
A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization



Abstract
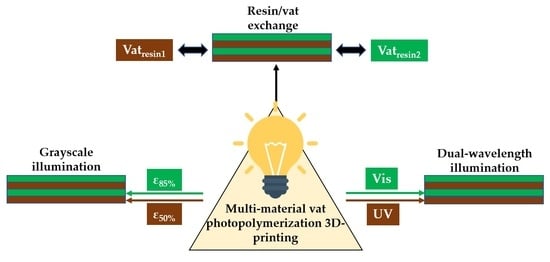
:
1. Introduction
2. Additive Manufacturing Technologies
2.1. Material Extrusion (Fused Filament Fabrication)
2.2. Direct Ink Writing
2.3. Selective Laser Sintering
2.4. Stereolithography
2.5. Digital Light Processing
2.6. Liquid Crystal Display
2.7. Continuous Liquid Interface Printing
2.8. Hot Lithography
2.9. Two-Photon Absorption 3D Printing
2.10. Volumetric 3D Printing
3. Multi-Material 3D Printing
4. Chemistry of Multi-Material 3D Printing via Vat Photopolymerization
4.1. Light-Triggered Reaction in Polymers
4.2. Photoinitiators

| Photoinitiators | Absorption Wavelength | Structure | Reference |
|---|---|---|---|
| Phenyl bis (2,4,6-trimethylbenzoyl) phosphine oxide (BAPO) | 295 nm, 370 nm | 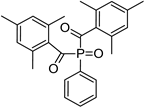 | [170,171,172,173,174] |
| 2-Hydroxy-2-methyl-1-phenyl-propan-1-one (Irgacure 1173) | 245 nm, 280 nm, 331 nm | 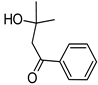 | [172] |
| Ethyl (2,4,6-trimethylbenzoyl) phosphine oxide (Irgacure TPO-L) | 275 nm, 379 nm | 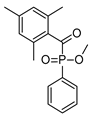 | [175,176] |
| 2,2-dimethoxy-e phenylacetohphenone (Irgacure 651) | 252 nm, 340 nm | 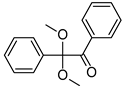 | [173,177,178] |
| Diphenyl (2,4,6 trimethylbenzoyl) phosphine oxide (Irgacure TPO) | 295 nm, 368 nm, 380 nm, 393 nm | 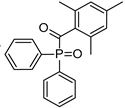 | [171,179,180] |
| Bis (4-methoxybenzoyl) diethylgermanium (Ivocerin) | 408 nm |  | [118,175,181] |
| Benzophenone | 253 nm | 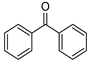 | [182] |
| Camphorquinone | 468 nm | 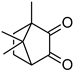 | [170,183,184] |
| 5-amino-2-benzyl-1H-benzo isoquinoline-1,3(2H)-dione (NDP2) | 417 nm | 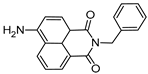 | [170] |
| 3-hydroxyflavone (3HF) | 370–470 nm | 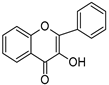 | [185] |
4.3. Light-Triggered Curing Reactions Exploited in Vat Photopolymerization 3D Printing
4.3.1. Chain-Growth Polymerization Reaction
Radical Polymerization

Cationic Polymerization
4.3.2. Step-Growth Polymerization
Thiol-ene and Thiol-yne Chemistry
4.3.3. Hybrid (Interpenetrating) Polymerization
Dual Curable Networks (Photothermal Sequential Curing)
Dual Photo-Curing System
4.4. Vat Photopolymerization of Hydrogels
5. Progress on Multi-Material Vat Photopolymerization 3D Printing
- Changing the resins during 3D printing in a sequential manner to form a multi-material structure.
- Printing one resin formulation while selectively activating photoreactions by changing the light source (layer-by-layer) during 3D printing.
- Printing one resin formulation while selectively varying the intensity of the light source (layer-by-layer) during 3D printing.
5.1. Multi-Vat Photopolymerization 3D Printing
5.2. Orthogonality-Mediated Multi-Material 3D Printing
5.3. Grayscale Photopolymerization 3D Printing
6. Applications of Multi-Material Vat Photopolymerization 3D Printing
6.1. Bio-Mimicking by Combining Soft and Hard Segments
6.2. Bio-Medical Applications
6.3. Stimuli-Responsive Behavior and Robotic Actuators
6.4. Multi-Material Structures with Self-Healing Functionality
7. Further Aspects of Multi-Material Vat Photopolymerization 3D Printing
7.1. Postprocessing of Multi-Material Vat Photopolymerization 3D Printing
7.2. Thermal and Mechanical Aspects of 3D Objects Fabricated via Multi-Material Vat Photopolymerization
7.3. Economical Aspects of Vat Photopolymerization 3D Printing
8. Outlook and Prospects
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, L.-Y.; Fu, J.; He, Y. A Review of 3D Printing Technologies for Soft Polymer Materials. Adv. Funct. Mater. 2020, 30, 2000187. [Google Scholar] [CrossRef]
- Kutz, M.; Peters, E.N. Handbook of Materials Selection: Plastics: Thermoplastics, Thermosets, and Elastomers; John Wiley & Sons, Ltd.: New York, NY, USA; Chichester, UK, 2002; ISBN 9780471359241. [Google Scholar]
- Shafranek, R.T.; Millik, S.C.; Smith, P.T.; Lee, C.-U.; Boydston, A.J.; Nelson, A. Stimuli-responsive materials in additive manufacturing. Prog. Polym. Sci. 2019, 93, 36–67. [Google Scholar] [CrossRef] [Green Version]
- Herzberger, J.; Sirrine, J.M.; Williams, C.B.; Long, T.E. Polymer Design for 3D Printing Elastomers: Recent Advances in Structure, Properties, and Printing. Prog. Polym. Sci. 2019, 97, 101144. [Google Scholar] [CrossRef]
- Brady, R.F. Comprehensive Desk Reference of Polymer Characterization and Analysis; American Chemical Society: Washington, DC, USA; Oxford University Press: Oxford, UK, 2003; ISBN 0841236658. [Google Scholar]
- Picard, M.; Mohanty, A.K.; Misra, M. Recent advances in additive manufacturing of engineering thermoplastics: Challenges and opportunities. RSC Adv. 2020, 10, 36058–36089. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.H. Handbook of Thermoset Plastics, 2nd ed.; Noyes Publications: Park Ridge, NJ, USA, 1999; ISBN 0815514212. [Google Scholar]
- McKeen, L.W. The Effect of Temperature and Other Factors on Plastics and Elastomers, 2nd ed.; Elsevier: Norwich, NY, USA, 2008; ISBN 9780815515685. [Google Scholar]
- McKeen, L.W. The Effect of UV Light and Weather on Plastics and Elastomers: Chapter 10 Elastomers and Rubbers, 4th ed.; William Andrew: Norwich, UK, 2019; ISBN 9780128172711. [Google Scholar]
- Wallin, T.J.; Pikul, J.; Shepherd, R.F. 3D printing of soft robotic systems. Nat. Rev. Mater. 2018, 3, 84–100. [Google Scholar] [CrossRef]
- Zhu, C.; Ninh, C.; Bettinger, C.J. Photoreconfigurable polymers for biomedical applications: Chemistry and macromolecular engineering. Biomacromolecules 2014, 15, 3474–3494. [Google Scholar] [CrossRef]
- Weerakoon-Ratnayake, K.M.; O’Neil, C.E.; Uba, F.I.; Soper, S.A. Thermoplastic nanofluidic devices for biomedical applications. Lab Chip 2017, 17, 362–381. [Google Scholar] [CrossRef] [Green Version]
- Góra, A.; Sahay, R.; Thavasi, V.; Ramakrishna, S. Melt-Electrospun Fibers for Advances in Biomedical Engineering, Clean Energy, Filtration, and Separation. Polym. Rev. 2011, 51, 265–287. [Google Scholar] [CrossRef]
- Rus, D.; Tolley, M.T. Design, fabrication and control of soft robots. Nature 2015, 521, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Shintake, J.; Cacucciolo, V.; Floreano, D.; Shea, H. Soft Robotic Grippers. Adv. Mater. 2018, 30, e1707035. [Google Scholar] [CrossRef] [Green Version]
- Cianchetti, M.; Laschi, C.; Menciassi, A.; Dario, P. Biomedical applications of soft robotics. Nat. Rev. Mater. 2018, 3, 143–153. [Google Scholar] [CrossRef]
- Terryn, S.; Brancart, J.; Lefeber, D.; van Assche, G.; Vanderborght, B. Self-healing soft pneumatic robots. Sci. Robot. 2017, 2, eaan4268. [Google Scholar] [CrossRef] [PubMed]
- Terryn, S.; Langenbach, J.; Roels, E.; Brancart, J.; Bakkali-Hassani, C.; Poutrel, Q.-A.; Georgopoulou, A.; George Thuruthel, T.; Safaei, A.; Ferrentino, P.; et al. A review on self-healing polymers for soft robotics. Mater. Today 2021, 47, 187–205. [Google Scholar] [CrossRef]
- Heo, J.S.; Eom, J.; Kim, Y.-H.; Park, S.K. Recent Progress of Textile-Based Wearable Electronics: A Comprehensive Review of Materials, Devices, and Applications. Small 2018, 14, 1703034. [Google Scholar] [CrossRef]
- Liu, Y.; Pharr, M.; Salvatore, G.A. Lab-on-Skin: A Review of Flexible and Stretchable Electronics for Wearable Health Monitoring. ACS Nano 2017, 11, 9614–9635. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, W. Wearable and flexible electronics for continuous molecular monitoring. Chem. Soc. Rev. 2019, 48, 1465–1491. [Google Scholar] [CrossRef]
- Reynaud, C.; Sommer, F.; Quet, C.; El Bounia, N.; Duc, T.M. Quantitative determination of Young’s modulus on a biphase polymer system using atomic force microscopy. Surf. Interface Anal. 2000, 30, 185–189. [Google Scholar] [CrossRef]
- Gu, G.X.; Libonati, F.; Wettermark, S.D.; Buehler, M.J. Printing nature: Unraveling the role of nacre’s mineral bridges. J. Mech. Behav. Biomed. Mater. 2017, 76, 135–144. [Google Scholar] [CrossRef]
- Dolinski, N.D.; Page, Z.A.; Callaway, E.B.; Eisenreich, F.; Garcia, R.V.; Chavez, R.; Bothman, D.P.; Hecht, S.; Zok, F.W.; Hawker, C.J. Solution Mask Liquid Lithography (SMaLL) for One-Step, Multimaterial 3D Printing. Adv. Mater. 2018, 30, e1800364. [Google Scholar] [CrossRef]
- Zorzetto, L.; Ruffoni, D. Wood-Inspired 3D-Printed Helical Composites with Tunable and Enhanced Mechanical Performance. Adv. Funct. Mater. 2019, 29, 1805888. [Google Scholar] [CrossRef] [Green Version]
