
Probing the Release of Bupropion and Naltrexone Hydrochloride Salts from Biopolymeric Matrices of Diverse Chemical Structures



Abstract
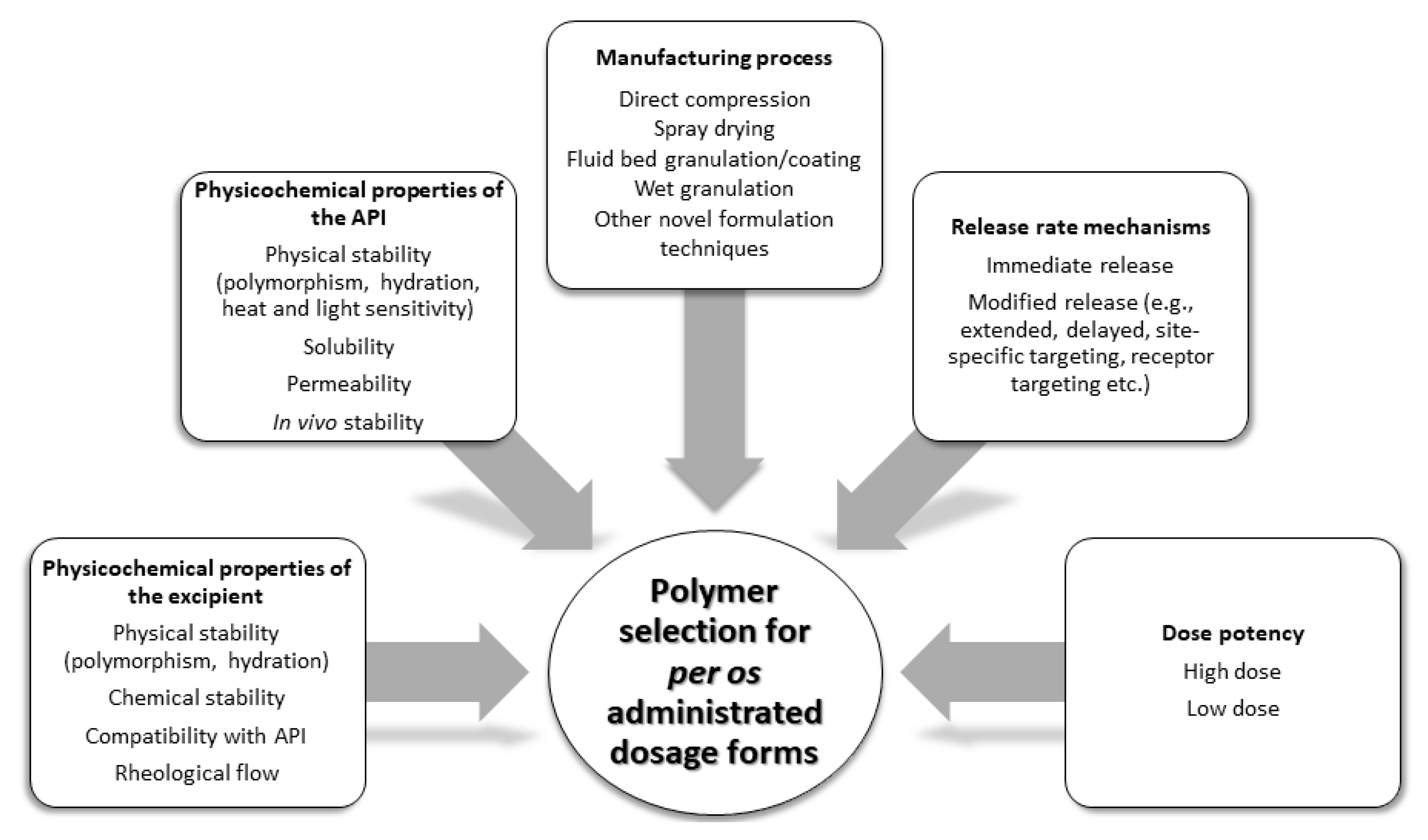
1. Introduction
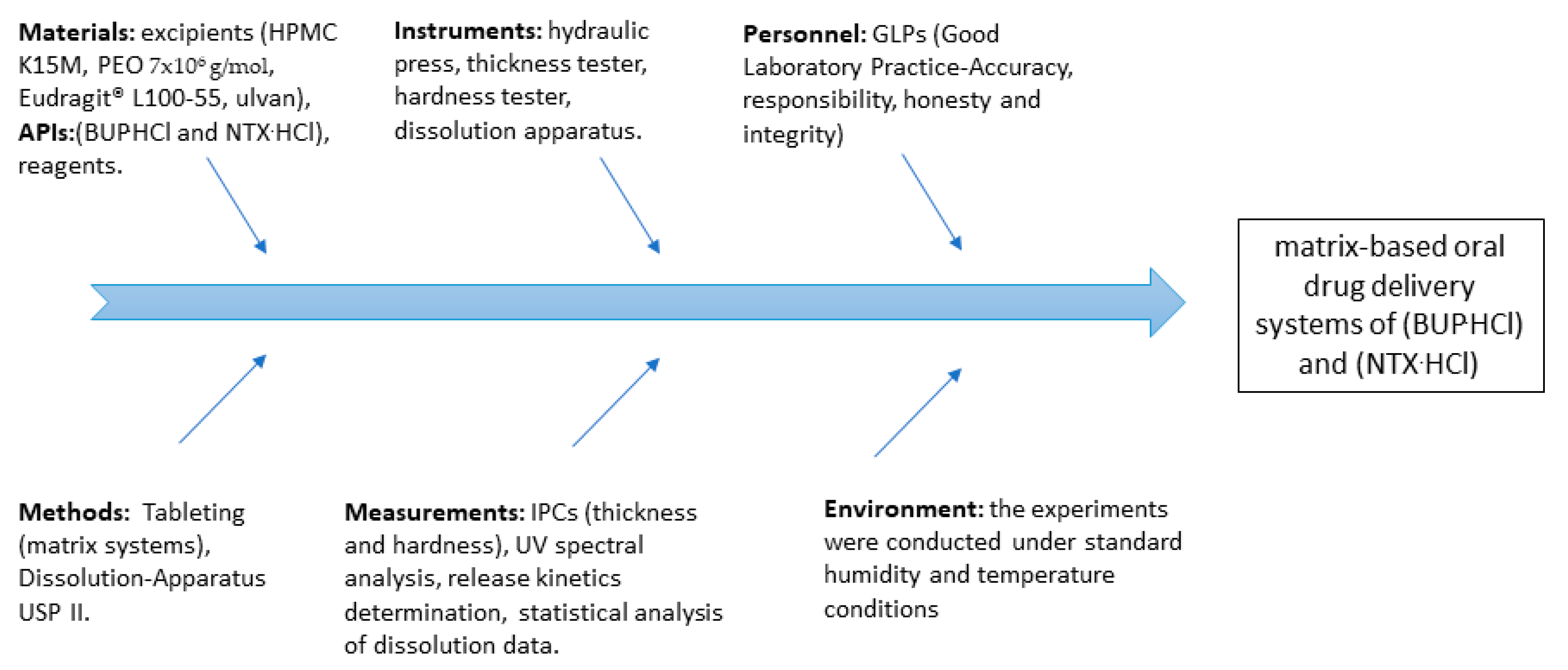
2. Materials and Methods
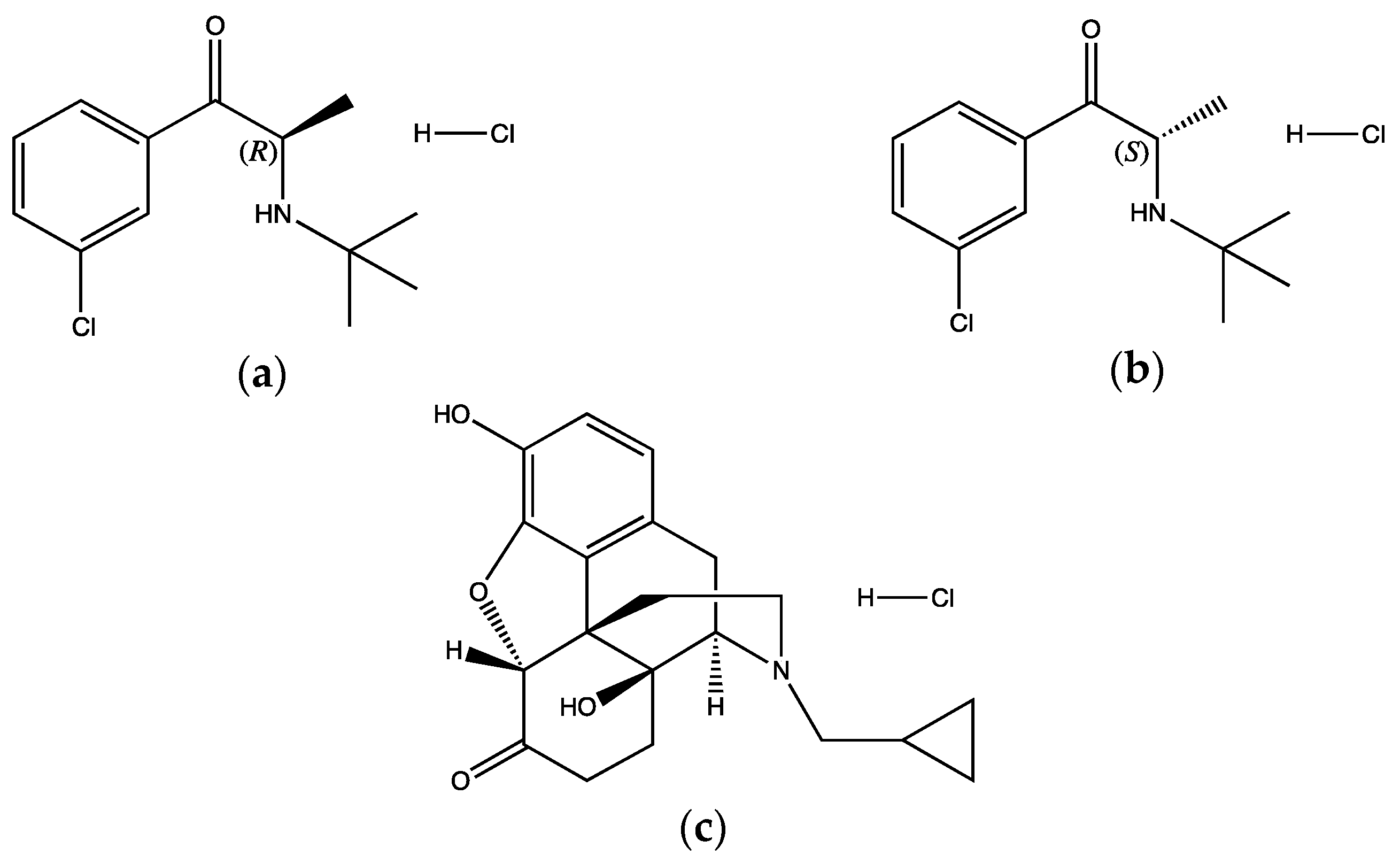
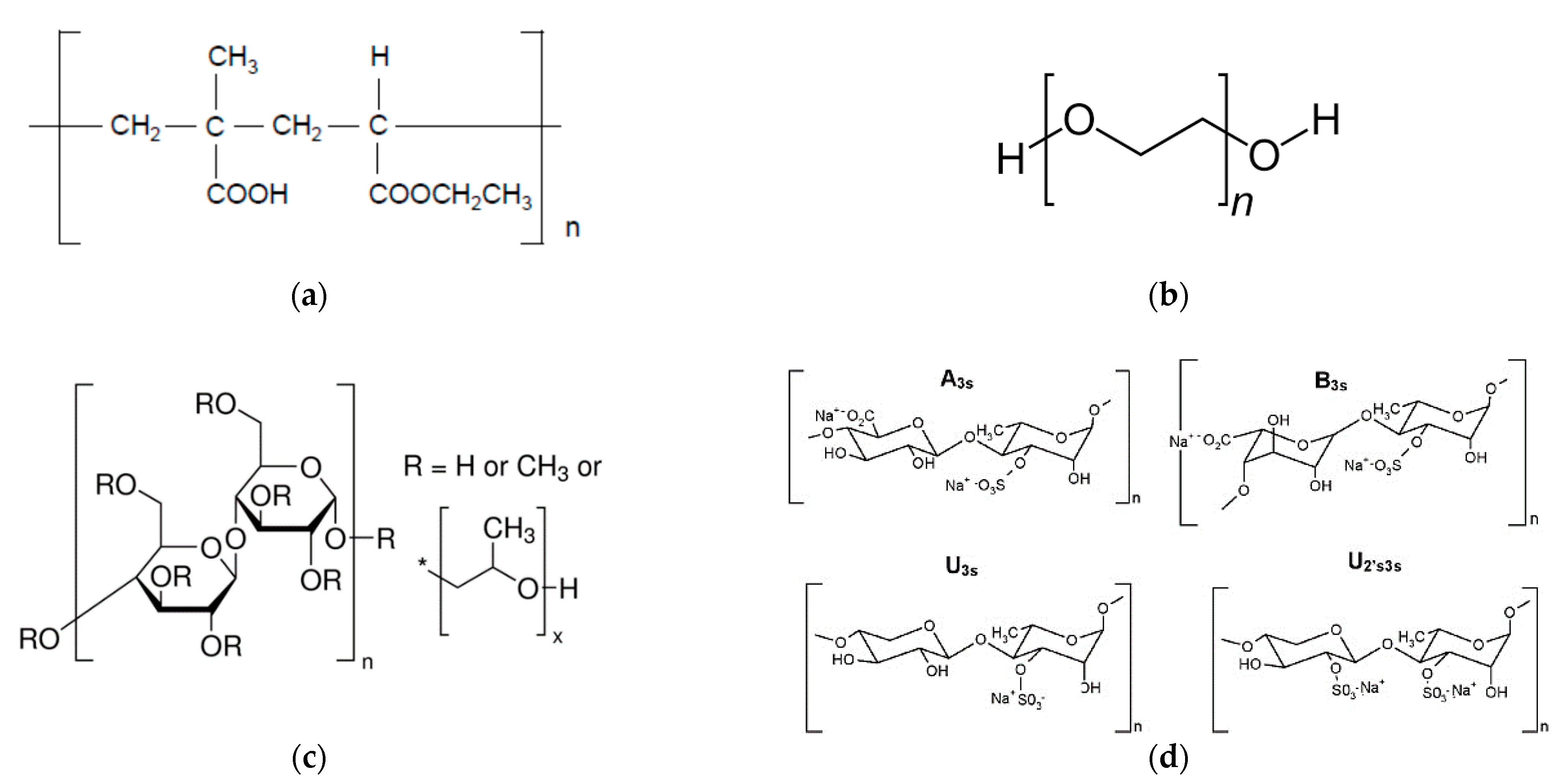
2.1. Materials
2.2. Methods
2.2.1. Preparation of BUP·HCl and NTX·HCl Modified Release Tablets
2.2.2. Post Compression Parameters
2.2.3. In Vitro Dissolution Studies
2.2.4. Methods to Compare Dissolution Profiles
2.2.5. Anova-Based Analysis
2.2.6. Model-Independent Analysis of Dissolution Profiles
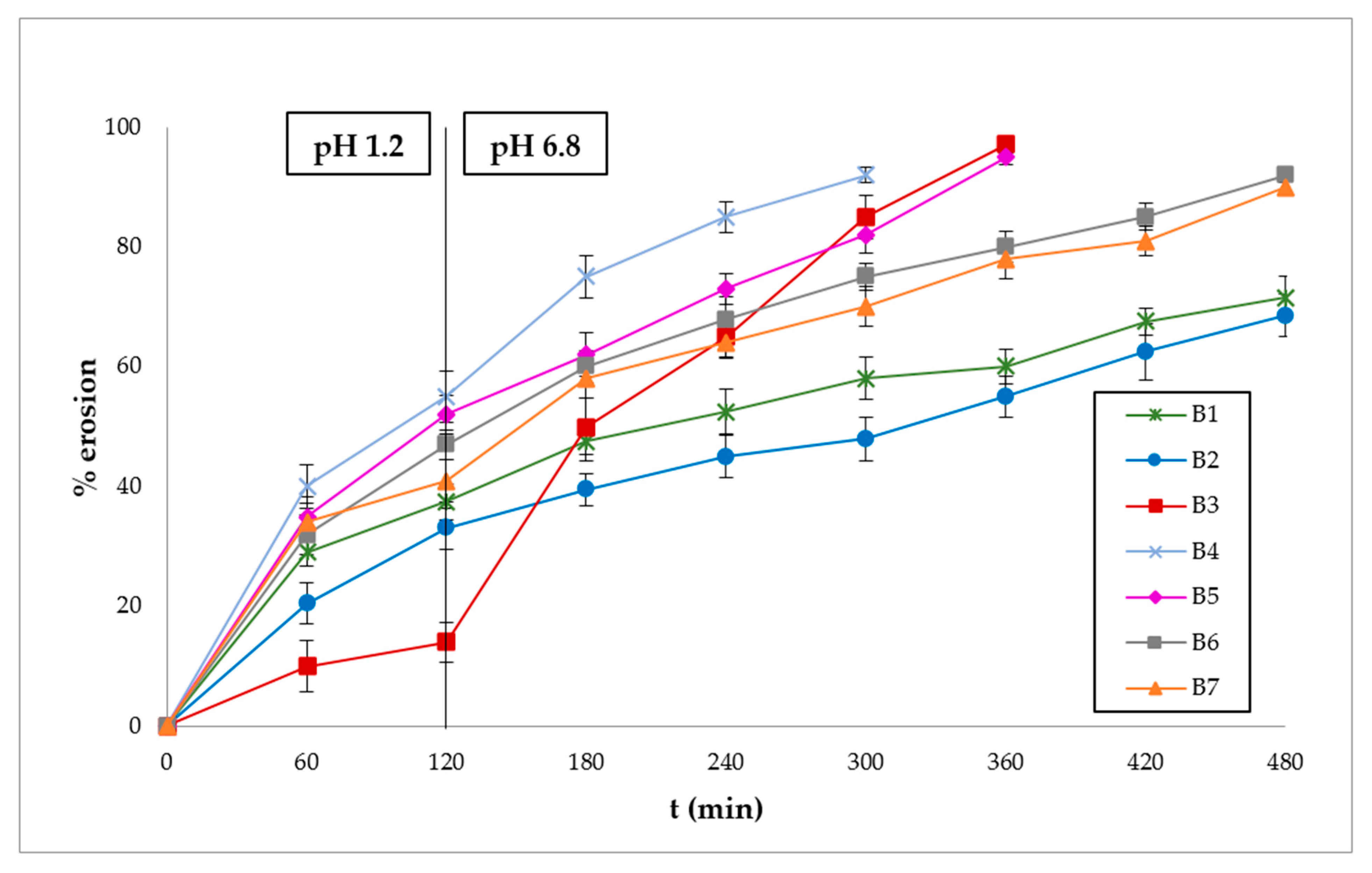
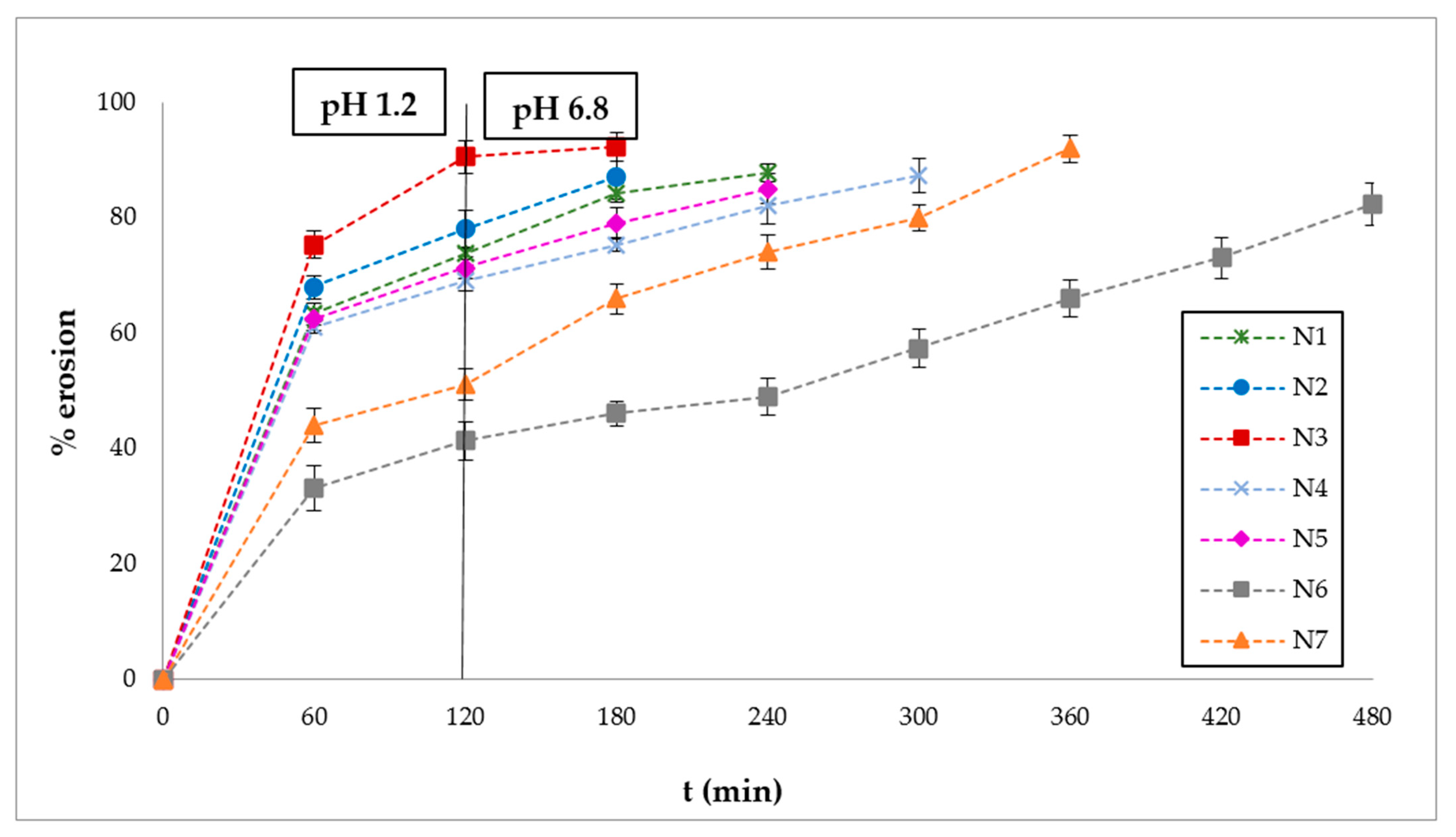
2.2.7. Polymer Erosion Studies
2.2.8. Disintegration Studies
3. Results
3.1. Post Compression Parameters
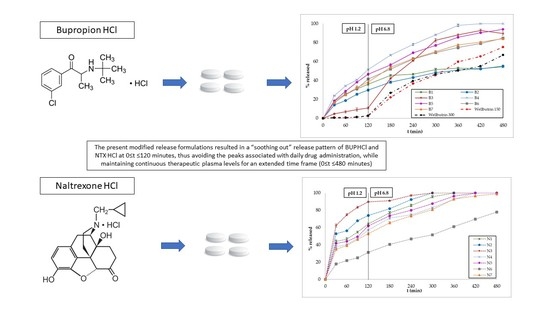
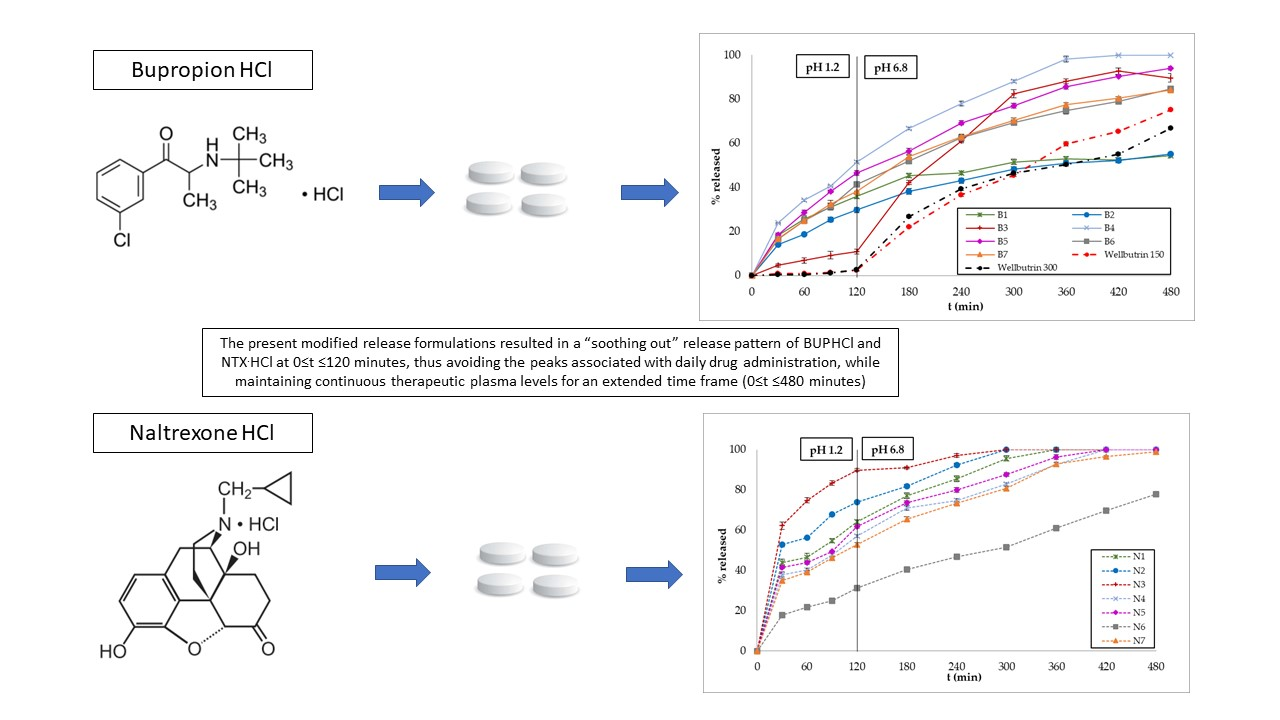
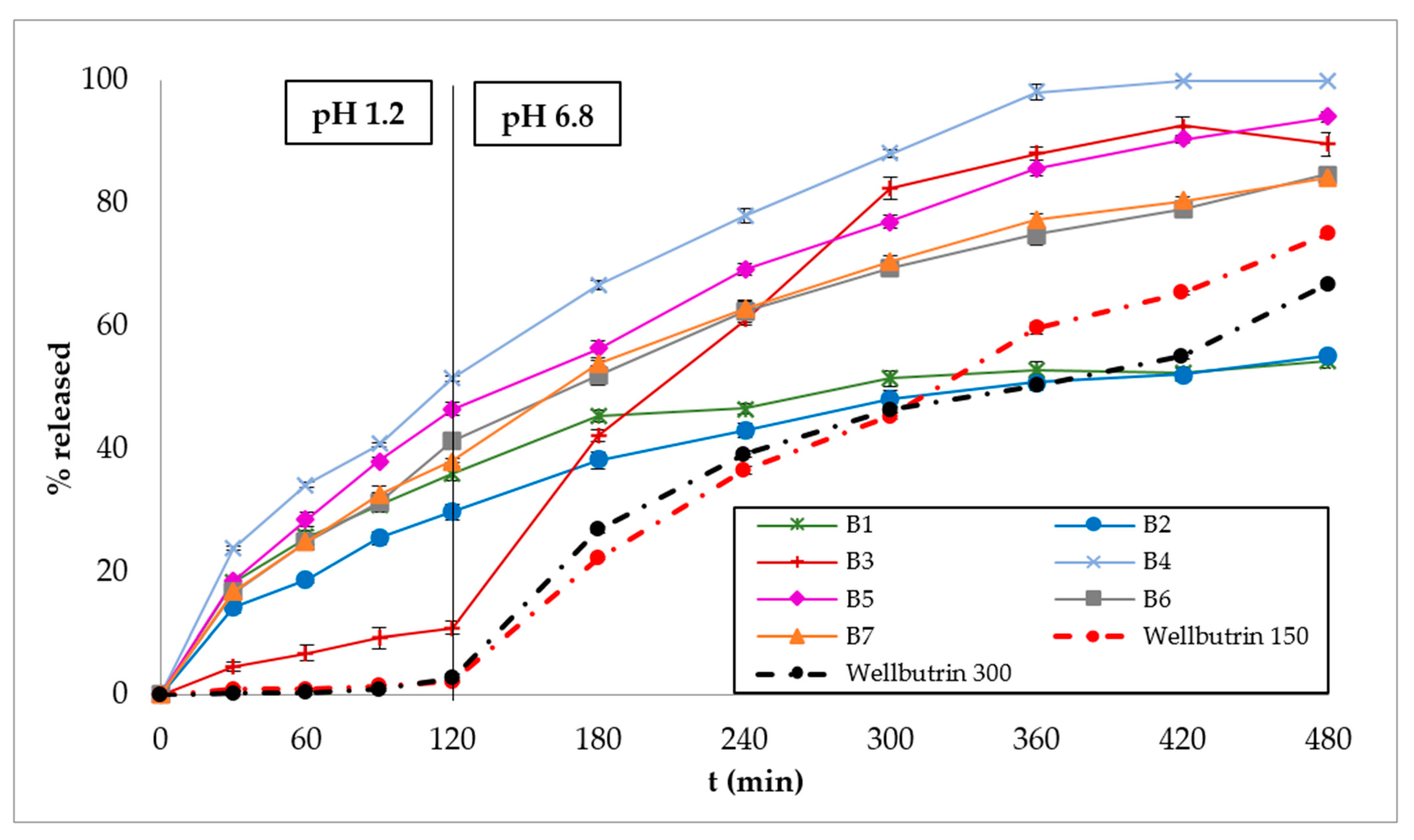
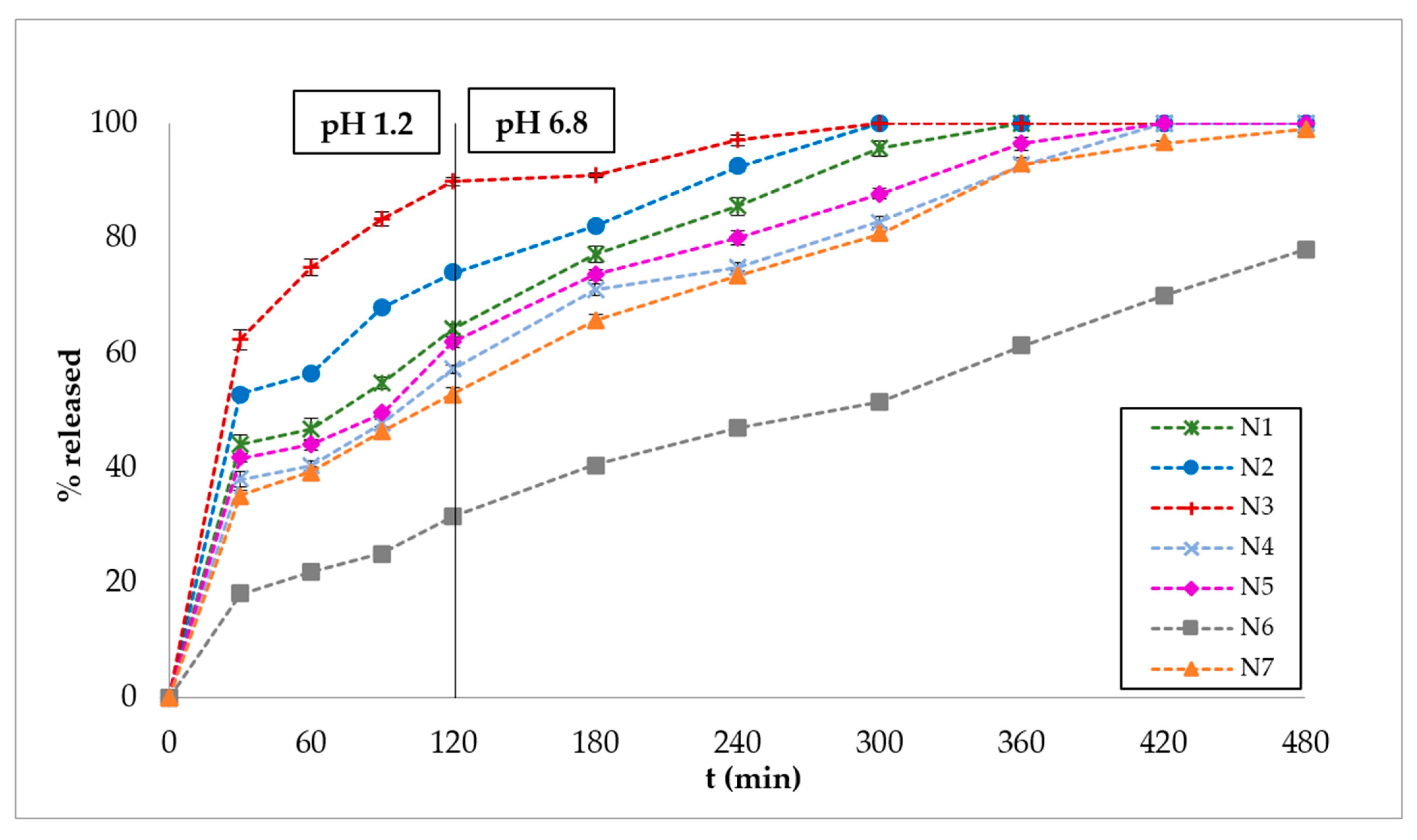
3.2. In Vitro Dissolution Studies
3.3. Polymer Erosion Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Connarn, J.N.; Flowers, S.; Kelly, M.; Luo, R.; Ward, K.M.; Harrington, G.; Moncion, I.; Kamali, M.; Mcinnis, M.; Feng, M.R.; et al. Pharmacokinetics and pharmacogenomics of bupropion in three different formulations with different release kinetics in healthy human volunteers. AAPS J. 2017, 19, 1513–1522. [Google Scholar] [CrossRef]
