timpu, D.; Oprea, V. Design-properties relationships of polyurethanes elastomers depending on different chain extenders structures. J. Polym. Res. 2019, 26, 1–15. [Google Scholar] [CrossRef]
- Tanzi, M.C.; Mantovani, D.; Petrini, P.; Guidoin, R.; Laroche, G. Chemical stability of polyether urethanes versus polycarbonate urethanes. J. Biomed. Mater. Res. 1997, 36, 550–559. [Google Scholar] [CrossRef]
- Meereboer, K.W.; Misra, M.; Mohanty, A.K. Review of recent advances in the biodegradability of polyhydroxyalkanoate (PHA) bioplastics and their composites. Green Chem. 2020, 22, 5519–5558. [Google Scholar] [CrossRef]
- Folino, A.; Karageorgiou, A.; Calabrò, P.S.; Komilis, D. Biodegradation of Wasted Bioplastics in Natural and Industrial Environments: A Review. Sustainability 2020, 12, 6030. [Google Scholar] [CrossRef]
- Villani, M.; Consonni, R.; Canetti, M.; Bertoglio, F.; Iervese, S.; Bruni, G.; Visai, L.; Iannace, S.; Bertini, F. Polyurethane-Based Composites: Efects of Antibacterial Fillers on the Physical-Mechanical Behavior of Thermoplastic Polyurethanes. Polymers 2020, 12, 362. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, J.F.; Pachekoski, W.M.; Vicino, J.R.D.L. Composition for Preparing a Biodegradable Polyurethane- Based Foam and a Biodegradable Polyurethane Foam. WO 2007/095713 Al, 24 February 2006.
- Lu, M.-Y.; Surányi, A.; Viskolcz, B.; Fiser, B. Molecular Design of Sugar-Based Polyurethanes. Croat. Chem. Acta 2018, 91, 299–307. [Google Scholar] [CrossRef]
- Javaid, M.A.; Zia, K.M.; Zafar, K.; Khosa, M.K.; Akrame, N.; Ajmal, M.; Imran, M.; Iqbal, M.N. Synthesis and molecular characterization of chitosan/starch blends based polyurethanes. Int. J. Biol. Macromol. 2020, 146, 243–252. [Google Scholar] [CrossRef]
- Javaid, M.A.; Zia, K.M.; Iqbal, A.; Ahmad, S.; Akramc, N.; Liu, X.; Nawaz, H.; Khosa, M.K.; Awais, M. Utilization of waxy corn starch as an efficient chain extender for the preparation of polyurethane elastomers. Int. J. Biol. Macromol. 2020, 148, 415–423. [Google Scholar] [CrossRef]
- Khan, M.S.J.; Khan, S.B.; Kamal, T.; Asiri, A.M. Agarose biopolymer coating on polyurethane sponge as host for catalytic silver metal nanoparticles. Polym. Test. 2019, 78, 105983. [Google Scholar] [CrossRef]
- Romero-Azogil, L.; Benito, E.; de Ilarduya, A.M.; García-Martín, M.G.; Galbis, J.A. Hydrolytic degradation of D-mannitol-based polyurethanes. Polym. Degrad. Stabil. 2018, 153, 262–271. [Google Scholar] [CrossRef]
- Gaikwad, M.S.; Gite, V.V.; Mahulikar, P.P.; Hundiwale, D.G.; Yemul, O.S. Eco-friendly polyurethane coatings from cottonseed and karanja oil. Prog. Org. Coat. 2015, 86, 164–172. [Google Scholar] [CrossRef]