- Dimas, L.S.; Bratzel, G.H.; Eylon, I.; Buehler, M.J. Tough Composites Inspired by Mineralized Natural Materials: Computation, 3D printing, and Testing. Adv. Funct. Mater. 2013, 23, 4629–4638. [Google Scholar] [CrossRef]
- Libonati, F.; Gu, G.X.; Qin, Z.; Vergani, L.; Buehler, M.J. Bone-Inspired Materials by Design: Toughness Amplification Observed Using 3D Printing and Testing. Adv. Eng. Mater. 2016, 18, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Suksangpanya, N.; Yaraghi, N.A.; Pipes, R.B.; Kisailus, D.; Zavattieri, P. Crack twisting and toughening strategies in Bouligand architectures. Int. J. Solids Struct. 2018, 150, 83–106. [Google Scholar] [CrossRef]
- Mueller, J.; Raney, J.R.; Shea, K.; Lewis, J.A. Architected Lattices with High Stiffness and Toughness via Multicore-Shell 3D Printing. Adv. Mater. 2018, 30, e1705001. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Chen, K.; Dunn, C.K.; Wu, J.; Li, V.C.F.; Qi, H.J. 3D Printing of Highly Stretchable, Shape-Memory, and Self-Healing Elastomer toward Novel 4D Printing. ACS Appl. Mater. Interfaces 2018, 10, 7381–7388. [Google Scholar] [CrossRef]
- Li, H.; Tan, Y.J.; Leong, K.F.; Li, L. 3D Bioprinting of Highly Thixotropic Alginate/Methylcellulose Hydrogel with Strong Interface Bonding. ACS Appl. Mater. Interfaces 2017, 9, 20086–20097. [Google Scholar] [CrossRef]
- Kokkinis, D.; Bouville, F.; Studart, A.R. 3D Printing of Materials with Tunable Failure via Bioinspired Mechanical Gradients. Adv. Mater. 2018, 30, e1705808. [Google Scholar] [CrossRef]
- Kim, D.; Park, K. Swelling and mechanical properties of superporous hydrogels of poly(acrylamide-co-acrylic acid)/polyethylenimine interpenetrating polymer networks. Polymer 2004, 45, 189–196. [Google Scholar] [CrossRef]
- Muralidharan, A.; Uzcategui, A.C.; McLeod, R.R.; Bryant, S.J. Stereolithographic 3D Printing for Deterministic Control over Integration in Dual-Material Composites. Adv. Mater. Technol. 2019, 4, 1900592. [Google Scholar] [CrossRef]
- Khorasani, M.; Ghasemi, A.; Rolfe, B.; Gibson, I. Additive manufacturing a powerful tool for the aerospace industry. Rapid Prototyp. J. 2022, 28, 87–100. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, Y.; Ramanujan, D.; Ramani, K.; Chen, Y.; Williams, C.B.; Wang, C.C.; Shin, Y.C.; Zhang, S.; Zavattieri, P.D. The status, challenges, and future of additive manufacturing in engineering. Comput.-Aided Des. 2015, 69, 65–89. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, L.; Li, Z.; Wang, S.; Shi, J.; Tang, W.; Li, N.; Yang, J. The recent development of vat photopolymerization: A review. Addit. Manuf. 2021, 48, 102423. [Google Scholar] [CrossRef]
- Gülcan, O.; Günaydın, K.; Tamer, A. The State of the Art of Material Jetting—A Critical Review. Polymers 2021, 13, 2829. [Google Scholar] [CrossRef] [PubMed]
- Tee, Y.L.; Tran, P.; Leary, M.; Pille, P.; Brandt, M. 3D Printing of polymer composites with material jetting: Mechanical and fractographic analysis. Addit. Manuf. 2020, 36, 101558. [Google Scholar] [CrossRef]
- Shahrubudin, N.; Lee, T.C.; Ramlan, R. An Overview on 3D Printing Technology: Technological, Materials, and Applications. Procedia Manuf. 2019, 35, 1286–1296. [Google Scholar] [CrossRef]
- Gibson, I.; Rosen, D.; Stucker, B.; Khorasani, M. Binder Jetting. In Additive Manufacturing Technologies; Springer: Cham, Switzerland, 2021; pp. 237–252. [Google Scholar]
- Dixit, N.; Sharma, V.; Kumar, P. Experimental investigations into abrasive flow machining (AFM) of 3D printed ABS and PLA parts. Rapid Prototyp. J. 2022, 28, 161–174. [Google Scholar] [CrossRef]
- Zhu, K.; Deng, Z.; Dai, S.; Yu, Y. Temperature-compensated constitutive model of fused filament fabrication 3D printed PLA materials with full extrusion temperatures. Rapid Prototyp. J. 2022, 28, 41–51. [Google Scholar] [CrossRef]
- Yan, R.; Wang, Y.; Luo, P.; Li, Y.; Lu, X. Fused filament fabrication of continuous optic fiber reinforced polylactic acid composites. Rapid Prototyp. J. 2022, 28, 766–776. [Google Scholar] [CrossRef]
- Arifvianto, B.; Wirawan, Y.B.; Salim, U.A.; Suyitno, S.; Mahardika, M. Effects of extruder temperatures and raster orientations on mechanical properties of the FFF-processed polylactic-acid (PLA) material. Rapid Prototyp. J. 2021, 27, 1761–1775. [Google Scholar] [CrossRef]
- Vanaei, H.R.; Shirinbayan, M.; Vanaei, S.; Fitoussi, J.; Khelladi, S.; Tcharkhtchi, A. Multi-scale damage analysis and fatigue behavior of PLA manufactured by fused deposition modeling (FDM). Rapid Prototyp. J. 2021, 27, 371–378. [Google Scholar] [CrossRef]
- Chatham, C.A.; Long, T.E.; Williams, C.B. A review of the process physics and material screening methods for polymer powder bed fusion additive manufacturing. Prog. Polym. Sci. 2019, 93, 68–95. [Google Scholar] [CrossRef]
- Singh, R.; Gupta, A.; Tripathi, O.; Srivastava, S.; Singh, B.; Awasthi, A.; Rajput, S.K.; Sonia, P.; Singhal, P.; Saxena, K.K. Powder bed fusion process in additive manufacturing: An overview. Mater. Today: Proc. 2020, 26, 3058–3070. [Google Scholar] [CrossRef]
- Sampson, K.L.; Deore, B.; Go, A.; Nayak, M.A.; Orth, A.; Gallerneault, M.; Malenfant, P.R.L.; Paquet, C. Multimaterial Vat Polymerization Additive Manufacturing. ACS Appl. Polym. Mater. 2021, 3, 4304–4324. [Google Scholar] [CrossRef]
- Pagac, M.; Hajnys, J.; Ma, Q.-P.; Jancar, L.; Jansa, J.; Stefek, P.; Mesicek, J. A Review of Vat Photopolymerization Technology: Materials, Applications, Challenges, and Future Trends of 3D Printing. Polymers 2021, 13, 598. [Google Scholar] [CrossRef] [PubMed]
- Nohut, S.; Schwentenwein, M. Vat Photopolymerization Additive Manufacturing of Functionally Graded Materials: A Review. J. Manuf. Mater. Process. 2022, 6, 17. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Z.; Zhang, J.; Wang, X.; Xu, Y.; Ding, N.; Peng, Z. Vat Photopolymerization 3D Printing of Advanced Soft Sensors and Actuators: From Architecture to Function. Adv. Mater. Technol. 2021, 6, 2001218. [Google Scholar] [CrossRef]
- Andreu, A.; Su, P.-C.; Kim, J.-H.; Ng, C.S.; Kim, S.; Kim, I.; Lee, J.; Noh, J.; Subramanian, A.S.; Yoon, Y.-J. 4D printing materials for vat photopolymerization. Addit. Manuf. 2021, 44, 102024. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, M.; Viswanathan, V.V.; Swart, B.; Shao, Y.; Wu, G.; Zhou, C. 3D printing technologies for electrochemical energy storage. Nano Energy 2017, 40, 418–431. [Google Scholar] [CrossRef]
- Salmi, M. Additive Manufacturing Processes in Medical Applications. Materials 2021, 14, 191. [Google Scholar] [CrossRef]
- Gross, B.; Lockwood, S.Y.; Spence, D.M. Recent Advances in Analytical Chemistry by 3D Printing. Anal. Chem. 2017, 89, 57–70. [Google Scholar] [CrossRef]
- Choi, J.-W.; Kim, H.-C.; Wicker, R. Multi-material stereolithography. J. Mater. Process. Technol. 2011, 211, 318–328. [Google Scholar] [CrossRef]
- Choi, J.-W.; MacDonald, E.; Wicker, R. Multi-material microstereolithography. Int. J. Adv. Manuf. Technol. 2010, 49, 543–551. [Google Scholar] [CrossRef]
- Hohnholz, A.; Obata, K.; Albrecht, D.; Koch, J.; Hohenhoff, G.; Suttmann, O.; Kaierle, S.; Overmeyer, L. Multimaterial bathless stereolithography using aerosol jet printing and UV laser based polymerization. J. Laser Appl. 2019, 31, 22301. [Google Scholar] [CrossRef]
- de Backer, W.; Bergs, A.P.; van Tooren, M.J. Multi-Axis Multi-Material Fused Filament Fabrication with Continuous Fiber Reinforcement. In 2018 AIAA/ASCE/AHS/ASC Structures, Structural Dynamics, and Materials Conference; American Institute of Aeronautics and Astronautics: Reston, VA, USA, 2018. [Google Scholar]
- Ge, Q.; Sakhaei, A.H.; Lee, H.; Dunn, C.K.; Fang, N.X.; Dunn, M.L. Multimaterial 4D Printing with Tailorable Shape Memory Polymers. Sci. Rep. 2016, 6, 31110. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.L.; Zhang, H.; Chong, T.H.; Yeong, W.Y. 3D Printing of Multilayered and Multimaterial Electronics: A Review. Adv. Electron. Mater. 2021, 7, 2100445. [Google Scholar] [CrossRef]
- Hu, K.; Zhao, P.; Li, J.; Lu, Z. High-resolution multiceramic additive manufacturing based on digital light processing. Addit. Manuf. 2022, 54, 102732. [Google Scholar] [CrossRef]