- Muralidhar, P.; Bhargav, E.; Srinath, B. Formulation and optimization of bupropion HCl microsponges by 2 3 factorial design. Int. J. Pharm. Sci. Res. 2017, 8, 1134–1144. [Google Scholar] [CrossRef]
- Foley, K.F.; De Santy, K.P.; Kast, R.E. Bupropion: Pharmacology and therapeutic applications. Expert Rev. Neurother. 2006, 6, 1249–1265. [Google Scholar] [CrossRef]
- Maccaroni, E.; Malpezzi, L.; Famulari, A.; Masciocchi, N. Structural and energetic aspects of a new bupropion hydrochloride polymorph. J. Pharm. Biomed. Anal. 2012, 60, 65–70. [Google Scholar] [CrossRef]
- Khan, S.R.; Berendt, R.T.; Ellison, C.D.; Ciavarella, A.B. Bupropion Hydrochloride. Profiles Drug Subst. Excip. Relat. Methodol. 2016, 41, 1–30. [Google Scholar] [CrossRef]
- Hollingsworth, E.B.; Kenney, B.T. Synthesis and Evaluation of the Antidepressant Activity of the Enantiomers of Bupropion. Chirality 1993, 5, 495–500. [Google Scholar] [CrossRef]
- Dwoskin, L.P.; Rauhut, A.S.; King-Pospisil, K.A.; Bardo, M.T. Review of the Pharmacology and Clinical Profile of Bupropion, an Antidepressant and Tobacco Use Cessation Agent. CNS Drug Rev. 2006, 12, 178–207. [Google Scholar] [CrossRef]
- Patel, K.; Allen, S.; Haque, M.N.; Angelescu, I.; Baumeister, D.; Tracy, D.K. Bupropion: A systematic review and meta-analysis of effectiveness as an antidepressant. Ther. Adv. Psychopharmacol. 2016, 6, 1–46. [Google Scholar] [CrossRef]