- Yin, S.; Xia, Y.; Jia, Q.; Hou, Z.-S.; Zhang, N. Preparation and properties of biomedical segmented polyurethanes based on poly(ether ester) and uniform-size diurethane diisocyanates. J. Biomat. Sci. Polym. Ed. 2017, 28, 119–138. [Google Scholar] [CrossRef]
- Špírková, M.; Hodan, J.; Kobera, L.; Kredatusová, J.; Kubies, D.; Machová, L.; Poręba, R.; Serkis, M.; Zhigunov, A.; Kotek, J. The influence of the length of the degradable segment on the functional properties and hydrolytic stability of multi-component polyurethane elastomeric films. Polym. Degrad. Stabil. 2017, 137, 216–228. [Google Scholar] [CrossRef]
- Aziz, M.S.; Elsoholy, M.G.; Saad, G.R. Preparation and Characterization of Bio-Based Polyurethanes Obtained From Castor Oil and Poly (3-Hydroxybutyrate) and Their Nanocomposites. Polym. Comp. 2018, 39, E489–E499. [Google Scholar] [CrossRef]
- Li, M.; Dong, Q.; Xiao, Y.; Du, Q.; Huselsteind, C.; Zhang, T.; He, X.; Tian, W.; Chen, Y. A biodegradable soy protein isolate-based waterborne polyurethane composite sponge for implantable tissue engineering. J. Mat. Sci. Mater. Med. 2020, 31, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alinejad, M.; Henry, C.; Nikafshar, S.; Gondaliya, A.; Bagheri, S.; Chen, N.; Singh, S.K.; Hodge, D.B.; Nejad, M. Lignin-Based Polyurethanes: Opportunities for Bio-Based Foams, Elastomers, Coatings and Adhesives. Polymers 2019, 11, 1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendels, S.; Averous, L. Biobased polyurethanes for biomedical applications. Bioact. Mater. 2021, 6, 1083–1106. [Google Scholar] [CrossRef]
- Lu, P.; Zhang, Y.; Jia, C.; Li, Y.; Zhang, M.; Mao, Z. Degradation of polyurethane coating materials from liquefied wheat straw for controlled release fertilizers. J. Appl. Polym. Sci. 2016. [Google Scholar] [CrossRef]
- Naureen, B.; Haseeb, A.S.M.A.; Basirun, W.J.; Muhamad, F. Recent advances in tissue engineering scaffolds based on polyurethane and modified polyurethane. Mater. Sci. Eng. C 2021, 118, 111228. [Google Scholar] [CrossRef]
- Zheng, T.; Chen, K.; Chen, W.; Wu, B.; Sheng, Y.; Xia, Y. Preparation and characterisation of polylactic acid modified polyurethane microcapsules for controlled-release of chlorpyrifos. J. Microencapsul. 2019, 36, 62–71. [Google Scholar] [CrossRef]
- Dong, H.; He, J.; Xiao, K.; Li, C. Temperature-sensitive polyurethane (TSPU) film incorporated with carvacrol and cinnamyl aldehyde: Antimicrobial activity, sustained release kinetics and potential use as food packaging for Cantonese-style moon cake. Int. J. Food Sci. Tech. 2020, 55, 293–302. [Google Scholar] [CrossRef]
- Brzeska, J.; Morawska, M.; Sikorska, W.; Tercjak, A.; Kowalczuk, M.; Rutkowska, M. Degradability of cross-linked polyurethanes based on synthetic polyhydroxybutyrate and modified with polylactide. Chem. Pap. 2017, 71, 2243–2251. [Google Scholar] [CrossRef] [Green Version]