- Khatri, B.; Lappe, K.; Noetzel, D.; Pursche, K.; Hanemann, T. A 3D-Printable Polymer-Metal Soft-Magnetic Functional Composite-Development and Characterization. Materials 2018, 11, 189. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.T.; Castro, K.; Bhattacharjee, N.; Folch, A. Digital Manufacturing of Selective Porous Barriers in Microchannels Using Multi-Material Stereolithography. Micromachines 2018, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Kowsari, K.; Akbari, S.; Wang, D.; Fang, N.X.; Ge, Q. High-Efficiency High-Resolution Multimaterial Fabrication for Digital Light Processing-Based Three-Dimensional Printing. 3D Print. Addit. Manuf. 2018, 5, 185–193. [Google Scholar] [CrossRef]
- Matsuzaki, R.; Kanatani, T.; Todoroki, A. Multi-material additive manufacturing of polymers and metals using fused filament fabrication and electroforming. Addit. Manuf. 2019, 29, 100812. [Google Scholar] [CrossRef]
- Rafiee, M.; Farahani, R.D.; Therriault, D. Multi-Material 3D and 4D Printing: A Survey. Adv. Sci. 2020, 7, 1902307. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.G.; Saiz, E.; Tirichenko, I.S.; García-Tuñón, E. Direct ink writing advances in multi-material structures for a sustainable future. J. Mater. Chem. A 2020, 8, 15646–15657. [Google Scholar] [CrossRef]
- Sachyani Keneth, E.; Lieberman, R.; Rednor, M.; Scalet, G.; Auricchio, F.; Magdassi, S. Multi-Material 3D Printed Shape Memory Polymer with Tunable Melting and Glass Transition Temperature Activated by Heat or Light. Polymers 2020, 12, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolini, A.; Kollmannsberger, S.; Rank, E. Additive manufacturing in construction: A review on processes, applications, and digital planning methods. Addit. Manuf. 2019, 30, 100894. [Google Scholar] [CrossRef]
- Beaman, J.J.; Bourell, D.L.; Seepersad, C.C.; Kovar, D. Additive Manufacturing Review: Early Past to Current Practice. J. Manuf. Sci. Eng. 2020, 142, 20. [Google Scholar] [CrossRef]
- Tan, L.J.; Zhu, W.; Zhou, K. Recent Progress on Polymer Materials for Additive Manufacturing. Adv. Funct. Mater. 2020, 30, 2003062. [Google Scholar] [CrossRef]
- Wu, H.; Fahy, W.P.; Kim, S.; Kim, H.; Zhao, N.; Pilato, L.; Kafi, A.; Bateman, S.; Koo, J.H. Recent developments in polymers/polymer nanocomposites for additive manufacturing. Prog. Mater. Sci. 2020, 111, 100638. [Google Scholar] [CrossRef]
- Ning, F.; Cong, W.; Qiu, J.; Wei, J.; Wang, S. Additive manufacturing of carbon fiber reinforced thermoplastic composites using fused deposition modeling. Compos. Part B Eng. 2015, 80, 369–378. [Google Scholar] [CrossRef]
- Tian, X.; Liu, T.; Yang, C.; Wang, Q.; Li, D. Interface and performance of 3D printed continuous carbon fiber reinforced PLA composites. Compos. Part A Appl. Sci. Manuf. 2016, 88, 198–205. [Google Scholar] [CrossRef]
- Fomina, N.; Sankaranarayanan, J.; Almutairi, A. Photochemical mechanisms of light-triggered release from nanocarriers. Adv. Drug Deliv. Rev. 2012, 64, 1005–1020. [Google Scholar] [CrossRef] [Green Version]
- Pape, M. Industrial applications of photochemistry. Pure Appl. Chem. 1975, 41, 535–558. [Google Scholar] [CrossRef] [Green Version]
- Chatani, S.; Kloxin, C.J.; Bowman, C.N. The power of light in polymer science: Photochemical processes to manipulate polymer formation, structure, and properties. Polym. Chem. 2014, 5, 2187–2201. [Google Scholar] [CrossRef] [Green Version]
- Bîrcă, A.; Gherasim, O.; Grumezescu, V.; Grumezescu, A.M. Introduction in thermoplastic and thermosetting polymers. In Materials for Biomedical Engineering; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Lin, C.-H.; Lin, Y.-M.; Lai, Y.-L.; Lee, S.-Y. Mechanical properties, accuracy, and cytotoxicity of UV-polymerized 3D printing resins composed of Bis-EMA, UDMA, and TEGDMA. J. Prosthet. Dent. 2020, 123, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Silva, J.; Vanat, P.; Marques-da-Silva, D.; Rodrigues, J.R.; Lagoa, R. Metal alginates for polyphenol delivery systems: Studies on crosslinking ions and easy-to-use patches for release of protective flavonoids in skin. Bioact. Mater. 2020, 5, 447–457. [Google Scholar] [CrossRef]
- Sun, A.; He, X.; Ji, X.; Hu, D.; Pan, M.; Zhang, L.; Qian, Z. Current research progress of photopolymerized hydrogels in tissue engineering. Chin. Chem. Lett. 2021, 32, 2117–2126. [Google Scholar] [CrossRef]
- Regehly, M.; Garmshausen, Y.; Reuter, M.; König, N.F.; Israel, E.; Kelly, D.P.; Chou, C.-Y.; Koch, K.; Asfari, B.; Hecht, S. Xolography for linear volumetric 3D printing. Nature 2020, 588, 620–624. [Google Scholar] [CrossRef]
- Hofmann, M. 3D Printing Gets a Boost and Opportunities with Polymer Materials. ACS Macro Lett. 2014, 3, 382–386. [Google Scholar] [CrossRef]
- Stansbury, J.W.; Idacavage, M.J. 3D printing with polymers: Challenges among expanding options and opportunities. Dent. Mater. 2016, 32, 54–64. [Google Scholar] [CrossRef]
- Zaneldin, E.; Ahmed, W.; Mansour, A.; Hassan, A.E. Dimensional Stability of 3D Printed Objects Made from Plastic Waste Using FDM: Potential Construction Applications. Buildings 2021, 11, 516. [Google Scholar] [CrossRef]
- Arefin, A.M.E.; Khatri, N.R.; Kulkarni, N.; Egan, P.F. Polymer 3D Printing Review: Materials, Process, and Design Strategies for Medical Applications. Polymers 2021, 13, 1499. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, A.; Jin, J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 2019, 1, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Steyrer, B.; Neubauer, P.; Liska, R.; Stampfl, J. Visible Light Photoinitiator for 3D-Printing of Tough Methacrylate Resins. Materials 2017, 10, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Li, D.; Jin, Z. Preliminary Investigation of Poly-Ether-Ether-Ketone Based on Fused Deposition Modeling for Medical Applications. Materials 2018, 11, 288. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Mazzoli, A. Selective laser sintering in biomedical engineering. Med. Biol. Eng. Comput. 2013, 51, 245–256. [Google Scholar] [CrossRef]
- Gibson, I.; Shi, D. Material properties and fabrication parameters in selective laser sintering process. Rapid Prototyp. J. 1997, 3, 129–136. [Google Scholar] [CrossRef]
- Goodridge, R.D.; Shofner, M.L.; Hague, R.; McClelland, M.; Schlea, M.R.; Johnson, R.B.; Tuck, C.J. Processing of a Polyamide-12/carbon nanofibre composite by laser sintering. Polym. Test. 2011, 30, 94–100. [Google Scholar] [CrossRef]
- Schmidleithner, C.; Kalaskar, D.M. Stereolithography. In 3D Printing; Cvetković, D., Ed.; InTech: Vienna, Austria, 2018; ISBN 978-1-78923-965-2. [Google Scholar]
- Wallin, T.J.; Pikul, J.H.; Bodkhe, S.; Peele, B.N.; Mac Murray, B.C.; Therriault, D.; McEnerney, B.W.; Dillon, R.P.; Giannelis, E.P.; Shepherd, R.F. Click chemistry stereolithography for soft robots that self-heal. J. Mater. Chem. B 2017, 5, 6249–6255. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, M.; Zhou, Z.; Gou, J.; Hui, D. 3D printing of polymer matrix composites: A review and prospective. Compos. Part B Eng. 2017, 110, 442–458. [Google Scholar] [CrossRef]
- Shaukat, U.; Rossegger, E.; Schlögl, S. Thiol–acrylate based vitrimers: From their structure–property relationship to the additive manufacturing of self-healable soft active devices. Polymer 2021, 231, 124110. [Google Scholar] [CrossRef]
- Rossegger, E.; Höller, R.; Reisinger, D.; Strasser, J.; Fleisch, M.; Griesser, T.; Schlögl, S. Digital light processing 3D printing with thiol–acrylate vitrimers. Polym. Chem. 2021, 12, 639–644. [Google Scholar] [CrossRef]
- Masahiro, I.; Tetsuya, K.; Hiroshi, Y.; Masaru, T. Liquid Crystal Display 3D Printing. U.S. Patent No. 6934000B1, 23 August 2005. [Google Scholar]
- Quan, H.; Zhang, T.; Xu, H.; Luo, S.; Nie, J.; Zhu, X. Photo-curing 3D printing technique and its challenges. Bioact. Mater. 2020, 5, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Chen, Y.; Hu, M.; Qin, S.; Liu, P. 4D printing of shape memory polymer via liquid crystal display (LCD) stereolithographic 3D printing. Mater. Res. Express 2020, 7, 105305. [Google Scholar] [CrossRef]
- Tumbleston, J.R.; Shirvanyants, D.; Ermoshkin, N.; Janusziewicz, R.; Johnson, A.R.; Kelly, D.; Chen, K.; Pinschmidt, R.; Rolland, J.P.; Ermoshkin, A.; et al. Additive manufacturing. Continuous liquid interface production of 3D objects. Science 2015, 347, 1349–1352. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. A robust and versatile photoinduced living polymerization of conjugated and unconjugated monomers and its oxygen tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. [Google Scholar] [CrossRef]
- Rapid 3D Continuous Printing of Casting Molds for Metals and other Materials. U.S. Patent No. 10,538,030, 21 January 2020.
