
Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of DES
2.3. MW Treatment Experiments
2.4. Experimental Design
2.5. Enzymatic Hydrolysis of PET Materials
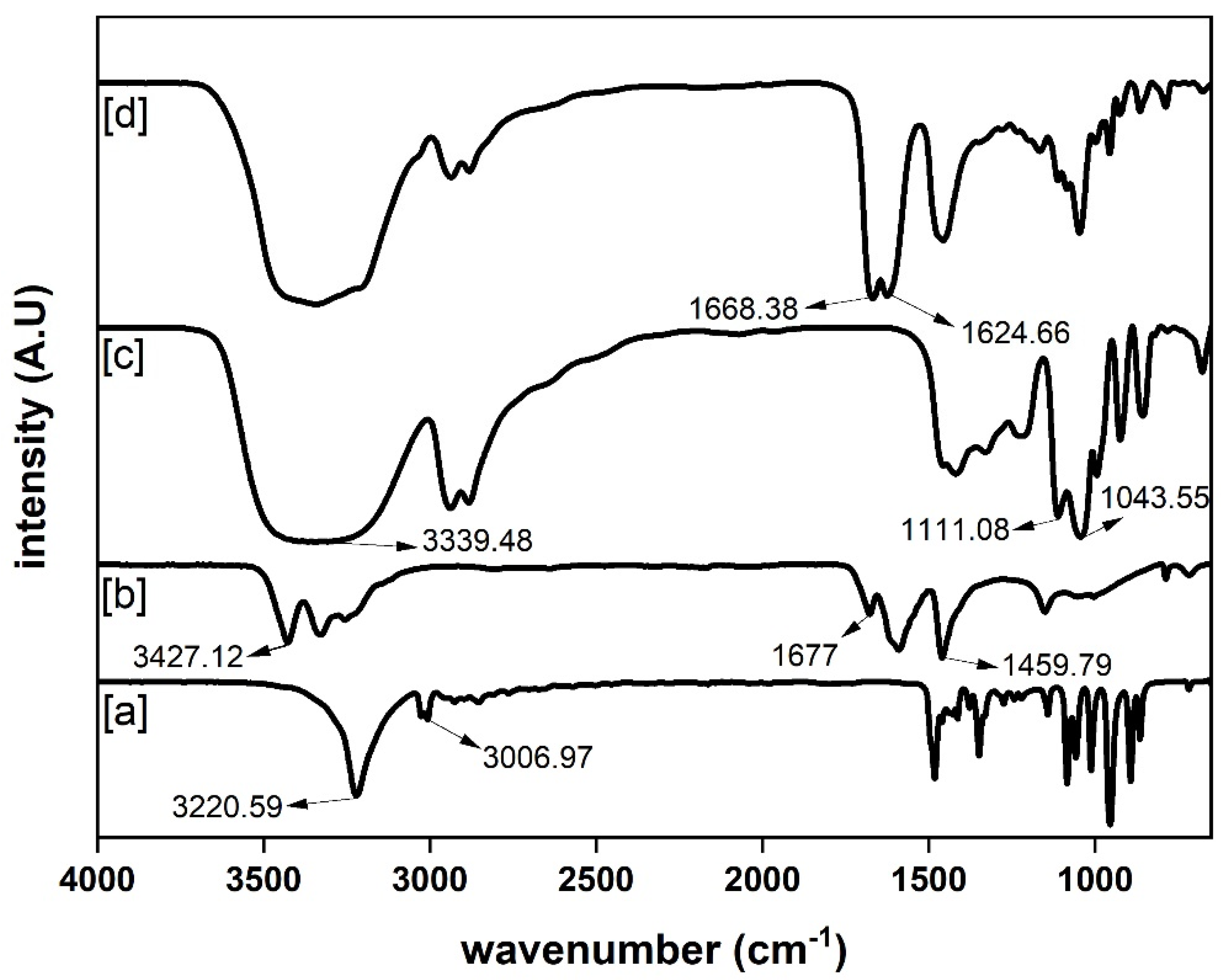
2.6. Instrumental Characterization
3. Results
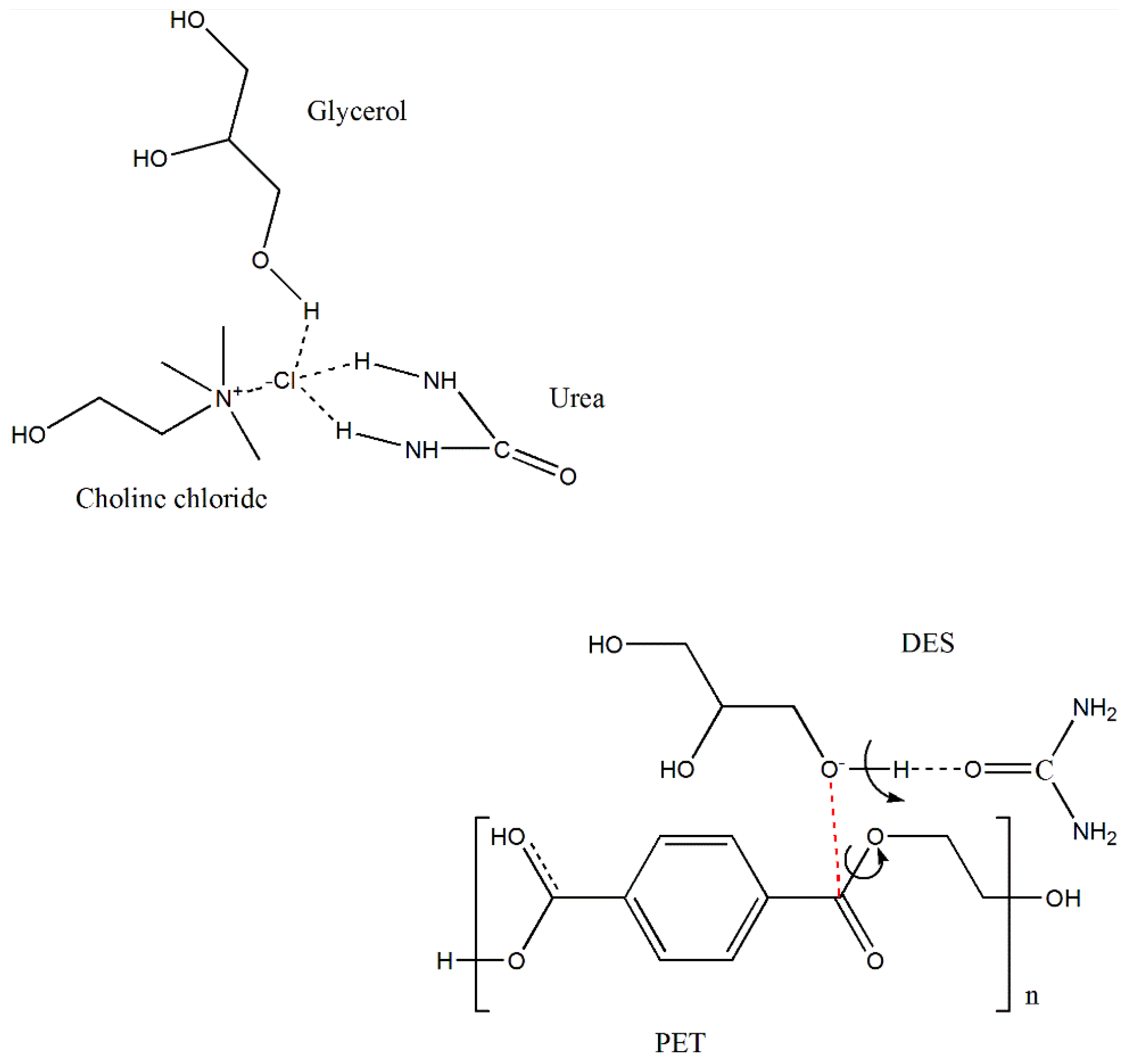
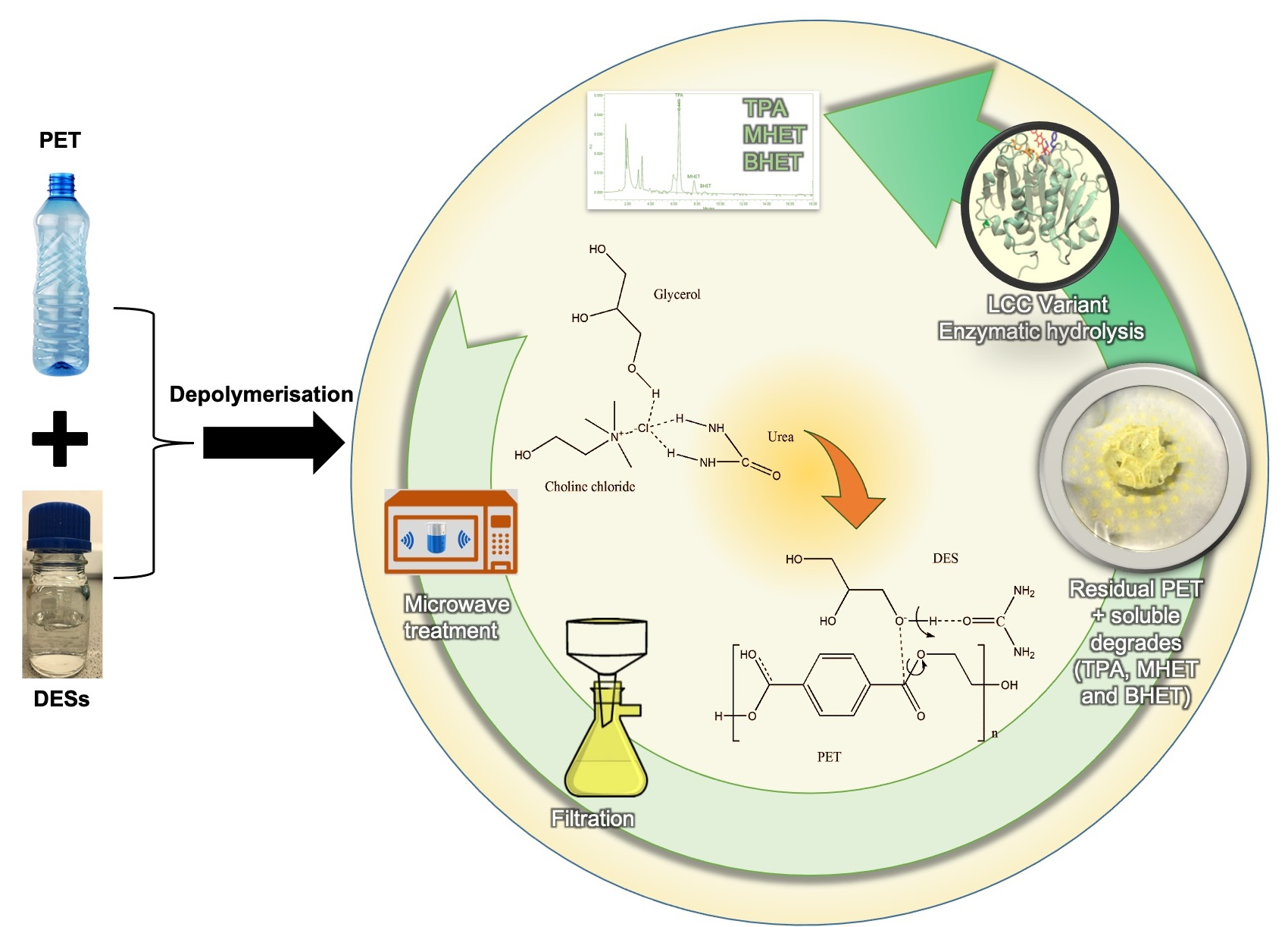
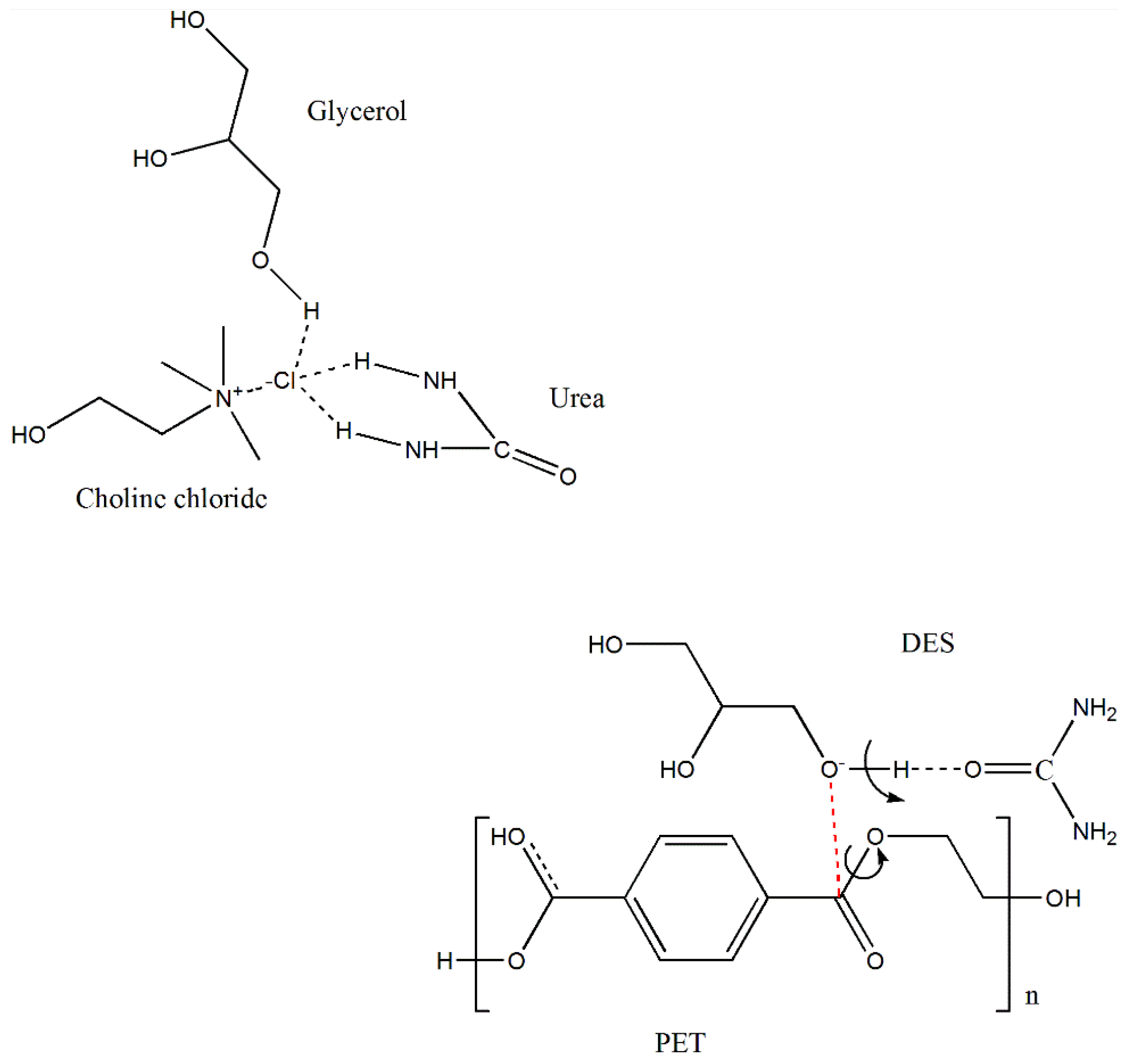
3.1. Properties of DES
3.2. Experimental Design Results
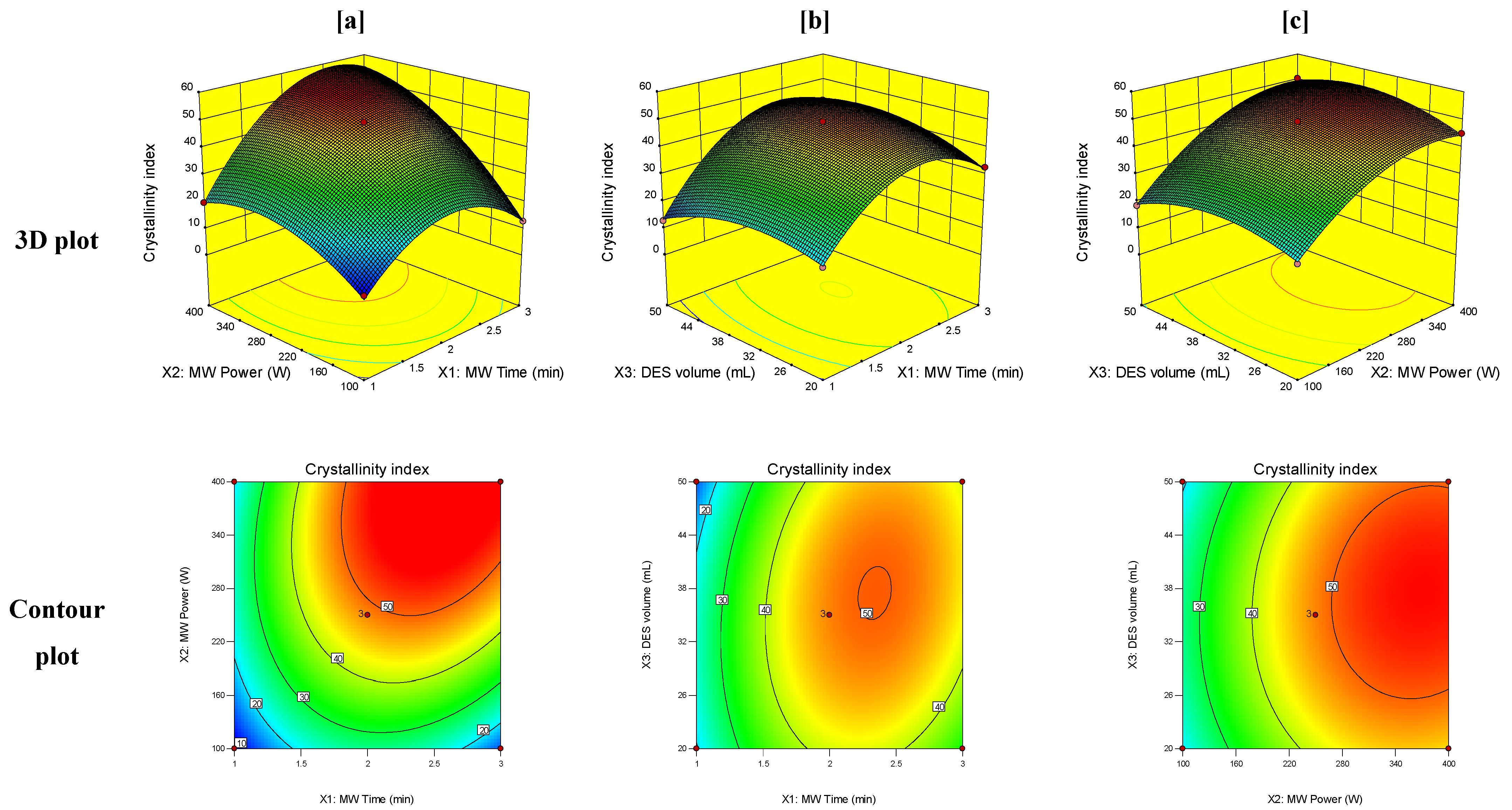
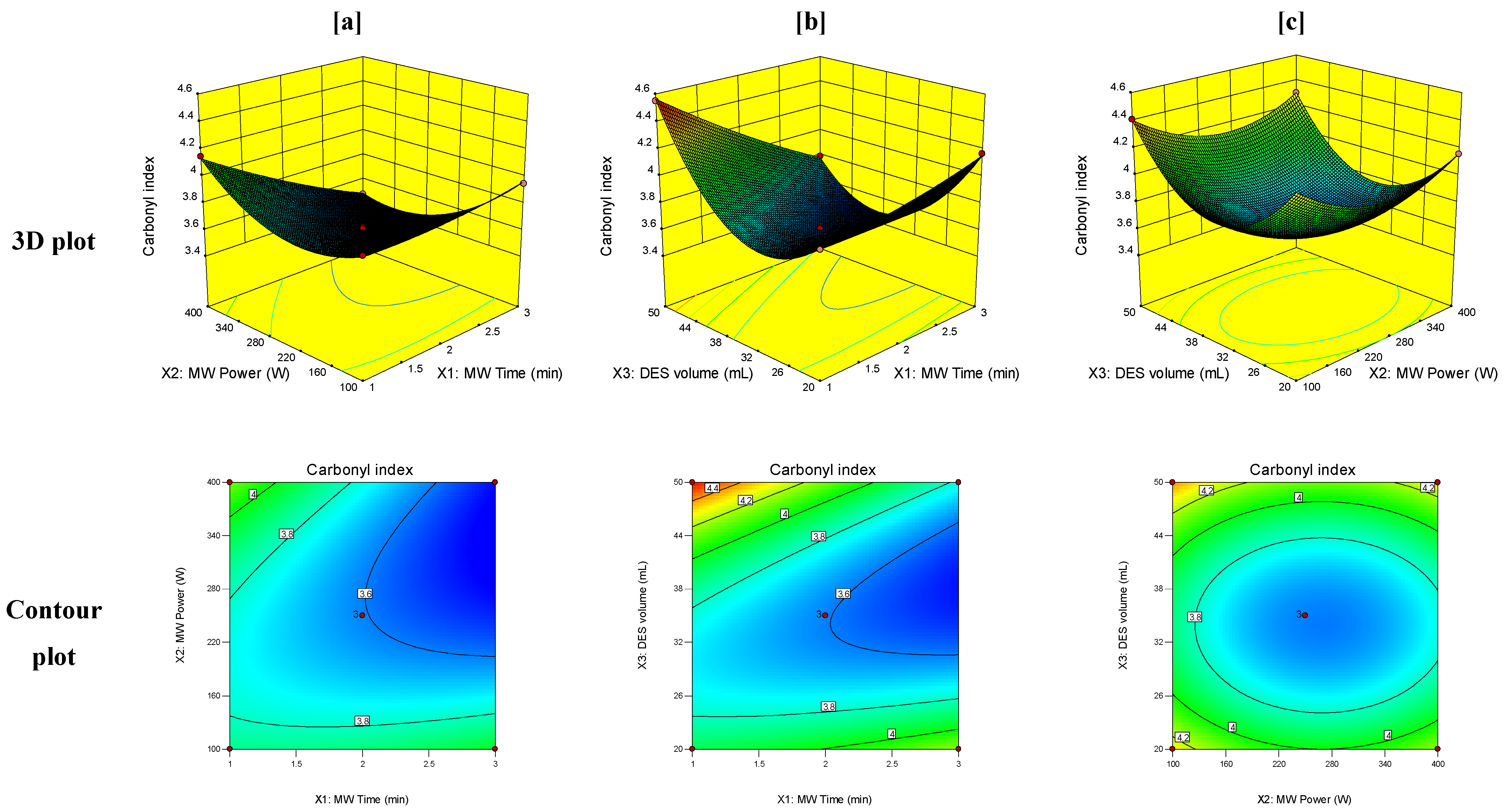
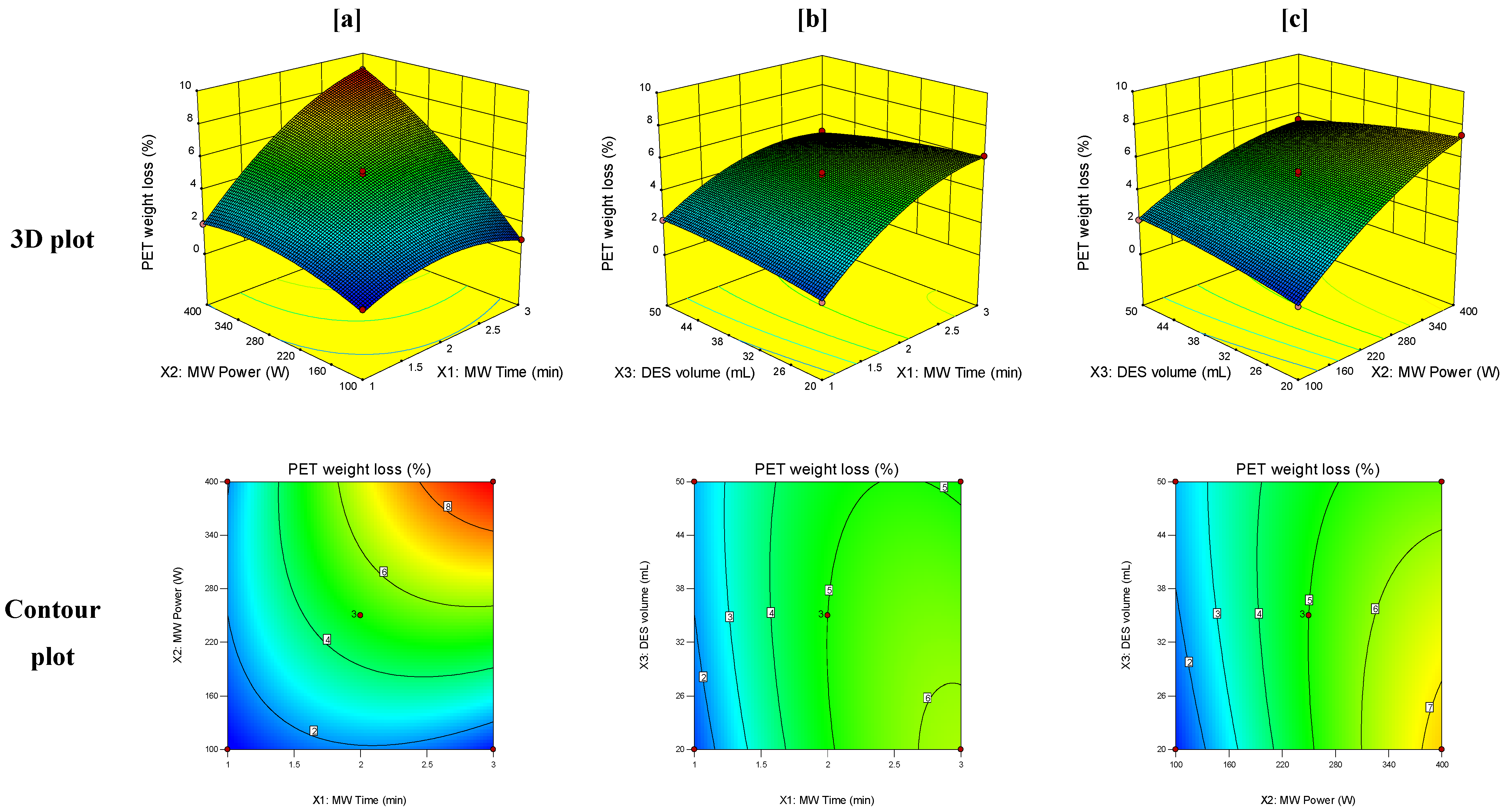
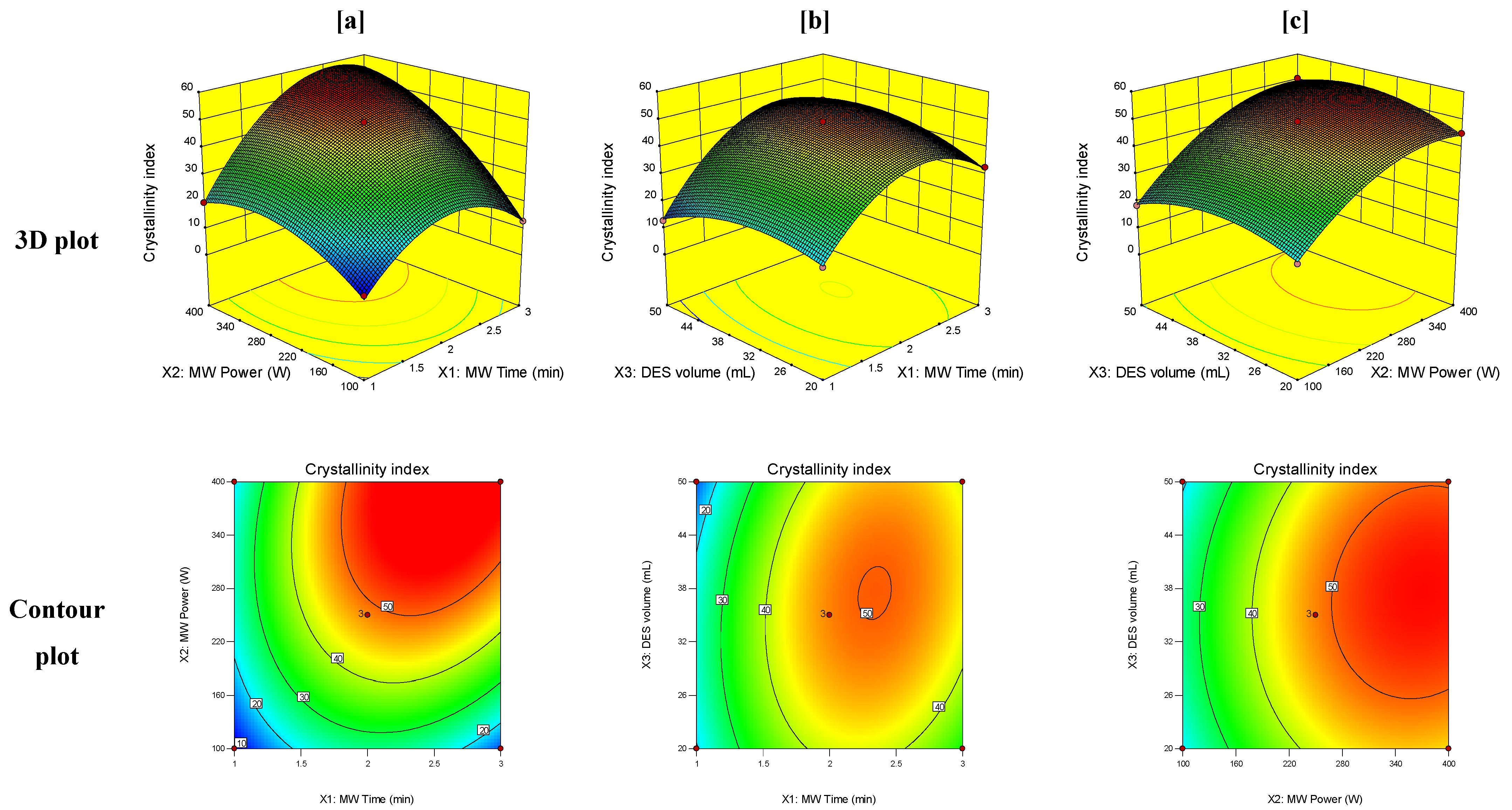
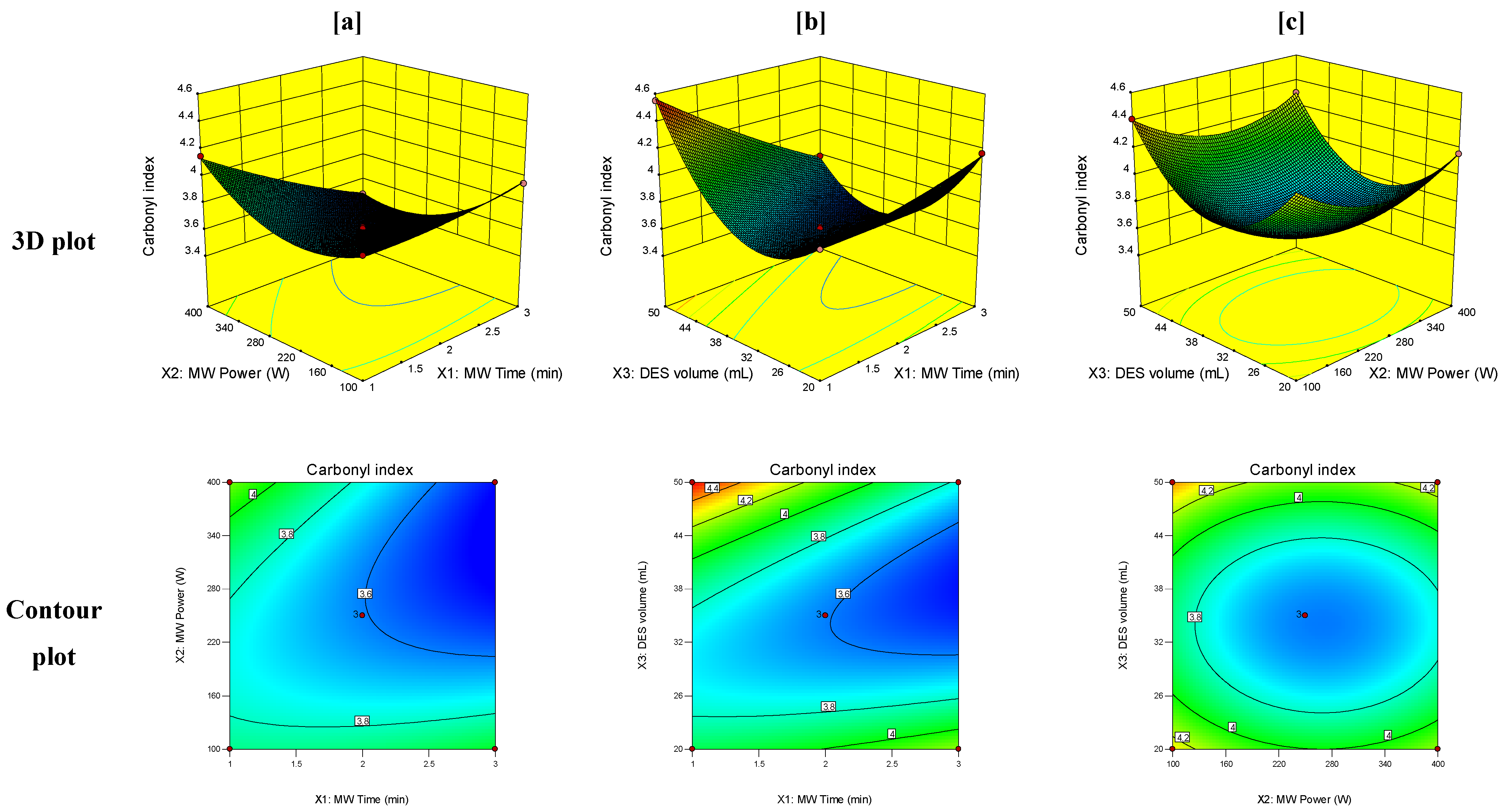
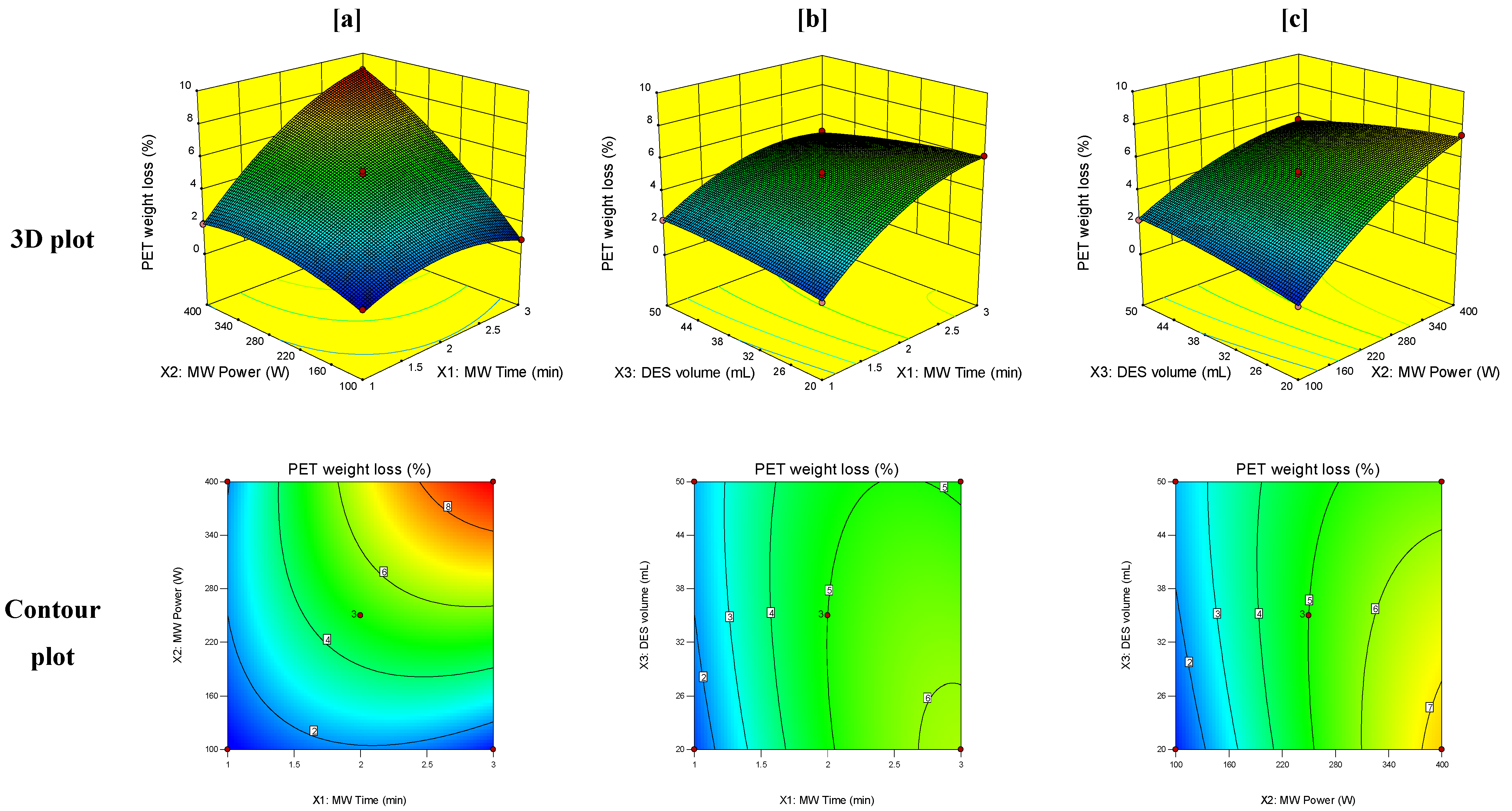
3.3. Response Surface Analysis
3.4. Optimization of the MW-Assisted DES Technique
3.5. Enzymatic Depolymerization of PET Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gómez, A.V.; Biswas, A.; Tadini, C.C.; Furtado, R.F.; Alves, C.R.; Cheng, H.N. Use of Natural Deep Eutectic Solvents for Polymerization and Polymer Reactions. J. Braz. Chem. Soc. 2019, 30, 717–726. [Google Scholar] [CrossRef]
- George, N.; Kurian, T. Recent Developments in the Chemical Recycling of Postconsumer Poly(Ethylene Terephthalate) Waste. Ind. Eng. Chem. Res. 2014, 53, 14185–14198. [Google Scholar] [CrossRef]
- Rorrer, N.A.; Nicholson, S.; Carpenter, A.; Biddy, M.J.; Grundl, N.J.; Beckham, G.T. Combining Reclaimed PET with Bio-Based Monomers Enables Plastics Upcycling. Joule 2019, 3, 1006–1027. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Hui, D.; Singh, R.; Ahuja, I.P.S.; Feo, L.; Fraternali, F. Recycling of Plastic Solid Waste: A State of Art Review and Future Applications. Compos. Part B Eng. 2017, 115, 409–422. [Google Scholar] [CrossRef]
- Filella, M. Antimony and PET Bottles: Checking Facts. Chemosphere 2020, 261, 127732. [Google Scholar] [CrossRef]