- Białkowska, A.; Bakar, M.; Marchut-Mikołajczyk, O. Biodegradation of linear and branched nonisocyanate condensation polyurethanes based on 2-hydroxy-naphthalene-6-sulfonic acid and phenol sulfonic acid. Polym. Degrad. Stabil. 2019, 159, 98–106. [Google Scholar] [CrossRef]
- Joy, Y.J.; Retna, A.M. A new insight of degradation and stability performance of polyurethane and its composites. Mater. Today Proc. 2018, 5, 6082–6089. [Google Scholar]
- Brzeska, J.; Tercjak, A.; Sikorska, W.; Kowalczuk, M.; Rutkowska, M. Predicted Studies of Branched and Cross-Linked Polyurethanes Based on Polyhydroxybutyrate with Polycaprolactone Triol in Soft Segments. Polymers 2020, 12, 1068. [Google Scholar] [CrossRef]
- Brzeska, J.; Tercjak, A.; Sikorska, W.; Kowalczuk, M.; Rutkowska, M. Morphology and Physicochemical Properties of Branched Polyurethane/Biopolymer Blends. Polymers 2020, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Christenson, E.M.; Patel, S.; Anderson, J.M.; Hiltner, A. Enzymatic degradation of poly(ether urethane) and poly(carbonate urethane) by cholesterol esterase. Biomaterials 2006, 27, 3920–3926. [Google Scholar] [CrossRef]
- Feng, Y.; Li, C. Study on oxidative degradation behaviors of polyesterurethane network. Polym. Degrad. Stabil. 2006, 91, 1711–1716. [Google Scholar] [CrossRef]
- Arslan, H.; Adamus, G.; Hazer, B.; Kowalczuk, M. Electrospray ionisation tandem mass spectrometry of poly[(R,S)-3-hydroxybutanoic acid] telechelics containing primary hydroxyl end groups. Rapid Commun. Mass Spectrom. 1999, 13, 2433–2438. [Google Scholar] [CrossRef]
- Gorna, K.; Gogolewski, S. Biodegradable polyurethanes for implants. II. In Vitro degradation and calcification of materials from poly(ε-caprolactone)–poly(ethylene oxide) diols and various chain extenders. J. Biomed. Mater. Res. 2002, 60, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Bil, M.; Hip, I.; Mrówka, P.; Święszkowski, W. Studies on enzymatic degradation of multifunctional composite consisting of chitosan microspheres and shape memory polyurethane matrix. Polym. Degrad. Stabil. 2020, 182, 109392. [Google Scholar] [CrossRef]
- Hong, J.H.; Jeon, H.J.; Yoo, J.H.; Yu, W.-R.; Youk, J.H. Synthesis and characterization of biodegradable poly(3-caprolactone-co-b-butyrolactone)-based polyurethane. Polym. Degrad. Stabil. 2007, 92, 1186–1192. [Google Scholar] [CrossRef]
- Heimowska, A.; Morawska, M.; Bocho-Janiszewska, A. Biodegradation of polylactide in the natural environment of fresh water. Pol. J. Commod. Sci. 2015, 4, 19–29. (In Polish) [Google Scholar]
- Norazlina, H.; Hadi, A.A.; Qurni, A.U.; Amri, M.; Mashelmie, S.; Kama, Y. Degradability studies of PLA nanocomposites under controlled water sorption and soil burial conditions. IOP Conf. Ser. Mater. Sci. Eng. 2018, 342, 1–10. [Google Scholar] [CrossRef]