- Janusziewicz, R.; Tumbleston, J.R.; Quintanilla, A.L.; Mecham, S.J.; DeSimone, J.M. Layerless fabrication with continuous liquid interface production. Proc. Natl. Acad. Sci. USA 2016, 113, 11703–11708. [Google Scholar] [CrossRef] [Green Version]
- Dall’Argine, C.; Hochwallner, A.; Klikovits, N.; Liska, R.; Stampf, J.; Sangermano, M. Hot-Lithography SLA-3D Printing of Epoxy Resin. Macromol. Mater. Eng. 2020, 305, 2000325. [Google Scholar] [CrossRef]
- Steyrer, B.; Busetti, B.; Harakály, G.; Liska, R.; Stampfl, J. Hot Lithography vs. room temperature DLP 3D-printing of a dimethacrylate. Addit. Manuf. 2018, 21, 209–214. [Google Scholar] [CrossRef]
- Pfaffinger, M. Hot Lithography—New Possibilities in Polymer 3D Printing. Laser Tech. J. 2018, 15, 45–47. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-B.; Kawata, S. Two-Photon Photopolymerization and 3D Lithographic Microfabrication. In NMR • 3D Analysis • Photopolymerization; Springer: Berlin/Heidelberg, Germany, 2004; pp. 169–273. [Google Scholar]
- Tanaka, T.; Sun, H.-B.; Kawata, S. Rapid sub-diffraction-limit laser micro/nanoprocessing in a threshold material system. Appl. Phys. Lett. 2002, 80, 312–314. [Google Scholar] [CrossRef]
- Sun, H.-B.; Takada, K.; Kim, M.-S.; Lee, K.-S.; Kawata, S. Scaling laws of voxels in two-photon photopolymerization nanofabrication. Appl. Phys. Lett. 2003, 83, 1104–1106. [Google Scholar] [CrossRef]
- Sun, H.-B.; Tanaka, T.; Kawata, S. Three-dimensional focal spots related to two-photon excitation. Appl. Phys. Lett. 2002, 80, 3673–3675. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-B.; Kawata, S. Two-Photon Laser Precision Microfabrication and Its Applications to Micro-Nano Devices and Systems. J. Lightwave Technol. 2003, 21, 624. [Google Scholar]
- Sun, H.-B.; Maeda, M.; Takada, K.; Chon, J.W.M.; Gu, M.; Kawata, S. Experimental investigation of single voxels for laser nanofabrication via two-photon photopolymerization. Appl. Phys. Lett. 2003, 83, 819–821. [Google Scholar] [CrossRef]
- Morikawa, J.; Orie, A.; Hashimoto, T.; Juodkazis, S. Thermal diffusivity in femtosecond-laser-structured micro-volumes of polymers. Appl. Phys. A 2010, 98, 551–556. [Google Scholar] [CrossRef]
- Takada, K.; Kaneko, K.; Li, Y.-D.; Kawata, S.; Chen, Q.-D.; Sun, H.-B. Temperature effects on pinpoint photopolymerization and polymerized micronanostructures. Appl. Phys. Lett. 2008, 92, 41902. [Google Scholar] [CrossRef]
- Takada, K.; Wu, D.; Chen, Q.-D.; Shoji, S.; Xia, H.; Kawata, S.; Sun, H.-B. Size-dependent behaviors of femtosecond laser-prototyped polymer micronanowires. Opt. Lett. 2009, 34, 566–568. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Chen, Q.D.; Niu, L.G.; Jiao, J.; Xia, H.; Song, J.F.; Sun, H.B. 100% Fill-Factor Aspheric Microlens Arrays (AMLA) With Sub-20-nm Precision. IEEE Photonics Technol. Lett. 2009, 21, 1535–1537. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Serbin, J.; Gu, M. Two-photon polymerisation for three-dimensional micro-fabrication. J. Photochem. Photobiol. A Chem. 2006, 181, 1–11. [Google Scholar] [CrossRef]
- Lee, K.-S.; Kim, R.H.; Yang, D.-Y.; Park, S.H. Advances in 3D nano/microfabrication using two-photon initiated polymerization. Prog. Polym. Sci. 2008, 33, 631–681. [Google Scholar] [CrossRef]
- Li, L.; Gattass, R.R.; Gershgoren, E.; Hwang, H.; Fourkas, J.T. Achieving lambda/20 resolution by one-color initiation and deactivation of polymerization. Science 2009, 324, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Chen, Q.-D.; Xia, H.; Sun, H.-B. Designable 3D nanofabrication by femtosecond laser direct writing. Nano Today 2010, 5, 435–448. [Google Scholar] [CrossRef]
- Wu, D.; Wu, S.-Z.; Niu, L.-G.; Chen, Q.-D.; Wang, R.; Song, J.-F.; Fang, H.-H.; Sun, H.-B. High numerical aperture microlens arrays of close packing. Appl. Phys. Lett. 2010, 97, 31109. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.E.; Bhattacharya, I.; Heidari, H.; Shusteff, M.; Spadaccini, C.M.; Taylor, H.K. Volumetric additive manufacturing via tomographic reconstruction. Science 2019, 363, 1075–1079. [Google Scholar] [CrossRef]
- Bernal, P.N.; Delrot, P.; Loterie, D.; Li, Y.; Malda, J.; Moser, C.; Levato, R. Volumetric Bioprinting of Complex Living-Tissue Constructs within Seconds. Adv. Mater. 2019, 31, e1904209. [Google Scholar] [CrossRef] [Green Version]
- Loterie, D.; Delrot, P.; Moser, C. High-resolution tomographic volumetric additive manufacturing. Nat. Commun. 2020, 11, 852. [Google Scholar] [CrossRef]
- Shusteff, M.; Browar, A.E.M.; Kelly, B.E.; Henriksson, J.; Weisgraber, T.H.; Panas, R.M.; Fang, N.X.; Spadaccini, C.M. One-step volumetric additive manufacturing of complex polymer structures. Sci. Adv. 2017, 3, eaao5496. [Google Scholar] [CrossRef] [Green Version]
- Yap, H.K.; Ng, H.Y.; Yeow, C.-H. High-Force Soft Printable Pneumatics for Soft Robotic Applications. Soft Robot. 2016, 3, 144–158. [Google Scholar] [CrossRef]
- Kim, K.; Park, J.; Suh, J.; Kim, M.; Jeong, Y.; Park, I. 3D printing of multiaxial force sensors using carbon nanotube (CNT)/thermoplastic polyurethane (TPU) filaments. Sens. Actuators A Phys. 2017, 263, 493–500. [Google Scholar] [CrossRef]
- Truby, R.L.; Lewis, J.A. Printing soft matter in three dimensions. Nature 2016, 540, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, G.; Qin, J.; Lowe, S.E.; Zhang, S.; Zhao, H.; Zhong, Y.L. Recent Progress of Direct Ink Writing of Electronic Components for Advanced Wearable Devices. ACS Appl. Electron. Mater. 2019, 1, 1718–1734. [Google Scholar] [CrossRef]
- Lewis, J.A. Direct Ink Writing of 3D Functional Materials. Adv. Funct. Mater. 2006, 16, 2193–2204. [Google Scholar] [CrossRef]
- Peyre, P.; Rouchausse, Y.; Defauchy, D.; Régnier, G. Experimental and numerical analysis of the selective laser sintering (SLS) of PA12 and PEKK semi-crystalline polymers. J. Mater. Process. Technol. 2015, 225, 326–336. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, X.; Chen, X.; He, Y.; Cheng, L.; Huo, M.; Yin, J.; Hao, F.; Chen, S.; Wang, P.; et al. Additive manufacturing of structural materials. Mater. Sci. Eng. R: Rep. 2021, 145, 100596. [Google Scholar] [CrossRef]
- Aramian, A.; Razavi, S.M.J.; Sadeghian, Z.; Berto, F. A review of additive manufacturing of cermets. Addit. Manuf. 2020, 33, 101130. [Google Scholar] [CrossRef]
- Waheed, S.; Cabot, J.M.; Macdonald, N.P.; Lewis, T.; Guijt, R.M.; Paull, B.; Breadmore, M.C. 3D printed microfluidic devices: Enablers and barriers. Lab Chip 2016, 16, 1993–2013. [Google Scholar] [CrossRef] [Green Version]
- Ligon, S.C.; Liska, R.; Stampfl, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef] [Green Version]
- Stampfl, J.; Baudis, S.; Heller, C.; Liska, R.; Neumeister, A.; Kling, R.; Ostendorf, A.; Spitzbart, M. Photopolymers with tunable mechanical properties processed by laser-based high-resolution stereolithography. J. Micromech. Microeng. 2008, 18, 125014. [Google Scholar] [CrossRef]
- Gong, H.; Bickham, B.P.; Woolley, A.T.; Nordin, G.P. Custom 3D printer and resin for 18 μm × 20 μm microfluidic flow channels. Lab Chip 2017, 17, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Bhaduri, B.; Mir, M.; Shkumatov, A.; Lee, M.K.; Popescu, G.; Kong, H.; Bashir, R. Bioprinting: High-Resolution Projection Microstereolithography for Patterning of Neovasculature (Adv. Healthcare Mater. 5/2016). Adv. Healthc. Mater. 2016, 5, 622. [Google Scholar] [CrossRef]
- Geng, Q.; Wang, D.; Chen, P.; Chen, S.-C. Ultrafast multi-focus 3-D nano-fabrication based on two-photon polymerization. Nat. Commun. 2019, 10, 2179. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Wang, D.; Nguyen, V.H.; Chang, Y.; Oakdale, J.S.; Chen, S.-C. Scalable submicrometer additive manufacturing. Science 2019, 366, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Bhattacharya, I.; Shusteff, M.; Panas, R.M.; Taylor, H.K.; Spadaccini, C.M. Computed Axial Lithography (CAL): Toward Single Step 3D Printing of Arbitrary Geometries. arXiv 2017, arXiv:1705.05893. [Google Scholar]
- Qiu, D.; Langrana, N.A. Void eliminating toolpath for extrusion-based multi-material layered manufacturing. Rapid Prototyp. J. 2002, 8, 38–45. [Google Scholar] [CrossRef]