- Khoonkari, M.; Haghighi, A.H.; Sefidbakht, Y.; Shekoohi, K.; Ghaderian, A. Chemical Recycling of PET Wastes with Different Catalysts. Int. J. Polym. Sci. 2015, 2015, 124524. [Google Scholar] [CrossRef] [Green Version]
- Yue, Q.F.; Xiao, L.F.; Zhang, M.L.; Bai, X.F. The Glycolysis of Poly(Ethylene Terephthalate) Waste: Lewis Acidic Ionic Liquids as High Efficient Catalysts. Polymers 2013, 5, 1258–1271. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, Y.; Lan, X.; Zhang, B.; Zhang, X.; Mu, T. Biomass-Derived γ-Valerolactone: Efficient Dissolution and Accelerated Alkaline Hydrolysis of Polyethylene Terephthalate. Green Chem. 2021, 23, 4065–4073. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, X.; Geng, Y.; Zhou, Q.; Lu, X.; Zhang, S. Deep Eutectic Solvents as Highly Active Catalysts for the Fast and Mild Glycolysis of Poly(Ethylene Terephthalate)(PET). Green Chem. 2015, 17, 2473–2479. [Google Scholar] [CrossRef]
- Rubio Arias, J.J.; Thielemans, W. Instantaneous Hydrolysis of PET Bottles: An Efficient Pathway for the Chemical Recycling of Condensation Polymers. Green Chem. 2021. [Google Scholar] [CrossRef]
- Nikolaivits, E.; Pantelic, B.; Azeem, M.; Taxeidis, G.; Babu, R.; Topakas, E.; Brennan Fournet, M.; Nikodinovic-Runic, J. Progressing Plastics Circularity: A Review of Mechano-Biocatalytic Approaches for Waste Plastic (Re)Valorization. Front. Bioeng. Biotechnol. 2021, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Musale, R.M.; Shukla, S.R. Deep Eutectic Solvent as Effective Catalyst for Aminolysis of Polyethylene Terephthalate (PET) Waste. Int. J. Plast. Technol. 2016, 20, 106–120. [Google Scholar] [CrossRef]
- Barnard, E.; Rubio Arias, J.J.; Thielemans, W. Chemolytic Depolymerisation of PET: A Review. Green Chem. 2021, 23, 3765–3789. [Google Scholar] [CrossRef]
- Imran, M.; Kim, B.K.; Han, M.; Cho, B.G.; Kim, D.H. Sub-and Supercritical Glycolysis of Polyethylene Terephthalate (PET) into the Monomer Bis (2-Hydroxyethyl) Terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693. [Google Scholar] [CrossRef]