- Scaffaro, R.; Maio, A.; Gulino, E.F.; Pitarresi, G. Lignocellulosic fillers and graphene nanoplatelets as hybrid reinforcement for polylactic acid: Effect on mechanical properties and degradability. Compos. Sci. Technol. 2020, 190, 1–12. [Google Scholar] [CrossRef]
- Tian, H.; Li, Z.; Lu, P.; Wang, Y.; Jia, C.; Wang, H.; Liu, Z.; Zhang, M. Starch and castor oil mutually modified, cross-linked polyurethane for improving the controlled release of urea. Carbohydr. Polym. 2021, 251, 1–10. [Google Scholar] [CrossRef]
- Mondal, S.; Martin, D. Hydrolytic degradation of segmented polyurethane copolymers for biomedical applications. Polym. Degrad. Stabil. 2012, 97, 1553–1561. [Google Scholar] [CrossRef]
- Alonso-Lerm, B.; Larraza, I.; Barandiaran, L.; Ugarte, L.; Saralegi, A.; Corcuera, M.A.; Perez-Jimenez, R.; Eceiza, A. Enzymatically produced cellulose nanocrystals as reinforcement for waterborne polyurethane and its applications. Carbohydr. Polym. 2021, 254, 1–12. [Google Scholar]
- Mehrbakhsh, E.; Rezaei, M.; Babaie, A.; Mohammadi, A.; Sofla, R.L.M. Physical and thermo-mechanical properties of shape memory polyurethane containing reversible chemical cross-links. J. Mech. Behav. Biomed. 2021, 116, 1–11. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, D.; Shi, W.; Li, S.; Fan, Z.; Tu, J.; Wang, W. Preparation and Properties of Novel Poly(propylene oxide)-block-Polylactide-Based Polyurethane Foams. Polym. Eng. Sci. 2013. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Brzeska, J.; Morawska, M.; Heimowska, A.; Sikorska, W.; Wałach, W.; Hercog, A.; Kowalczuk, M.; Rutkowska, M. The influence of chemical structure on thermal properties and surface morphology of polyurethane materials. Chem. Pap. 2018, 72, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, B.; Wei, B.; Wei, Y.; Yao, J.; Zhang, H.; Chen, X.; Shao, Z. Enhanced compatibility between poly(lactic acid) and poly (butylene adipate-co-terephthalate) by incorporation of N-halamine epoxy precursor. Int. J. Biol. Macromol. 2020, 165, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Rychter, P.; Lewicka, K.; Rogacz, D. Environmental usefulness of PLA/PEG blends for controlled-release systems of soil-applied herbicides. J. Appl. Polym. Sci. 2019, 1–16. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Producer | Chemical | Producer |
|---|---|---|---|
| ß-butyrolactone | Aldrich, St. Luis, MO, USA | Tin(II) octanoate (OSn) | Alfa Aestar, Karlsruhe, Germany |
| ß-18-crown-6 complex | Aldrich, St. Luis, MO, USA | Poly([D,L]-lactide) (PLA), Mn 18,000–28,000 | Aldrich, Steinheim, Germany |
| 2-iodoethanol and 2-bromoethanol | Aldrich, St. Luis, MO, USA | Pea starch (St) | HengshuiFuqiao Starch Co., Hengshui, China |
| Poly(ε-caprolactone)diol (PCLdiol),Mn 1900 | Aldrich, St. Luis, MO, USA | Chitosan (Ch), Mn 171,000, degree of deacetylation 97% | MRI Gdynia, Gdynia, Poland |