- Singh, S.; Singh, G.; Prakash, C.; Ramakrishna, S. Current status and future directions of fused filament fabrication. J. Manuf. Process. 2020, 55, 288–306. [Google Scholar] [CrossRef]
- Singh, S.; Singh, N.; Gupta, M.; Prakash, C.; Singh, R. Mechanical feasibility of ABS/HIPS-based multi-material structures primed by low-cost polymer printer. Rapid Prototyp. J. 2019, 25, 152–161. [Google Scholar] [CrossRef]
- Khatri, B.; Lappe, K.; Habedank, M.; Mueller, T.; Megnin, C.; Hanemann, T. Fused Deposition Modeling of ABS-Barium Titanate Composites: A Simple Route towards Tailored Dielectric Devices. Polymers 2018, 10, 666. [Google Scholar] [CrossRef] [Green Version]
- Nötzel, D.; Eickhoff, R.; Hanemann, T. Fused Filament Fabrication of Small Ceramic Components. Materials 2018, 11, 1463. [Google Scholar] [CrossRef] [Green Version]
- Hanemann, T.; Syperek, D.; Nötzel, D. 3D Printing of ABS Barium Ferrite Composites. Materials 2020, 13, 1481. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kumar, R.; Farina, I.; Colangelo, F.; Feo, L.; Fraternali, F. Multi-Material Additive Manufacturing of Sustainable Innovative Materials and Structures. Polymers 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C.G.; Meier, W. Stimuli-responsive polymers and their applications in nanomedicine. Biointerphases 2012, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Fouassier, J.-P.; Rabek, J.F. Radiation Curing in Polymer Science and Technology: Fundamentals and Methods; Elsevier Applied Science: London, UK; New York, NY, USA, 1993; ISBN 9781851669295. [Google Scholar]
- Wardle, B. Principles and Applications of Photochemistry; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Schnabel, W. Polymers and Light: Fundamentals and Technical Applications; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Kagan, J. Organic Photochemistry: Principles and Applications; Academic Press: London, UK, 1993. [Google Scholar]
- Mäntele, W.; Deniz, E. UV-VIS absorption spectroscopy: Lambert-Beer reloaded. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 173, 965–968. [Google Scholar] [CrossRef]
- Jasinski, F.; Zetterlund, P.B.; Braun, A.M.; Chemtob, A. Photopolymerization in dispersed systems. Prog. Polym. Sci. 2018, 84, 47–88. [Google Scholar] [CrossRef] [Green Version]
- Wayner, D.D.M.; Clark, K.B.; Rauk, A.; Yu, D.; Armstrong, D.A. C−H Bond Dissociation Energies of Alkyl Amines: Radical Structures and Stabilization Energies. J. Am. Chem. Soc. 1997, 119, 8925–8932. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Schnabel, W. On the reactivity of phosphonyl radicals towards olefinic compounds. Makromol. Chem. 1985, 186, 1811–1823. [Google Scholar] [CrossRef]
- Mondschein, R.J.; Kanitkar, A.; Williams, C.B.; Verbridge, S.S.; Long, T.E. Polymer structure-property requirements for stereolithographic 3D printing of soft tissue engineering scaffolds. Biomaterials 2017, 140, 170–188. [Google Scholar] [CrossRef]
- Davidson, R.S. The Chemistry of Excited Complexes: A Survey of Reactions. In Advances in Physical Organic Chemistry Volume 19; Elsevier: Amsterdam, The Netherlands, 1983; pp. 1–130. ISBN 9780120335190. [Google Scholar]
- Hutchison, J.; Lambert, M.C.; Ledwith, A. Role of semi-pinacol radicals in the benzophenone-photoinitiated polymerization of methyl methacrylate. Polymer 1973, 14, 250–254. [Google Scholar] [CrossRef]
- Allen, N.S. Photoinitiators for UV and visible curing of coatings” mechanisms and properties. J. Photochem. Photobiol. A Chem. 1996, 100, 101–107. [Google Scholar] [CrossRef]
- Zhang, J.; Dumur, F.; Xiao, P.; Graff, B.; Bardelang, D.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Structure Design of Naphthalimide Derivatives: Toward Versatile Photoinitiators for Near-UV/Visible LEDs, 3D Printing, and Water-Soluble Photoinitiating Systems. Macromolecules 2015, 48, 2054–2063. [Google Scholar] [CrossRef]
- Wu, G.-H.; Hsu, S. Review: Polymeric-Based 3D Printing for Tissue Engineering. J. Med. Biol. Eng. 2015, 35, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Cullen, A.T.; Price, A.D. Digital light processing for the fabrication of 3D intrinsically conductive polymer structures. Synth. Met. 2018, 235, 34–41. [Google Scholar] [CrossRef]
- Gou, M.; Qu, X.; Zhu, W.; Xiang, M.; Yang, J.; Zhang, K.; Wei, Y.; Chen, S. Bio-inspired detoxification using 3D-printed hydrogel nanocomposites. Nat. Commun. 2014, 5, 3774. [Google Scholar] [CrossRef] [Green Version]
- Schafer, K.J.; Hales, J.M.; Balu, M.; Belfield, K.D.; van Stryland, E.W.; Hagan, D.J. Two-photon absorption cross-sections of common photoinitiators. J. Photochem. Photobiol. A Chem. 2004, 162, 497–502. [Google Scholar] [CrossRef]
- Dilla, R.A.; M Motta, C.M.; Snyder, S.R.; Wilson, J.A.; Wesdemiotis, C.; Becker, M.L. Synthesis and 3D Printing of PEG-Poly(propylene fumarate) Diblock and Triblock Copolymer Hydrogels. ACS Macro Lett. 2018, 7, 1254–1260. [Google Scholar] [CrossRef]
- Huang, T.Q.; Qu, X.; Liu, J.; Chen, S. 3D printing of biomimetic microstructures for cancer cell migration. Biomed. Microdevices 2014, 16, 127–132. [Google Scholar] [CrossRef]
- Gauvin, R.; Chen, Y.-C.; Lee, J.W.; Soman, P.; Zorlutuna, P.; Nichol, J.W.; Bae, H.; Chen, S.; Khademhosseini, A. Microfabrication of complex porous tissue engineering scaffolds using 3D projection stereolithography. Biomaterials 2012, 33, 3824–3834. [Google Scholar] [CrossRef] [Green Version]
- Mann, B.K.; Gobin, A.S.; Tsai, A.T.; Schmedlen, R.H.; West, J.L. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: Synthetic ECM analogs for tissue engineering. Biomaterials 2001, 22, 3045–3051. [Google Scholar] [CrossRef]
- Ravve, A. Light-Associated Reactions of Synthetic Polymers; Springer: New York, NY, USA, 2006; ISBN 9780387364148. [Google Scholar]
- Lin, H.; Zhang, D.; Alexander, P.G.; Yang, G.; Tan, J.; Cheng, A.W.-M.; Tuan, R.S. Application of visible light-based projection stereolithography for live cell-scaffold fabrication with designed architecture. Biomaterials 2013, 34, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauk, S.; Liska, R. Photoinitiators with Functional Groups, 8. Macromol. Rapid Commun. 2005, 26, 1687–1692. [Google Scholar] [CrossRef]
- Park, H.K.; Shin, M.; Kim, B.; Park, J.W.; Lee, H. A visible light-curable yet visible wavelength-transparent resin for stereolithography 3D printing. NPG Asia Mater. 2018, 10, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Warner, J.; Soman, P.; Zhu, W.; Tom, M.; Chen, S. Design and 3D Printing of Hydrogel Scaffolds with Fractal Geometries. ACS Biomater. Sci. Eng. 2016, 2, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gu, Y.; Liu, X.; Zhang, L.; Cheng, Z.; Zhu, X. Metal-Free Atom Transfer Radical Polymerization of Methyl Methacrylate with ppm Level of Organic Photocatalyst. Macromol. Rapid Commun. 2017, 38, 1600461. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Dumur, F.; Garra, P.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Carbazole Scaffold Based Photoinitiator/Photoredox Catalysts: Toward New High Performance Photoinitiating Systems and Application in LED Projector 3D Printing Resins. Macromolecules 2017, 50, 2747–2758. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bulut, U. Dual photo- and thermally initiated cationic polymerization of epoxy monomers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6750–6764. [Google Scholar] [CrossRef]
- Michaudel, Q.; Kottisch, V.; Fors, B.P. Cationic Polymerization: From Photoinitiation to Photocontrol. Angew. Chem. Int. Ed Engl. 2017, 56, 9670–9679. [Google Scholar] [CrossRef]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar] [CrossRef]
- Saleh, T.A. (Ed.) Polymer Hybrid Materials and Nanocomposites: Fundamentals and Applications/Tawfik Abdo Saleh; William Andrew: Norwich, UK, 2019; ISBN 9780128132944. [Google Scholar]
- McKeen, L.W. (Ed.) Introduction to Plastics and Polymers. In The Effect of UV Light and Weather on Plastics and Elastomers, 4th ed.; William Andrew: Norwich, UK, 2019; pp. 1–20. ISBN 9780128164570. [Google Scholar]