- Choi, S.; Choi, H.M. Eco-Friendly, Expeditious Depolymerization of PET in the Blend Fabrics by Using a Bio-Based Deep Eutectic Solvent under Microwave Irradiation for Composition Identification. Fibers Polym. 2019, 20, 752–759. [Google Scholar] [CrossRef]
- Attallah, O.A.; Janssens, A.; Azeem, M.; Fournet, M.B. Fast, High Monomer Yield from Post-Consumer Polyethylene Terephthalate via Combined Microwave and Deep Eutectic Solvent Hydrolytic Depolymerization. ACS Sustain. Chem. Eng. 2021. [Google Scholar] [CrossRef]
- Kawai, F.; Kawabata, T.; Oda, M. Current Knowledge on Enzymatic PET Degradation and Its Possible Application to Waste Stream Management and Other Fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Alammar, A.; Fulop, Z.; Pulido, B.A.; Nunes, S.P.; Szekely, G. Hydrophobic Thin Film Composite Nanofiltration Membranes Derived Solely from Sustainable Sources. Green Chem. 2021, 23, 1175–1184. [Google Scholar] [CrossRef]
- Attallah, O.A.; Mojicevic, M.; Garcia, E.L.; Azeem, M.; Chen, Y.; Asmawi, S.; Brenan Fournet, M. Macro and Micro Routes to High Performance Bioplastics: Bioplastic Biodegradability and Mechanical and Barrier Properties. Polymers 2021, 13, 2155. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Zimmermann, W. Biocatalysis as a Green Route for Recycling the Recalcitrant Plastic Polyethylene Terephthalate. Microb. Biotechnol. 2017, 10, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Pestana, S.C.; Machado, J.N.; Pinto, R.D.; Ribeiro, B.D.; Marrucho, I.M. Natural Eutectic Solvents for Sustainable Recycling of Poly (Ethyleneterephthalate): Closing the Circle. Green Chem. 2021, 23, 9460–9464. [Google Scholar] [CrossRef]
- Kawai, F. The Current State of Research on PET Hydrolyzing Enzymes Available for Biorecycling. Catalysts 2021, 11, 206. [Google Scholar] [CrossRef]