- Fava, M.; Rush, A.J.; Thase, M.E.; Clayton, A.; Stahl, S.M.; Pradko, J.F.; Johnston, J.A. 15 Years of Clinical Experience With Bupropion HCl: From Bupropion to Bupropion SR to Bupropion XL. J. Clin. Psychiatry 2005, 7, 106–113. [Google Scholar] [CrossRef]
- Damaj, M.I.; Carroll, F.I.; Eaton, J.B.; Navarro, H.A.; Blough, B.E.; Mirza, S.; Lukas, R.J.; Martin, B.R. Enantioselective Effects of Hydroxy Metabolites of Bupropion on Behavior and on Function of Monoamine Transporters and Nicotinic Receptors. Mol. Pharmacol. 2004, 66, 675–682. [Google Scholar] [CrossRef]
- Cha, K.-H.; Lee, N.; Kim, M.-S.; Kim, J.-S.; Park, H.J.; Park, J.; Cho, W.; Hwang, S.-J. Development and Optimization of a Novel Sustained-release Tablet Formulation for Bupropion Hydrochloride using Box-Behnken Design. J. Pharm. Investig. 2010, 40, 313–319. [Google Scholar] [CrossRef]
- Settle, E.C. Bupropion sustained release: Side effect profile. J. Clin. Psychiatry 1998, 59, 32–36. [Google Scholar] [PubMed]
- Jefferson, J.W.; Pradko, J.F.; Muir, K.T. Bupropion for major depressive disorder: Pharmacokinetic and formulation considerations. Clin. Ther. 2005, 27, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Dayton, H.B. Naloxone, naltrexone and related noroxymorphones. Adv. in Biochem. Psychopharmacol. 1974, 8, 33–43. [Google Scholar]