- Hasirci, V.; Huri, P.Y.; Tanir, T.E.; Eke, G.; Hasirci, N. 1.22 Polymer Fundamentals: Polymer Synthesis. In Comprehensive biomaterials II, 2nd ed.; Ducheyne, P., Grainger, D.W., Healy, K.E., Hutmacher, D.W., Kirkpatrick, C.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 478–506. ISBN 9780081006924. [Google Scholar]
- Tulig, T.J.; Tirrell, M. Molecular theory of the Trommsdorff effect. Macromolecules 1981, 14, 1501–1511. [Google Scholar] [CrossRef]
- Tulig, T.J.; Tirrell, M. On the onset of the Trommsdorff effect. Macromolecules 1982, 15, 459–463. [Google Scholar] [CrossRef]
- Mishra, M.K.; Yagci, Y. Handbook of Radical Vinyl Polymerization; Marcel Dekker: New York, NY, USA, 1998; ISBN 9780824794644. [Google Scholar]
- Ligon-Auer, S.C.; Schwentenwein, M.; Gorsche, C.; Stampfl, J.; Liska, R. Toughening of photo-curable polymer networks: A review. Polym. Chem. 2016, 7, 257–286. [Google Scholar] [CrossRef]
- Tanzi, M.C.; Farè, S.; Candiani, G. Foundations of Biomaterials Engineering: Organization, Structure, and Properties of Materials; Academic Press: New York, NY, USA, 2019; ISBN 978-0-08-101034-1. [Google Scholar]
- Gloeckner, P. Radiation Curing: Coatings and Printing Inks; Vincentz Network: Hannover, Germany, 2008; ISBN 9783866309074. [Google Scholar]
- Colombani, D. Chain-growth control in free radical polymerization. Prog. Polym. Sci. 1997, 22, 1649–1720. [Google Scholar] [CrossRef]
- Cowie, J.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780429125546. [Google Scholar]
- Odian, G.G. Principles of Polymerization, 4th ed.; Wiley: New York, NY, USA; Chichester, UK, 2004; ISBN 9780471274001. [Google Scholar]
- Young, R.J.; Lovell, P.A. Introduction to Polymers, 3rd ed.; CRC Press: Hoboken, NJ, USA, 2011; ISBN 9781439894156. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953; ISBN 9780801401343. [Google Scholar]
- Braun, D. Polymer Synthesis: Theory and Practice Fundamentals, Methods, Experiments, 5th ed.; Springer: Heidelberg, Germany; New York, NY, USA, 2013; ISBN 9783642289798. [Google Scholar]
- Bamford, C.H.; Jenkins, A.D.; Symons, M.C.R.; Townsend, M.G. Trapped radicals in heterogeneous vinyl polymerization. J. Polym. Sci. 1959, 34, 181–198. [Google Scholar] [CrossRef]
- Stille, J.K. Step-growth polymerization. J. Chem. Educ. 1981, 58, 862. [Google Scholar] [CrossRef] [Green Version]
- Rossegger, E.; Höller, R.; Reisinger, D.; Fleisch, M.; Strasser, J.; Wieser, V.; Griesser, T.; Schlögl, S. High resolution additive manufacturing with acrylate based vitrimers using organic phosphates as transesterification catalyst. Polymer 2021, 221, 123631. [Google Scholar] [CrossRef]
- Decker, C.; Nguyen Thi Viet, T.; Pham Thi, H. Photoinitiated cationic polymerization of epoxides. Polym. Int. 2001, 50, 986–997. [Google Scholar] [CrossRef]
- Sangermano, M.; Rodriguez, D.; Gonzalez, M.C.; Laurenti, E.; Yagci, Y. Visible Light Induced Cationic Polymerization of Epoxides by Using Multiwalled Carbon Nanotubes. Macromol. Rapid Commun. 2018, 39, e1800250. [Google Scholar] [CrossRef]
- Rudin, A.; Choi, P. (Eds.) Ionic and Coordinated Polymerizations. In The Elements of Polymer Science and Engineering, 3rd ed.; Elsevier/AP: San Diego, CA, USA, 2013; pp. 449–493. ISBN 9780123821782. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W. Photoinitiated cationic polymerization with triarylsulfonium salts. J. Polym. Sci. Part A Polym. Chem. 1996, 34, 3231–3253. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Complex triarylsulfonium salt photoinitiators. I. The identification, characterization, and syntheses of a new class of triarylsulfonium salt photoinitiators. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 2677–2695. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Garra, P.; Schmitt, M.; Toufaily, J.; Hamieh, T.; Graff, B.; Fouassier, J.P.; Dumur, F.; Lalevée, J. 3-Hydroxyflavone and N-Phenylglycine in High Performance Photoinitiating Systems for 3D Printing and Photocomposites Synthesis. Macromolecules 2018, 51, 4633–4641. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Dumur, F.; Garra, P.; Toufaily, J.; Hamieh, T.; Goubard, F.; Bui, T.-T.; Graff, B.; Gigmes, D.; Pierre Fouassier, J.; et al. Azahelicenes as visible light photoinitiators for cationic and radical polymerization: Preparation of photoluminescent polymers and use in high performance LED projector 3D printing resins. J. Polym. Sci. A Polym. Chem. 2017, 55, 1189–1199. [Google Scholar] [CrossRef]
- Zhang, J.; Dumur, F.; Xiao, P.; Graff, B.; Gigmes, D.; Pierre Fouassier, J.; Lalevée, J. Aminothiazonaphthalic anhydride derivatives as photoinitiators for violet/blue LED-Induced cationic and radical photopolymerizations and 3D-Printing resins. J. Polym. Sci. A Polym. Chem. 2016, 54, 1189–1196. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Lara, D.M.; Noirbent, G.; Dumur, F.; Toufaily, J.; Hamieh, T.; Bui, T.-T.; Goubard, F.; Graff, B.; Gigmes, D.; et al. Carbazole Derivatives with Thermally Activated Delayed Fluorescence Property as Photoinitiators/Photoredox Catalysts for LED 3D Printing Technology. Macromolecules 2017, 50, 4913–4926. [Google Scholar] [CrossRef]
- Steinmann, B.; Wolf, J.; Schulthess, A.; Hunziker, M. Photosensitive Compositions. U.S. Patent No. 5476748A, 19 December 1995. [Google Scholar]
- Steinmann, B.; Schulthess, A. Liquid, Radiation-Curable Composition, Especially for Stereolithography. U.S. Patent No. 5972563A, 26 October 1999. [Google Scholar]
- Al Mousawi, A.; Poriel, C.; Dumur, F.; Toufaily, J.; Hamieh, T.; Fouassier, J.P.; Lalevée, J. Zinc Tetraphenylporphyrin as High Performance Visible Light Photoinitiator of Cationic Photosensitive Resins for LED Projector 3D Printing Applications. Macromolecules 2017, 50, 746–753. [Google Scholar] [CrossRef]
- Crivello, J.V.; Liu, S. Photoinitiated cationic polymerization of epoxy alcohol monomers. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 389–401. [Google Scholar] [CrossRef]
- Weems, A.C.; Pérez-Madrigal, M.M.; Arno, M.C.; Dove, A.P. 3D Printing for the Clinic: Examining Contemporary Polymeric Biomaterials and Their Clinical Utility. Biomacromolecules 2020, 21, 1037–1059. [Google Scholar] [CrossRef]
- Fan, D.; Staufer, U.; Accardo, A. Engineered 3D Polymer and Hydrogel Microenvironments for Cell Culture Applications. Bioengineering 2019, 6, 113. [Google Scholar] [CrossRef] [Green Version]
- Carothers, W.H. Polymers and polyfunctionality. Trans. Faraday Soc. 1936, 32, 39. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Kade, M.J.; Burke, D.J.; Hawker, C.J. The power of thiol-ene chemistry. J. Polym. Sci. A Polym. Chem. 2010, 48, 743–750. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. Engl. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lee, T.Y.; Roper, T. Thiol-enes: Chemistry of the past with promise for the future. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5301–5338. [Google Scholar] [CrossRef]
- Rohit, K.R.; Ujwaldev, S.M.; Krishnan, K.K.; Anilkumar, G. Recent Developments and Perspectives in the Zinc-Catalysed Michael Addition. Asian J. Org. Chem. 2018, 7, 85–102. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Husár, B.; Ligon, S.C.; Wutzel, H.; Hoffmann, H.; Liska, R. The formulator’s guide to anti-oxygen inhibition additives. Prog. Org. Coat. 2014, 77, 1789–1798. [Google Scholar] [CrossRef]
- McNair, O.D.; Janisse, A.P.; Krzeminski, D.E.; Brent, D.E.; Gould, T.E.; Rawlins, J.W.; Savin, D.A. Impact properties of thiol-ene networks. ACS Appl. Mater. Interfaces 2013, 5, 11004–11013. [Google Scholar] [CrossRef] [PubMed]
- Senyurt, A.F.; Wei, H.; Phillips, B.; Cole, M.; Nazarenko, S.; Hoyle, C.E.; Piland, S.G.; Gould, T.E. Physical and Mechanical Properties of Photopolymerized Thiol−Ene/Acrylates. Macromolecules 2006, 39, 6315–6317. [Google Scholar] [CrossRef]
- Senyurt, A.F.; Hoyle, C.E.; Wei, H.; Piland, S.G.; Gould, T.E. Thermal and Mechanical Properties of Cross-Linked Photopolymers Based on Multifunctional Thiol−Urethane Ene Monomers. Macromolecules 2007, 40, 3174–3182. [Google Scholar] [CrossRef]