- Ali, S.S.; Elsamahy, T.; Koutra, E.; Kornaros, M.; El-Sheekh, M.; Abdelkarim, E.A.; Zhu, D.; Sun, J. Degradation of Conventional Plastic Wastes in the Environment: A Review on Current Status of Knowledge and Future Perspectives of Disposal. Sci. Total Environ. 2021, 771, 144719. [Google Scholar] [CrossRef] [PubMed]
- Donelli, I.; Taddei, P.; Smet, P.F.; Poelman, D.; Nierstrasz, V.A.; Freddi, G. Enzymatic Surface Modification and Functionalization of PET: A Water Contact Angle, FTIR, and Fluorescence Spectroscopy Study. Biotechnol. Bioeng. 2009, 103, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, P.; Gräsing, D.; Bielytskyi, P.; Zimmermann, W.; Matysik, J.; Wei, R.; Song, C. UV Pretreatment Impairs the Enzymatic Degradation of Polyethylene Terephthalate. Front. Microbiol. 2020, 11, 689. [Google Scholar] [CrossRef]
- Falah, W.; Chen, F.J.; Zeb, B.S.; Hayat, M.T.; Mahmood, Q.; Ebadi, A.; Toughani, M.; Li, E.Z. Polyethylene Terephthalate Degradation by Microalga Chlorella Vulgaris along with Pretreatment. Mater. Plast. 2020, 57, 260–270. [Google Scholar] [CrossRef]
- Quartinello, F.; Vajnhandl, S.; Volmajer Valh, J.; Farmer, T.J.; Vončina, B.; Lobnik, A.; Herrero Acero, E.; Pellis, A.; Guebitz, G.M. Synergistic Chemo-Enzymatic Hydrolysis of Poly(Ethylene Terephthalate) from Textile Waste. Microb. Biotechnol. 2017, 10, 1376–1383. [Google Scholar] [CrossRef]
- Gong, J.; Kong, T.; Li, Y.; Li, Q.; Li, Z.; Zhang, J. Biodegradation of Microplastic Derived from Poly(Ethylene Terephthalate) with Bacterial Whole-Cell Biocatalysts. Polymers 2018, 10, 1326. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fu, W.; Lu, X.; Zhou, Q.; Zhang, S. Lewis Acid-Base Synergistic Catalysis for Polyethylene Terephthalate Degradation by 1,3-Dimethylurea/Zn(OAc)2 Deep Eutectic Solvent. ACS Sustain. Chem. Eng. 2019, 7, 3292–3300. [Google Scholar] [CrossRef]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis-Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Marichamy, S.; Stalin, B.; Ravichandran, M.; Sudha, G.T. Optimization of Machining Parameters of EDM for α-β Brass Using Response Surface Methodology. Mater. Today Proc. 2020, 24, 1400–1409. [Google Scholar] [CrossRef]