- Caraballo, I.; Melgoza, L.M.; Alvarez-Fuentes, J.; Soriano, M.C.; Rabasco, A.M. Design of controlled release inert matrices of naltrexone hydrochloride based on percolation concepts. Int. J. Pharm. 1999, 181, 23–30. [Google Scholar] [CrossRef]
- Akala, E.O.; Wiriyacoonkasem, P.; Pan, G. Studies on in vitro availability, degradation, and thermal properties of naltrexone-loaded biodegradable microspheres. Drug Dev. Ind. Pharm. 2011, 37, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Fuller, B.E.; Rieckmann, T.; McCarty, D.; Smith, K.W.; Levine, H. Adoption of naltrexone to treat alcohol dependence. J. Subst. Abuse Treat. 2005, 28, 273–280. [Google Scholar] [CrossRef]
- Kirchmayer, U.; Davoli, M.; Verster, A. Naltrexone maintenance treatment for opioid dependence. The Cochrane Library 2003, 2, 1–17. [Google Scholar] [CrossRef]
- Goonoo, N.; Bhaw-Luximon, A.; Ujoodha, R.; Jhugroo, A.; Hulse, G.K.; Jhurry, D. Naltrexone: A review of existing sustained drug delivery systems and emerging nano-based systems. J. Control. Release 2014, 183, 154–166. [Google Scholar] [CrossRef]
- Meyer, M.C.; Straughn, A.B.; Lo, M.W.; Schary, W.L.; Whitney, C.C. Bioequivalence, dose-proportionality, and pharmacokinetics of naltrexone after oral administration. J. Clin. Psychiatry 1984, 45, 15–19. [Google Scholar]
- Ornellas, T.; Chavez, B. Naltrexone SR/bupropion SR (Contrave): A new approach to weight loss in obese adults. P & T 2011, 36, 255–362. [Google Scholar]