- Oesterreicher, A.; Wiener, J.; Roth, M.; Moser, A.; Gmeiner, R.; Edler, M.; Pinter, G.; Griesser, T. Tough and degradable photopolymers derived from alkyne monomers for 3D printing of biomedical materials. Polym. Chem. 2016, 7, 5169–5180. [Google Scholar] [CrossRef]
- Oesterreicher, A.; Moser, A.; Edler, M.; Griesser, H.; Schlögl, S.; Pichelmayer, M.; Griesser, T. Investigating Photocurable Thiol-Yne Resins for Biomedical Materials. Macromol. Mater. Eng. 2017, 302, 1600450. [Google Scholar] [CrossRef]
- Sycks, D.G.; Wu, T.; Park, H.S.; Gall, K. Tough, stable spiroacetal thiol-ene resin for 3D printing. J. Appl. Polym. Sci. 2018, 135, 46259. [Google Scholar] [CrossRef]
- Moazzen, K.; Rossegger, E.; Alabiso, W.; Shaukat, U.; Schlögl, S. Role of Organic Phosphates and Phosphonates in Catalyzing Dynamic Exchange Reactions in Thiol-Click Vitrimers. Macromol. Chem. Phys. 2021, 222, 2100072. [Google Scholar] [CrossRef]
- Rossegger, E.; Moazzen, K.; Fleisch, M.; Schlögl, S. Locally controlling dynamic exchange reactions in 3D printed thiol-acrylate vitrimers using dual-wavelength digital light processing. Polym. Chem. 2021, 12, 3077–3083. [Google Scholar] [CrossRef]
- Lowe, A.B.; Hoyle, C.E.; Bowman, C.N. Thiol-yne click chemistry: A powerful and versatile methodology for materials synthesis. J. Mater. Chem. 2010, 20, 4745. [Google Scholar] [CrossRef]
- Hoogenboom, R. Thiol-yne chemistry: A powerful tool for creating highly functional materials. Angew. Chem. Int. Ed Engl. 2010, 49, 3415–3417. [Google Scholar] [CrossRef]
- Yao, B.; Sun, J.; Qin, A.; Tang, B.Z. Thiol-yne click polymerization. Chin. Sci. Bull. 2013, 58, 2711–2718. [Google Scholar] [CrossRef] [Green Version]
- Roppolo, I.; Frascella, F.; Gastaldi, M.; Castellino, M.; Ciubini, B.; Barolo, C.; Scaltrito, L.; Nicosia, C.; Zanetti, M.; Chiappone, A. Thiol–yne chemistry for 3D printing: Exploiting an off-stoichiometric route for selective functionalization of 3D objects. Polym. Chem. 2019, 10, 5950–5958. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Sims, E.A.; Anseth, K.S.; Bowman, C.N. Reaction Rates and Mechanisms for Radical, Photoinitated Addition of Thiols to Alkynes, and Implications for Thiol−Yne Photopolymerizations and Click Reactions. Macromolecules 2010, 43, 4113–4119. [Google Scholar] [CrossRef]
- Chan, J.W.; Shin, J.; Hoyle, C.E.; Bowman, C.N.; Lowe, A.B. Synthesis, Thiol−Yne “Click” Photopolymerization, and Physical Properties of Networks Derived from Novel Multifunctional Alkynes. Macromolecules 2010, 43, 4937–4942. [Google Scholar] [CrossRef]
- Oesterreicher, A.; Gorsche, C.; Ayalur-Karunakaran, S.; Moser, A.; Edler, M.; Pinter, G.; Schlögl, S.; Liska, R.; Griesser, T. Exploring Network Formation of Tough and Biocompatible Thiol-yne Based Photopolymers. Macromol. Rapid Commun. 2016, 37, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Beigi, S.; Yeganeh, H.; Atai, M. Evaluation of fracture toughness and mechanical properties of ternary thiol-ene-methacrylate systems as resin matrix for dental restorative composites. Dent. Mater. 2013, 29, 777–787. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.K.; Cramer, N.B.; Bowman, C.N. Oxygen inhibition in thiol–acrylate photopolymerizations. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 2007–2014. [Google Scholar] [CrossRef]
- Sangermano, M.; Cerrone, M.; Colucci, G.; Roppolo, I.; Acosta Ortiz, R. Preparation and characterization of hybrid thiol-ene/epoxy UV-thermal dual-cured systems. Polym. Int. 2010, 59, 1046–1051. [Google Scholar] [CrossRef]
- Yu, R.; Yang, X.; Zhang, Y.; Zhao, X.; Wu, X.; Zhao, T.; Zhao, Y.; Huang, W. Three-Dimensional Printing of Shape Memory Composites with Epoxy-Acrylate Hybrid Photopolymer. ACS Appl. Mater. Interfaces 2017, 9, 1820–1829. [Google Scholar] [CrossRef]
- Morancho, J.M.; Fernández-Francos, X.; Ramis, X.; Salla, J.M.; Serra, À. Photocuring and thermal post-curing of a cycloaliphatic epoxide resin with a trithiol and a vinyl epoxy compound. J. Anal. Calorim. 2015, 121, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, R.A.; Urbina, B.A.P.; Santos, R.G.; Duarte, L.B.; Valdez, A.E.G.; Soucek, M.D. Development of Hybrid Polymeric Materials Based on Thiol-Ene/Cationic Formulations. Macromol. Mater. Eng. 2008, 293, 731–739. [Google Scholar] [CrossRef]
- Li, W.; Delpouve, N.; Araujo, S.; Batteux, F.; Bobo, E.; Saiter, J.-M.; Tan, L.; Negahban, M. Controlling properties of acrylate/epoxy interpenetrating polymer networks by premature termination of radical polymerization of acrylate. Polym. Eng. Sci. 2019, 59, 1729–1738. [Google Scholar] [CrossRef]
- Konuray, O.; Di Donato, F.; Sangermano, M.; Bonada, J.; Tercjak, A.; Fernandez-Francos, X.; Serra, A.; Ramis, X. Dual-curable stereolithography resins for superior thermomechanical properties. Express Polym. Lett. 2020, 14, 881–894. [Google Scholar] [CrossRef]
- Kuang, X.; Zhao, Z.; Chen, K.; Fang, D.; Kang, G.; Qi, H.J. High-Speed 3D Printing of High-Performance Thermosetting Polymers via Two-Stage Curing. Macromol. Rapid Commun. 2018, 39, e1700809. [Google Scholar] [CrossRef]
- Griffini, G.; Invernizzi, M.; Levi, M.; Natale, G.; Postiglione, G.; Turri, S. 3D-printable CFR polymer composites with dual-cure sequential IPNs. Polymer 2016, 91, 174–179. [Google Scholar] [CrossRef]
- Decker, C.; Nguyen Thi Viet, T.; Decker, D.; Weber-Koehl, E. UV-radiation curing of acrylate/epoxide systems. Polymer 2001, 42, 5531–5541. [Google Scholar] [CrossRef]
- Jian, Y.; He, Y.; Sun, Y.; Yang, H.; Yang, W.; Nie, J. Thiol–epoxy/thiol–acrylate hybrid materials synthesized by photopolymerization. J. Mater. Chem. C 2013, 1, 4481. [Google Scholar] [CrossRef]
- Konuray, O.; Sola, A.; Bonada, J.; Tercjak, A.; Fabregat-Sanjuan, A.; Fernández-Francos, X.; Ramis, X. Cost-Effectively 3D-Printed Rigid and Versatile Interpenetrating Polymer Networks. Materials 2021, 14, 4544. [Google Scholar] [CrossRef] [PubMed]
- Lantean, S.; Roppolo, I.; Sangermano, M.; Pirri, C.; Chiappone, A. Development of New Hybrid Acrylic/Epoxy DLP-3D Printable Materials. Inventions 2018, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-X.; Li, Y.; Zhong, J.; Luo, Z.; Gong, C.-R.; Zheng, Y.-Q.; Peng, S.; Yu, L.-M.; Wu, L.; Xu, Y. High-Performance Cyanate Ester Resins with Interpenetration Networks for 3D Printing. ACS Appl. Mater. Interfaces 2020, 12, 38682–38689. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, M.; Ji, M.; Kuang, X.; Qi, H.J.; Wang, T. Recyclable thermosetting polymers for digital light processing 3D printing. Mater. Des. 2021, 197, 109189. [Google Scholar] [CrossRef]
- Champeau, M.; Heinze, D.A.; Viana, T.N.; de Souza, E.R.; Chinellato, A.C.; Titotto, S. 4D Printing of Hydrogels: A Review. Adv. Funct. Mater. 2020, 30, 1910606. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Chu, P.K.; Gelinsky, M. 3D printing of hydrogels: Rational design strategies and emerging biomedical applications. Mater. Sci. Eng. R Rep. 2020, 140, 100543. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Berglund, C.M.; Lee, J.Y.; Polacheck, W.J.; Tsui, Y.; Bonassar, L.J.; Kirby, B.J. Methods for photocrosslinking alginate hydrogel scaffolds with high cell viability. Tissue Eng. Part C Methods 2011, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ma, X.; Sun, S.; Su, H.; Tan, T. A new photocrosslinkable hydrogel based on a derivative of polyaspartic acid for the controlled release of ketoprofen. Polym. Bull. 2010, 64, 623–632. [Google Scholar] [CrossRef]
- Lim, K.S.; Schon, B.S.; Mekhileri, N.V.; Brown, G.C.J.; Chia, C.M.; Prabakar, S.; Hooper, G.J.; Woodfield, T.B.F. New Visible-Light Photoinitiating System for Improved Print Fidelity in Gelatin-Based Bioinks. ACS Biomater. Sci. Eng. 2016, 2, 1752–1762. [Google Scholar] [CrossRef]
- Tytgat, L.; van Damme, L.; van Hoorick, J.; Declercq, H.; Thienpont, H.; Ottevaere, H.; Blondeel, P.; Dubruel, P.; van Vlierberghe, S. Additive manufacturing of photo-crosslinked gelatin scaffolds for adipose tissue engineering. Acta Biomater. 2019, 94, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Elvin, C.M.; Vuocolo, T.; Brownlee, A.G.; Sando, L.; Huson, M.G.; Liyou, N.E.; Stockwell, P.R.; Lyons, R.E.; Kim, M.; Edwards, G.A.; et al. A highly elastic tissue sealant based on photopolymerised gelatin. Biomaterials 2010, 31, 8323–8331. [Google Scholar] [CrossRef]