- Kadhom, M.A.; Abdullah, G.H.; Al-Bayati, N. Studying Two Series of Ternary Deep Eutectic Solvents (Choline Chloride–Urea–Glycerol) and (Choline Chloride–Malic Acid–Glycerol), Synthesis and Characterizations. Arab. J. Sci. Eng. 2017, 42, 1579–1589. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.-L.; Texier, H.; Gavalda, S.; et al. An Engineered PET Depolymerase to Break down and Recycle Plastic Bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Djapovic, M.; Milivojevic, D.; Ilic-Tomic, T.; Lješević, M.; Nikolaivits, E.; Topakas, E.; Maslak, V.; Nikodinovic-Runic, J. Synthesis and Characterization of Polyethylene Terephthalate (PET) Precursors and Potential Degradation Products: Toxicity Study and Application in Discovery of Novel PETases. Chemosphere 2021, 275, 130005. [Google Scholar] [CrossRef]
- Chelliah, A.; Subramaniam, M.; Gupta, R.; Gupta, A. Evaluation on the Thermo-Oxidative Degradation of PET Using Prodegradant Additives. Indian J. Sci. Technol. 2017, 10, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, C.; Chi, H.; Li, L.; Lan, T.; Han, P.; Chen, H.; Qin, Y. Development of Antimicrobial Packaging Film Made from Poly(Lactic Acid) Incorporating Titanium Dioxide and Silver Nanoparticles. Molecules 2017, 22, 1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosse, C.; Bourdet, A.; Ernault, E.; Esposito, A.; Delpouve, N.; Delbreilh, L.; Thiyagarajan, S.; Knoop, R.J.I.; Dargent, E. Determination of the Equilibrium Enthalpy of Melting of Two-Phase Semi-Crystalline Polymers by Fast Scanning Calorimetry. Thermochim. Acta 2019, 677, 67–78. [Google Scholar] [CrossRef]