- Swinyard, E.A. Analgesic and Antipyretics. In Remington’s Pharmaceutical Sciences, 18th ed.; Mack Publishing Company: Northampton, Pennsylvania, PA, USA, 1990; p. 1107. [Google Scholar]
- Majee, S.B.; Avlani, D.; Biswas, G.R. Pharmacological, pharmaceutical, cosmetic and diagnostic applications of sulfated polysaccharides from marine algae and bacteria. Afr. J. Pharm. Pharmacol. 2017, 11, 68–77. [Google Scholar] [CrossRef]
- Cardoso, M.J.; Costa, R.R.; Mano, J.F. Marine origin polysaccharides in drug delivery systems. Mar. Drugs 2016, 14, 34. [Google Scholar] [CrossRef]
- Thirumurugan, G.; Dhanaraju, M.D. Marine Polysaccharides as Multifunctional Marine Polysaccharides as Multifunctional Pharmaceutical Excipients Pharmaceutical Excipients. Biol. Act. Appl. Mar. Polysacch. 2017, 129–143. [Google Scholar] [CrossRef]
- Tziveleka, L.A.; Ioannou, E.; Roussis, V. Ulvan, a bioactive marine sulphated polysaccharide as a key constituent of hybrid biomaterials: A review. Carbohydr. Polym. 2019, 218, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Gao, X.; Cheng, C.; Liu, C.; Wang, Q.; Han, X. The Structural Characteristics of Seaweed Polysaccharides and Their Application in Gel Drug Delivery Systems. Mar. Drugs 2020, 18, 658. [Google Scholar] [CrossRef] [PubMed]