| Poly(ε-caprolactone)triol (PCLtriol), Mn 900 | Aldrich, St. Luis, MO, USA | Phosphate buffer (pH 7.01) | Chempur, Piekary Śląskie, Poland |
| 4,4′-methylene dicyclohexyl diisocyanate (H12MDI) | Aldrich, St. Luis, MO, USA | H2O2 (30%) | Chempur, Piekary Śląskie, Poland |
| 1,4-butanediol (1,4-BD) | Aldrich, Steinheim, Germany | CoCl2 | Chempur, Piekary Śląskie, Poland |
| N,N’-dimethylformamide (DMF) | POCh, Gliwice, Poland |
| Sample | Percentage of Oligomerol in Soft Segments [wt.%] | Percentage of Biopolymer in Blend [wt.%] | ||||
|---|---|---|---|---|---|---|
| R,S-PHB | PCLtriol | PCLdiol | Ch | PLA | St | |
| PUR 0/5 | 0 | 5 | 95 | 0 | 0 | 0 |
| PUR 10/5 | 10 | 5 | 85 | 0 | 0 | 0 |
| PUR 20/5 | 20 | 5 | 75 | 0 | 0 | 0 |
| PUR 30/5 | 30 | 5 | 65 | 0 | 0 | 0 |
| PUR 0/15 | 0 | 15 | 85 | 0 | 0 | 0 |
| PUR 10/15 | 10 | 15 | 75 | 0 | 0 | 0 |
| PUR 20/15 | 20 | 15 | 65 | 0 | 0 | 0 |
| PUR 15/20 | 15 | 20 | 65 | 0 | 0 | 0 |
| PUR 30/20 | 30 | 20 | 50 | 0 | 0 | 0 |
| PUR 45/20 | 45 | 20 | 35 | 0 | 0 | 0 |
| PUR 10/5+Ch1.5 | 10 | 5 | 85 | 1.5 | 0 | 0 |
| PUR 10/5+Ch2.5 | 10 | 5 | 85 | 2.5 | 0 | 0 |
| PUR 10/5+PLA | 10 | 5 | 85 | 0 | 5 | 0 |
| PUR 20/5+Ch2.5 | 20 | 5 | 75 | 2.5 | 0 | 0 |
| PUR 20/5+PLA | 20 | 5 | 75 | 0 | 5 | 0 |
| PUR 20/5+St | 20 | 5 | 75 | 0 | 0 | 2.5 |
| PUR 20/15+PLA | 20 | 15 | 65 | 0 | 5 | 0 |
| Sample | Mn[Da] | Mw/Mn | Mn Reduction After Hydrolysis [%] |
|---|---|---|---|
| PUR 0/5 * | 62 000 | 4.1 | - |
| PUR 0/5 H * | 49 000 | 3.4 | 21.0 |
| PUR 20/5 | 27 000 | 2.2 | - |
| PUR 20/5 H | 9 700 | 2.6 | 64.1 |
| PUR 30/5 | 17 500 | 2.2 | - |
| PUR 30/5 H | 5 000 | 2.2 | 71.4 |
| PUR 20/5+PLA | 26 000 | 2.4 | - |
| PUR 20/5+PLA H | 5 900 | 2.5 | 77.3 |
| Sample | Tg [°C] | Tm [°C] | ΔH [J/g] |
|---|---|---|---|
| PUR 0/5 | −43.1 | 47.8 | 20.9 |
| PUR 0/5 H | −41.6 | 44.5/49.6 | 16.3 |
| PUR 10/5 | −24.5 | 47.5 | 27.4 |
| PUR 10/5 H | −16.0 | 44.0/56.0 | 52.6 |
| PUR 20/5 | −18.0 | 50.4 | 25.8 |
| PUR 20/5 H | −14.2 | 54.9 | 32.5 |
| PUR 30/5 | −19.7 | 47.7 | 12.8 |
| PUR 30/5 H | −14.0 | 44.4/55.2 | 15.8 |
| PUR 0/15 | −45.3 | 46.2 | 0.9 |
| PUR 0/15 H | −42.1 | 42.9 | 0.3 |
| PUR 10/15 | −36.6 | 47.3/57.3 | 5.5 |
| PUR 10/15 H | −24.0 | 42.3/55.7 | 17.7 |
| PUR 20/15 | −33.0 | 51.1 | 0.8 |
| PUR 20/15 H | −14.0 | 47.0/57.0 | 23.8 |
| PUR 15/20 | −39.4 | 47.1 | 7.7 |
| PUR 15/20 H | −22.4 | 45.0/56.9 | 12.6 |
| PUR 30/20 | −11.5 | 47.5 | 10.1 |
| PUR 30/20 H | −9.5 | 44.7/56.0 | 10.0 |
| PUR 45/20 | −8.9 | 47.7 | 3.6 |
| PUR 45/20 H | −2.4 | 44.2/57.2 | 6.9 |
| Sample | Tg [°C] | Tm [°C] | ΔH [J/g] |
|---|---|---|---|
| PUR 10/5 | −24.5 | 47.5 | 27.4 |
| PUR 10/5 H | −16.0 | 44.0/56.0 | 52.6 |
| PUR 10/5+Ch1.5 | −32.5 | 50.8 | 17.3 |