- Alomar, M.K.; Hayyan, M.; Alsaadi, M.A.; Akib, S.; Hayyan, A.; Hashim, M.A. Glycerol-Based Deep Eutectic Solvents: Physical Properties. J. Mol. Liq. 2016, 215, 98–103. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, X.; Tang, S.; Lu, X.; Zhang, X.; Zhang, S. Urea as an Efficient and Reusable Catalyst for the Glycolysis of Poly (Ethylene Terephthalate) Wastes and the Role of Hydrogen Bond in This Process. Green Chem. 2012, 14, 2559–2566. [Google Scholar] [CrossRef]
- Sert, E.; Yılmaz, E.; Atalay, F. Chemical Recycling of Polyethlylene Terephthalate by Glycolysis Using Deep Eutectic Solvents. J. Polym. Environ. 2019, 27, 2956–2962. [Google Scholar] [CrossRef]
- Choi, H.M.; Cho, J.Y. Microwave-Mediated Rapid Tailoring of PET Fabric Surface by Using Environmentally-Benign, Biodegradable Urea-Choline Chloride Deep Eutectic Solvent. Fibers Polym. 2016, 17, 847–856. [Google Scholar] [CrossRef]
- Çabuk, H.; Yılmaz, Y.; Yıldız, E. A Vortex-Assisted Deep Eutectic Solvent-Based Liquid-Liquid Microextraction for the Analysis of Alkyl Gallates in Vegetable Oils. Acta Chim. Slov. 2019, 66, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Beltrán-Sanahuja, A.; Casado-Coy, N.; Simó-Cabrera, L.; Sanz-Lázaro, C. Monitoring Polymer Degradation under Different Conditions in the Marine Environment. Environ. Pollut. 2020, 259, 113836. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Choi, H.M.; Oh, K.W. Rapid Hydrophilic Modification of Poly(Ethylene Terephthalate) Surface by Using Deep Eutectic Solvent and Microwave Irradiation. Text. Res. J. 2016, 86, 1318–1327. [Google Scholar] [CrossRef]
- Winter, R. Method and Apparatus for Recycling of Organic Waste Preferably Used Tires Using Microwave Technique. Eur. Pat. Appl. 2013, 1, 1–23. [Google Scholar]
- Formela, K.; Hejna, A.; Zedler; Colom, X.; Cañavate, J. Microwave Treatment in Waste Rubber Recycling – Recent Advances and Limitations. Express Polym. Lett. 2019, 13, 565–588. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | ||||
|---|---|---|---|---|
| Independent Variables | −1 | 0 | 1 | Constrains |