- Robic, A.; Gaillard, C.; Sassi, J.F.; Lerat, Y.; Lahaye, M. Ultrastructure of ulvan: A polysaccharide from green seaweeds. Biopolymers 2009, 91, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, M.; Robic, A. Structure and Functional Properties of Ulvan, a Polysaccharide from Green Seaweeds. Biomacromolecules 2007, 8, 1765–1774. [Google Scholar] [CrossRef]
- Vlachou, M.; Kikionis, S.; Siamidi, A.; Tragou, K.; Ioannou, E.; Roussis, V.; Tsotinis, A. Modified in vitro release of melatonin loaded in nanofibrous electrospun mats incorporated into mono-layered and three-layered tablets. J. Pharm. Sci. 2019, 108, 970–976. [Google Scholar] [CrossRef]
- Abdul, S.; Poddar, S.S. A flexible technology for modified release of drugs: Multi layered tablets. J. Control Release 2004, 97, 393–405. [Google Scholar] [CrossRef]
- Kamboj, S.; Saroha, K.; Goel, M.; Madhu, C. Sustained release drug delivery system: An overview. Pharm. Res. 2012, 8, 169–186. [Google Scholar]
- Krishna, A.V.; Kumaran, K.S.; Meena, A. Development and in vitro characteristics of bupropion HCl sustained release matrix tablets one. IJNTPS 2015, 5, 172–177. [Google Scholar]
- Bankole, A.J. Naltrexone long-acting formulation in the treatment of alcohol dependence. Ther. Clin. Risk Manag. 2007, 3, 741–749. [Google Scholar]
- Revision of Monograph on Tablets: Final Text for Addition to The International Pharmacopoeia. Available online: https://www.who.int/medicines/publications/pharmacopoeia/Tabs-GeneralMono-rev-FINAL_31032011.pdf (accessed on 17 April 2021).