| PUR 10/5+Ch1.5 H | - | 2.4 | 66.6 |
| PUR 10/5+Ch2.5 | −36.7 | 52.5 | 13.1 |
| PUR 10/5+Ch2.5 H | - | 7.1 | 79.4 |
| PUR 20/5 | −18.0 | 50.4 | 25.8 |
| PUR 20/5 H | −14.2 | 54.9 | 32.5 |
| PUR 20/5+Ch2.5 | −24.9 | 51.8 | 13.1 |
| PUR 20/5+Ch2.5 H | −24.1 | 45.0 | 18.7 |
| PUR 20/5+St | −26.7 | 51.1 | 11.9 |
| PUR 20/5+St H | - | 3.3 | 115.5 |
| Sample | Tg [°C] | Tm [°C] | ΔH [J/g] |
|---|---|---|---|
| PUR 10/5 | −24.5 | 47.5 | 27.4 |
| PUR 10/5 H | −16.0 | 44.0/56.0 | 52.6 |
| PUR 10/5+PLA | −35.3 | 48.9/79.4 | 19.9 |
| PUR 10/5+PLA H | - | 7.3 | 79.2 |
| PUR 20/5 | −18.0 | 50.4 | 25.8 |
| PUR 20/5 H | −14.2 | 54.9 | 32.5 |
| PUR 20/5+PLA | −29.5 | 50.1 | 10.1 |
| PUR 20/5+PLA H | −15.0 | 45.0/54.0 | 27.1 |
| PUR 20/15 | −33.0 | 51.1 | 0.8 |
| PUR 20/15 H | −14.0 | 47.0/57.0 | 23.8 |
| PUR 20/15+PLA | −32.6 | 49.7 | 0.2 |
| PUR 20/15+PLA H | −15.0 | 47.0/52.0 | 19.3 |
| Sample | The Weight Changes [%] with Time of Incubation [Weeks] | |||
|---|---|---|---|---|
| 1 | 2 | 7 | 11 | |
| PUR 0/5 | −0.2 | −0.1 | −0.4 | −0.8 |
| PUR 10/5 | −1.3 | 0.1 | −1.9 | disintegration |
| PUR 20/5 | −1 | disintegration | ||
| PUR 30/5 | −2.1 | disintegration | ||
| PUR 0/15 | 0.0 | −0.1 | −0.5 | −1.2 |
| PUR 10/15 | 0.1 | −0.1 | −1 | −38.1 |
| PUR 20/15 | 0.1 | 0.0 | −1.3 | disintegration |
| PUR 15/20 | −1.1 | −1.6 | −2.1 | −40 |
| PUR 30/20 | −1.7 | −1.7 | −5.6 | disintegration |
| PUR 45/20 | −2 | −2 | −13 | disintegration |
| PUR 10/5+Ch1.5 | −2 | disintegration | ||
| PUR 10/5+Ch2.5 | −5.4 | disintegration | ||
| PUR 10/5+PLA | 0.2 | −0.1 | −12.7 | disintegration |
| PUR 20/5+Ch2.5 | −29.3 | disintegration | ||
| PUR 20/5+PLA | −6.1 | disintegration | ||
| PUR 20/5+St | −6.3 | disintegration | ||
| PUR 20/15+PLA | −0.7 | −1.1 | −5 | disintegration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzeska, J.; Tercjak, A.; Sikorska, W.; Mendrek, B.; Kowalczuk, M.; Rutkowska, M. Degradability of Polyurethanes and Their Blends with Polylactide, Chitosan and Starch. Polymers 2021, 13, 1202. https://doi.org/10.3390/polym13081202
Brzeska J, Tercjak A, Sikorska W, Mendrek B, Kowalczuk M, Rutkowska M. Degradability of Polyurethanes and Their Blends with Polylactide, Chitosan and Starch. Polymers. 2021; 13(8):1202. https://doi.org/10.3390/polym13081202
Chicago/Turabian StyleBrzeska, Joanna, Agnieszka Tercjak, Wanda Sikorska, Barbara Mendrek, Marek Kowalczuk, and Maria Rutkowska. 2021. "Degradability of Polyurethanes and Their Blends with Polylactide, Chitosan and Starch" Polymers 13, no. 8: 1202. https://doi.org/10.3390/polym13081202
APA StyleBrzeska, J., Tercjak, A., Sikorska, W., Mendrek, B., Kowalczuk, M., & Rutkowska, M. (2021). Degradability of Polyurethanes and Their Blends with Polylactide, Chitosan and Starch. Polymers, 13(8), 1202. https://doi.org/10.3390/polym13081202