| X1: MW time (min) | 1 | 2 | 3 | In the range |
| X2: MW power (W) | 100 | 250 | 400 | In the range |
| X3: Volume of DES (mL) | 20 | 35 | 50 | In the range |
| Independent Variable | Dependent Variable | |||||
|---|---|---|---|---|---|---|
| Run | X1 (min) | X2 (W) | X3 (mL) | Y1 | Y2 | Y3 (%) |
| 1 | 3 | 400 | 35 | 54.10 | 3.47 | 8.90 |
| 2 | 2 | 100 | 50 | 18.57 | 4.41 | 2.17 |
| 3 | 2 | 250 | 35 | 48.30 | 3.61 | 4.80 |
| 4 | 1 | 250 | 20 | 19.00 | 3.91 | 1.20 |
| 5 | 3 | 250 | 50 | 41.67 | 3.79 | 4.91 |
| 6 | 2 | 400 | 20 | 45.07 | 4.16 | 7.39 |
| 7 | 2 | 250 | 35 | 47.80 | 3.62 | 5.00 |
| 8 | 2 | 400 | 50 | 50.29 | 4.30 | 5.63 |
| 9 | 3 | 100 | 35 | 12.83 | 3.95 | 0.90 |
| 10 | 3 | 250 | 20 | 32.79 | 4.17 | 6.20 |
| 11 | 2 | 100 | 20 | 20.21 | 4.28 | 0.94 |
| 12 | 1 | 400 | 35 | 19.86 | 4.15 | 1.89 |
| 13 | 1 | 250 | 50 | 12.86 | 4.55 | 2.20 |
| 14 | 2 | 250 | 35 | 49.32 | 3.59 | 5.20 |
| 15 | 1 | 100 | 35 | 9.52 | 3.87 | 0.75 |
| Fitting Model | Factors | Coefficient | p-Value | ANOVA |
|---|---|---|---|---|
| PET crystallinity index (Y1) | Intercept | 48.47 | F = 276.92, R2 = 0.9944, Model p-value < 0.0001, p-value of lack of fit = 0.228 | |
| X1 | 10.02 | <0.0001 | ||
| X2 | 13.52 | <0.0001 | ||
| X3 | 0.79 | 0.1290 | ||
| X1X2 | 7.73 | <0.0001 | ||
| X1X3 | 3.76 | 0.0017 | ||
| X2X3 | 1.71 | 0.0385 | ||
| X12 | −15.68 | <0.0001 | ||
| X22 | −8.72 | <0.0001 | ||
| X32 | −6.22 | 0.0002 | ||
| PET carbonyl index (Y2) | Intercept | 3.61 | F = 461.34, R2 = 0.9966, Model p-value < 0.0001, p-value of lack of fit = 0.355 | |
| X1 | −0.14 | <0.0001 | ||
| X2 | −0.054 | <0.0001 | ||
| X3 | 0.066 | 0.0005 | ||
| X1X2 | −0.19 | 0.0002 | ||
| X1X3 | −0.25 | <0.0001 | ||
| X2X3 | 2.5 × 10−3 | <0.0001 | ||
| X12 | 0.035 | 0.8048 | ||
| X22 | 0.22 | 0.0165 | ||
| X32 | 0.46 | <0.0001 | ||
| Weight loss of PET at T0 of degradation (Y3) | Intercept | 5.00 | F = 264.71, R2 = 0.9941, Model p-value < 0.0001, p-value of lack of fit = 0.537 | |