- Vlachou, M.; Kikionis, S.; Siamidi, A.; Kyriakou, S.; Tsotinis, A.; Ioannou, E.; Roussis, V. Development and Characterization of Eudragit®-Based Electrospun Nanofibrous Mats and Their Formulation into Nanofiber Tablets for the Modified Release of Furosemide. Pharmaceutics 2019, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Podczeck, F. Comparison of in vitro dissolution profiles by calculating mean dissolution time (MDT) or mean residence time (MRT). Int. J. Pharmaceut. 1993, 97, 1–3. [Google Scholar] [CrossRef]
- Ritger, P.; Peppas, N. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.P. In vitro dissolution profile comparison-statistics and analysis of the similarity factor, f2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Efentakis, M.; Iliopoulou, A.; Siamidi, A. Effect of core size and excipients on the lag time and drug release from a pulsatile drug delivery system. Drug Dev. Ind. Pharm. 2011, 37, 113–120. [Google Scholar] [CrossRef]
- Markl, D.; Zeitler, J.A. A Review of Disintegration Mechanisms and Measurement Techniques. Pharm. Res. 2017, 34, 890–917. [Google Scholar] [CrossRef] [PubMed]
- Kambham, V.; Kothapalli Bonnoth, C. Development of stavudine sustained release tablets: In-vitro studies. Futur. J. Pharm. Sci. 2016, 2, 37–42. [Google Scholar] [CrossRef]
- Jain, A.K.; Söderlind, E.; Viridén, A.; Schung, B.; Abrahamsson, B.; Knopke, C.; Tajarobi, F.; Blume, H.; Anschutz, M.; Welinder, A.; et al. The influence of hydroxypropyl methylcellulose (HPMC) molecular weight, concentration and effect of food on in vivo erosion behavior of HPMC matrix tablets. J. Control. Release 2014, 187, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Mahaparale, P. Effect of Hydrophilic and Hydrophobic Polymer on Sustained Release Matrix Tablet of Bupropion Hydrochloride. Int. J. Pharm. Sci. Res. 2013, 4, 2835–2842. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Murthy, T.K.; Pai, M.R.; Mehta, P.R.; Chowdary, P.B. Controlled release formulation of Tramadol Hydrochloride using hydrophilic & hydrophobic matrix system. AAPS Pharm. Science Tech. 2003, 4, 18–23. [Google Scholar] [CrossRef]
- Makhija, S.N.; Vavia, P.R. Investigated swellable as well as nonswellable polymers for sustaining the release of Venlafaxine. Eur. J. Pharm. Biopharm. 2004, 54, 9. [Google Scholar] [CrossRef]
- Li, F.-Q.; Hu, J.-H.; Deng, J.-X.; Su, H.; Xu, S.; Liu, J.-Y. In vitro controlled release of sodium ferulate from Compritol 888 ATO-based matrix tablets. Int. J. Pharm. 2006, 324, 152–157. [Google Scholar] [CrossRef]
- Maggi, L.; Bruni, R.; Conte, U. High molecular weight polyethylene oxides (PEOs) as an alternative to HPMC in controlled release dosage forms. Int. J. Pharm. 2000, 195, 229–238. [Google Scholar] [CrossRef]
- Maggi, L.; Segale, L.; Torre, M.L.; Ochoa, M.E.; Conte, U. Dissolution behaviour of hydrophilic matrix tablets containing two different polyethylene oxides (PEOs) for the controlled release of a water-soluble drug. Dimensionality study. Biomaterials 2002, 23, 1113–1119. [Google Scholar] [CrossRef]
- Wu, N.; Wang, L.-S.; Tan, D.C.-W.; Moochhala, S.M.; Yang, Y.-Y. Mathematical modeling and in vitro study of controlled drug release via a highly swellable and dissoluble polymer matrix: Polyethylene oxide with high molecular weights. J. Control. Release 2005, 102, 569–581. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, M.-J.; Yoon, H.; Shim, C.-R.; Ko, H.-A.; Cho, S.-A.; Lee, D.; Khang, G. Enhanced dissolution rate of celecoxib using PVP and/or HPMC based solid dispersions prepared by spray drying method. Int. J. Pharm. Investig. 2013, 43, 205–213. [Google Scholar] [CrossRef]
- Vlachou, M.; Tragou, K.; Siamidi, A.; Kikionis, S.; Chatzianagnostou, A.L.; Mitsopoulos, A.; Ioannou, E.; Roussis, V.; Tsotinis., A. Modified in vitro release of the chronobiotic hormone melatonin from matrix tablets based on the marine sulfated polysaccharide ulvan. J. Drug Deliv. Sci. Technol. 2018, 44, 41–48. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Bupropion. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/444#section=Melting-Point (accessed on 12 December 2020).
- Naltrexone. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Naltrexone#section=Melting-Point (accessed on 12 December 2020).
- Vlachou, M.; Siamidi, A.; Goula, E.; Georgas, P.; Pippa, N.; Karalis, V.; Sentoukas, T.; Pispas, S. Probing the release of the chronobiotic hormone melatonin from hybrid calcium alginate hydrogel beads. Acta Pharm. 2020, 70, 527–538. [Google Scholar] [CrossRef]
- McKinney, A.A.; Tollefson, G.D.; Soltero, R.; Dunzo, T.E. Sustained Release Formulation of Naltrexone. U.S. Patent 8,916,195 B2, 4 June 2007. [Google Scholar]
- Ghimire, M.; Hodges, L.A.; Band, J.; O’Mahony, B.; McInnes, F.J.; Mullen, A.B.; Stevens, H.N.E. In-vitro and in-vivo erosion profiles of hydroxypropylmethylcellulose (HPMC) matrix tablets. J. Control. Release 2010, 147, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Lamoudi, L.; Chaumeil, J.C.; Daoud, K. Swelling, erosion and drug release characteristics of Sodium Diclofenac from heterogeneous matrix tablets. J. Drug Deliv. Sci. Technol. 2016, 31, 93–100. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK; Chicago, IL, USA, 2009. [Google Scholar]
- Kar, R.K.; Mohapatra, S.; Barik, B.B. Design and characterization of controlled release matrix tablets of Zidovudine. Asian J. Pharm. Clin. Res. 2009, 2, 54–61. [Google Scholar]
- Khamanga, S.M.; Walker, R.B. Evaluation of rate of swelling and erosion of verapamil (VRP) sustained-release matrix tablets. Drug Dev. Ind. Pharm. 2006, 32, 1139–1148. [Google Scholar] [CrossRef]
- Efentakis, M.; Pagoni, I.; Vlachou, M.; Avgoustakis, K. Dimensional changes, gel layer evolution and drug release studies in hydrophilic matrices loaded with drugs of different solubility. Int. J. Pharm. 2007, 339, 66–75. [Google Scholar] [CrossRef]
- Chaibva, F.A.; Khamanga, S.M.M.; Walker, R.B. Swelling, erosion and drug release characteristics of salbutamol sulfate from hydroxypropyl methylcellulose-based matrix tablets. Drug Dev. Ind. Pharm. 2010, 36, 1497–1510. [Google Scholar] [CrossRef]
- Li, H.; Hardy, R.J.; Gu, X. Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS Pharm. Sci. Tech. 2008, 9, 437–443. [Google Scholar] [CrossRef]
- Zhang, F.; Meng, F.; Lubach, J.; Koleng, J.; Watson, N.A. Properties and mechanisms of drug release from matrix tablets containing poly(ethylene oxide) and poly(acrylic acid) as release retardants. Eur. J. Pharm. Biopharm. 2016, 105, 97–105. [Google Scholar] [CrossRef]
- Apovian, C.M. Naltrexone/bupropion for the treatment of obesity and obesity with Type 2 diabetes. Future Cardiol. 2016, 12, 129–138. [Google Scholar] [CrossRef]
- Yanovski, S.Z.; Yanovski, J.A. Naltrexone extended release plus bupropion extended release for treatment of obesity. JAMA 2015, 313, 1213–1214. [Google Scholar] [CrossRef]
- Verpeut, J.L.; Bello, N.T. Drug safety evaluation of naltrexone/bupropion for the treatment of obesity. Expert Opin Drug Saf. 2014, 13, 831–841. [Google Scholar] [CrossRef]
- Dhillon, S.; Yang, L.P.H.; Curran, M.P. A Review of its Use in the Management of Major Depressive Disorder. Adis. Drug Eval. 2008, 68, 653–689. [Google Scholar]
- Modesto-Lowe, V.; Van Kirk, J. Clinical uses of naltrexone: A review of the evidence. Exp. Clin. Psychopharmacol. 2002, 10, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Dart, R.C. Medical Toxicology, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; p. 854. [Google Scholar]
- RxList. Available online: https://www.rxlist.com/naltrexone-hydrochloride-drug.htm (accessed on 17 April 2021).