| X1 | 1.86 | <0.0001 | ||
| X2 | 2.38 | <0.0001 | ||
| X3 | −0.10 | 0.2060 | ||
| X1X2 | 1.72 | <0.0001 | ||
| X1X3 | −0.57 | 0.0023 | ||
| X2X3 | −0.75 | 0.0007 | ||
| X12 | −1.15 | 0.0001 | ||
| X22 | −0.74 | 0.0008 | ||
| X32 | −0.23 | 0.0825 |
| Independent Variable | Optimized Level | ||
|---|---|---|---|
| X1: MW time (min) | 3.0 | ||
| X2: MW power (W) | 260 | ||
| X3: Volume of DES (mL) | 20.0 | ||
| Over all desirability | 0.59 | ||
| Dependent variables | Desirability | Expected | Observed |
| Y1: PET crystallinity index | Minimize | 33.39 | 32.98 |
| Y2: PET carbonyl index | Maximize | 4.14 | 4.22 |
| Y3: PET weight loss (%) | Maximize | 6.47 | 6.25 |
| Material | TPA (μΜ) | MHET (μΜ) | BHET (μΜ) |
|---|---|---|---|
| Untreated: Control | 0.55 ± 0.04 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Untreated: LCCv | 521.13 ± 23.22 | 287.04 ± 7.63 | 7.07 ± 0.36 |
| Treated: Control | 0.83 ± 0.04 | 0.20 ± 0.00 | 0.00 ± 0.00 |
| Treated: LCCv | 384.79 ± 4.91 | 158.83 ± 4.52 | 4.21 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attallah, O.A.; Azeem, M.; Nikolaivits, E.; Topakas, E.; Fournet, M.B. Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polymers 2022, 14, 109. https://doi.org/10.3390/polym14010109
Attallah OA, Azeem M, Nikolaivits E, Topakas E, Fournet MB. Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polymers. 2022; 14(1):109. https://doi.org/10.3390/polym14010109
Chicago/Turabian StyleAttallah, Olivia A., Muhammad Azeem, Efstratios Nikolaivits, Evangelos Topakas, and Margaret Brennan Fournet. 2022. "Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment" Polymers 14, no. 1: 109. https://doi.org/10.3390/polym14010109
APA StyleAttallah, O. A., Azeem, M., Nikolaivits, E., Topakas, E., & Fournet, M. B. (2022). Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polymers, 14(1), 109. https://doi.org/10.3390/polym14010109