- Billes, S.K.; Sinnayah, P.; Cowley, M.A. Naltrexone/bupropion for obesity: An investigational combination pharmacotherapy for weight loss. Pharmacol Res. 2014, 84, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heer, D. Novel Excipients As Different Polymers: A Review. J. Drug Deliv. Ther. 2013, 3, 202–207. [Google Scholar] [CrossRef]
- Saeidipour, F.; Mansourpour, Z.; Mortazavian, E. Rafiee-Tehrani N and Rafiee-Tehrani M: New comprehensive mathematical model for HPMC-MCC based matrices to design oral controlled release systems. Eur. J. Pharm. Biopharm. 2017, 121, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Aparna, G.; Vijay, S.; Lalit, S. Process variable studies for preparation of optimized system for bupropion hydrochloride using CCD. J. Drug Deliv. Ther. 2019, 9, 281–290. [Google Scholar] [CrossRef]
- Joshi, M. Role of Eudragit in Targeted Drug Delivery. Int. J. Curr. Pharm. Res. 2013, 5, 58–62. [Google Scholar]
- Dhawan, S.; Varma, M.; Sinha, V.R. High molecular weight poly(ethylene oxide)-based drug delivery systems. Part I: Hydrogels and hydrophilic matrix systems. Pharm. Technol. 2005, 29, 72–80. [Google Scholar]
- Dhawan, S.; Dhawan, K.; Varma, M.; Sinha, V.R. Applications of poly(ethylene oxide) in drug delivery systems. Part II. Pharm. Technol. 2005, 29, 82–96. [Google Scholar]
- Bottenberg, P.; Cleymaet, R.; De Muynck, C.; Remon, J.P.; Coomans, D.; Michotte, Y.; Slop, D. Development and testing of bioadhesive, fluoride-containing slow-release tablets for oral use. J. Pharm. Pharmacol. 1991, 43, 457–464. [Google Scholar] [CrossRef]
- Tukaram, B.N.; Rajagopalan, I.V.; Shartchandra, P.S.I. The Effects of Lactose, Microcrystalline Cellulose and Dicalcium Phosphate on Swelling and Erosion of Compressed HPMC Matrix Tablets: Texture Analyzer. IJPR 2010, 9, 349–358. [Google Scholar]
- Cunha, L.; Grenha, A. Sulfated Seaweed Polysaccharides as Multifunctional Materials in Drug Delivery Applications. Marine Drugs 2016, 14, 1–41. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | B1 | B2 | B3 | B4 | B5 | B6 | B7 | N1 | N2 | N3 | N4 | N5 | N6 | N7 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BUP HCl | 75 | 75 | 75 | 75 | 75 | 75 | 75 | - | - | - | - | - | - | - |
| NTX HCl | - | - | - | - | - | - | - | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Eudragit® L100-55 | 48 | 75 | 123 | 48 | 75 | - | - | 74 | 116 | 190 | 74 | 116 | - | - |
| HPMC K15M | 75 | 48 | - | - | - | - | - | 116 | 74 | - | - | - | - | - |
| PEO (7 × 106) | - | - | - | 75 | 48 | 123 | 75 | - | - | - | 116 | 74 | 190 | 116 |
| Ulvan | - | - | - | - | - | - | 48 | - | - | - | - | - | - | 74 |
| Mg. Stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| TOTAL | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
| Formulations | MDT | t20% | t50% | t90% | n | Mean % D.E. |
|---|---|---|---|---|---|---|
| B1 | 98.86 | 37 | 284 | * | 0.36 | 39.56 |
| B2 | 129.50 | 65 | 350 | * | 0.44 | 36.53 |
| B3 | 194.58 | 138 | 210 | 399 | ** | 51.97 |
| B4 | 133.56 | 23 | 114 | 315 | 0.61 | 71.03 |
| B5 | 145.93 | 32 | 137 | 393 | 0.61 | 62.81 |
| B6 | 146.72 | 35 | 159 | * | 0.66 | 55.76 |
| B7 | 144.99 | 45 | 167 | * | 0.67 | 56.75 |
| Wellbutrin® XR 150 mg | 232.29 | 173 | 320 | * | 0.51 | 33.73 |
| Wellbutrin® XR 300 mg | 217.44 | 163 | 354 | * | 0.75 | 31.74 |
| N1 | 101.14 | 16 | 72 | 271 | 0.33 | 78.74 |
| N2 | 78.93 | 13 | 70 | 269 | ** | 83.67 |
| N3 | 50.60 | 10 | 24 | 131 | ** | 89.42 |
| N4 | 124.16 | 16 | 98 | 342 | 0.31 | 72.21 |
| N5 | 113.18 | 14 | 94 | 318 | 0.29 | 75.28 |
| N6 | 169.97 | 46 | 283 | * | 0.56 | 45.60 |
| N7 | 130.82 | 15 | 118 | 346 | 0.39 | 70.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siamidi, A.; Dedeloudi, A.; Vlachou, M. Probing the Release of Bupropion and Naltrexone Hydrochloride Salts from Biopolymeric Matrices of Diverse Chemical Structures. Polymers 2021, 13, 1456. https://doi.org/10.3390/polym13091456
Siamidi A, Dedeloudi A, Vlachou M. Probing the Release of Bupropion and Naltrexone Hydrochloride Salts from Biopolymeric Matrices of Diverse Chemical Structures. Polymers. 2021; 13(9):1456. https://doi.org/10.3390/polym13091456
Chicago/Turabian StyleSiamidi, Angeliki, Aikaterini Dedeloudi, and Marilena Vlachou. 2021. "Probing the Release of Bupropion and Naltrexone Hydrochloride Salts from Biopolymeric Matrices of Diverse Chemical Structures" Polymers 13, no. 9: 1456. https://doi.org/10.3390/polym13091456
APA StyleSiamidi, A., Dedeloudi, A., & Vlachou, M. (2021). Probing the Release of Bupropion and Naltrexone Hydrochloride Salts from Biopolymeric Matrices of Diverse Chemical Structures. Polymers, 13(9), 1456. https://doi.org/10.3390/polym13091456