- Niu, Y.; Yang, T.; Ke, R.; Wang, C. Preparation and Characterization of pH-Responsive Sodium Alginate/Humic Acid/Konjac Hydrogel for L-Ascorbic Acid Controlled Release; American Scientific Publishers: Stevenson Ranch, CA, USA, 2019. [Google Scholar]
- Xu, Y.; Wu, X.; Wang, S.; Yang, C.; Li, Y.; Cao, Y. Hydroxyapatite Nanoparticle-Crosslinked Peptide Hydrogels for Three-Dimensional Culture and Differentiation of MC3T3-E1 Osteoblasts; American Scientific Publishers: Stevenson Ranch, CA, USA, 2019. [Google Scholar]
- He, X.; Zhu, B.; Xie, W.; He, Y.; Song, J.; Zhang, Y.; Sun, C.; Li, H.; Tang, Q.; Sun, X.; et al. Amelioration of imiquimod-induced psoriasis-like dermatitis in mice by DSW therapy inspired hydrogel. Bioact. Mater. 2021, 6, 299–311. [Google Scholar] [CrossRef]
- Li, Y.; Rodrigues, J.M.; Tomás, H. Injectable and biodegradable hydrogels: Gelation, biodegradation and biomedical applications. Chem. Soc. Rev. 2012, 41, 2193–2221. [Google Scholar] [CrossRef]
- Bertlein, S.; Brown, G.; Lim, K.S.; Jungst, T.; Boeck, T.; Blunk, T.; Tessmar, J.; Hooper, G.J.; Woodfield, T.B.F.; Groll, J. Thiol-Ene Clickable Gelatin: A Platform Bioink for Multiple 3D Biofabrication Technologies. Adv. Mater. 2017, 29, 1703404. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, E.; Imai, K.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997, 272, 2446–2451. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.-H.; Torgersen, J.; Saf, R.; Mühleder, S.; Pucher, N.; Ligon, S.C.; Holnthoner, W.; Redl, H.; Ovsianikov, A.; Stampfl, J.; et al. Three-dimensional microfabrication of protein hydrogels via two-photon-excited thiol-vinyl ester photopolymerization. J. Polym. Sci. A Polym. Chem. 2013, 51, 4799–4810. [Google Scholar] [CrossRef] [Green Version]
- Sušec, M.; Liska, R.; Russmüller, G.; Kotek, J.; Krajnc, P. Microcellular open porous monoliths for cell growth by thiol-ene polymerization of low-toxicity monomers in high internal phase emulsions. Macromol. Biosci. 2015, 15, 253–261. [Google Scholar] [CrossRef]
- Highley, C.B.; Prestwich, G.D.; Burdick, J.A. Recent advances in hyaluronic acid hydrogels for biomedical applications. Curr. Opin. Biotechnol. 2016, 40, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Highley, C.B.; Rodell, C.B.; Sun, W.; Burdick, J.A. 3D Printing of Shear-Thinning Hyaluronic Acid Hydrogels with Secondary Cross-Linking. ACS Biomater. Sci. Eng. 2016, 2, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Burdick, J.A.; Kobler, J.; Clifton, R.J.; Rosowski, J.J.; Zeitels, S.M.; Langer, R. Synthesis and Characterization of in Situ Cross-Linkable Hyaluronic Acid-Based Hydrogels with Potential Application for Vocal Fold Regeneration. Macromolecules 2004, 37, 3239–3248. [Google Scholar] [CrossRef]
- Qin, X.-H.; Gruber, P.; Markovic, M.; Plochberger, B.; Klotzsch, E.; Stampfl, J.; Ovsianikov, A.; Liska, R. Enzymatic synthesis of hyaluronic acid vinyl esters for two-photon microfabrication of biocompatible and biodegradable hydrogel constructs. Polym. Chem. 2014, 5, 6523–6533. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Soliman, B.G.; Alcala-Orozco, C.R.; Li, J.; Vis, M.A.M.; Santos, M.; Wise, S.G.; Levato, R.; Malda, J.; Woodfield, T.B.F.; et al. Rapid Photocrosslinking of Silk Hydrogels with High Cell Density and Enhanced Shape Fidelity. Adv. Healthc. Mater. 2020, 9, e1901667. [Google Scholar] [CrossRef]
- Kim, S.H.; Yeon, Y.K.; Lee, J.M.; Chao, J.R.; Lee, Y.J.; Seo, Y.B.; Sultan, M.T.; Lee, O.J.; Lee, J.S.; Yoon, S.; et al. Precisely printable and biocompatible silk fibroin bioink for digital light processing 3D printing. Nat. Commun. 2018, 9, 1620. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, K.; Zhao, S.; Zhang, C.; Li, J.; Yang, H.; Liu, X.; Yin, X.; Chen, D.; Xu, W.; et al. Photopolymerized maleilated chitosan/methacrylated silk fibroin micro/nanocomposite hydrogels as potential scaffolds for cartilage tissue engineering. Int. J. Biol. Macromol. 2018, 108, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, J.; Zhu, Q.; Yu, C.; Yin, J.; Zheng, L.; Li, A. Mannosylated PEGylated-Polyethyleneimine as Efficient CpG Oligodeoxynucleotide Carriers for Efficient Dendritic Cell Targeting Delivery and Activation; American Scientific Publishers: Stevenson Ranch, CA, USA, 2019. [Google Scholar]
- Serra, T.; Ortiz-Hernandez, M.; Engel, E.; Planell, J.A.; Navarro, M. Relevance of PEG in PLA-based blends for tissue engineering 3D-printed scaffolds. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 38, 55–62. [Google Scholar] [CrossRef]
- Bédouet, L.; Pascale, F.; Moine, L.; Wassef, M.; Ghegediban, S.H.; Nguyen, V.-N.; Bonneau, M.; Labarre, D.; Laurent, A. Intra-articular fate of degradable poly(ethyleneglycol)-hydrogel microspheres as carriers for sustained drug delivery. Int. J. Pharm. 2013, 456, 536–544. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Wang, X.; Wang, Y.; Zhang, Q.; Cheng, Y. A Polydopamine Nanoparticle-Knotted Poly(ethylene glycol) Hydrogel for On-Demand Drug Delivery and Chemo-photothermal Therapy. Chem. Mater. 2017, 29, 1370–1376. [Google Scholar] [CrossRef]
- Weber, L.M.; Lopez, C.G.; Anseth, K.S. Effects of PEG hydrogel crosslinking density on protein diffusion and encapsulated islet survival and function. J. Biomed. Mater. Res. Part A 2009, 90, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mautner, A.; Qin, X.; Wutzel, H.; Ligon, S.C.; Kapeller, B.; Moser, D.; Russmueller, G.; Stampfl, J.; Liska, R. Thiol-ene photopolymerization for efficient curing of vinyl esters. J. Polym. Sci. A Polym. Chem. 2013, 51, 203–212. [Google Scholar] [CrossRef]
- Moradi, L.; Vasei, M.; Dehghan, M.M.; Majidi, M.; Farzad Mohajeri, S.; Bonakdar, S. Regeneration of meniscus tissue using adipose mesenchymal stem cells-chondrocytes co-culture on a hybrid scaffold: In vivo study. Biomaterials 2017, 126, 18–30. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, K.; Zhou, D.; Yang, H.; Liu, X.; Yin, X.; Xu, W.; Zhou, Y.; Xiao, P. High-Performance Photopolymerized Poly(vinyl alcohol)/Silica Nanocomposite Hydrogels with Enhanced Cell Adhesion. ACS Appl. Mater. Interfaces 2018, 10, 27692–27700. [Google Scholar] [CrossRef] [PubMed]
- Billiet, T.; Vandenhaute, M.; Schelfhout, J.; van Vlierberghe, S.; Dubruel, P. A review of trends and limitations in hydrogel-rapid prototyping for tissue engineering. Biomaterials 2012, 33, 6020–6041. [Google Scholar] [CrossRef]
- Manapat, J.Z.; Chen, Q.; Ye, P.; Advincula, R.C. 3D Printing of Polymer Nanocomposites via Stereolithography. Macromol. Mater. Eng. 2017, 302, 1600553. [Google Scholar] [CrossRef]
- Maruo, S.; Ikuta, K.; Ninagawa, T. Multi-polymer microstereolithography for hybrid opto-MEMS. In The 14th IEEE international Conference on Micro Electro Mechanical Systems. Technical Digest MEMS 2001 14th IEEE International Conference on Micro Electro Mechanical Systems, Interlaken, Switzerland, 21–25 January 2001; IEEE: New York, NY, USA, 2001; pp. 151–154. ISBN 0-7803-5998-4. [Google Scholar]
- Wicker, R.; Medina, F.; Elkins, C. Multiple Material Micro-Fabrication: Extending Stereolithography to Tissue Engineering and Other Novel Applications; The University of Texas at Austin: Austin, TX, USA, 2004. [Google Scholar]
- Zhou, C.; Chen, Y.; Yang, Z.; Khoshnevis, B. Digital material fabrication using mask-image-projection-based stereolithography. Rapid Prototyp. J. 2013, 19, 153–165. [Google Scholar] [CrossRef]
- Matte, C.-D.; Pearson, M.; Trottier-Cournoyer, F.; Dafoe, A.; Kwok, T.H. Automated storage and active cleaning for multi-material digital-light-processing printer. Rapid Prototyp. J. 2019, 25, 864–874. [Google Scholar] [CrossRef]
- Bártolo, P. Stereolithography: Materials, Processes and Applications; Springer: New York, NY, USA, 2011; ISBN 9780387929040. [Google Scholar]
- Inamdar, A.; Magana, M.; Medina, F.; Grajeda, Y.; Wicker, R. Development of an Automated Multiple Material Stereolithography Machine; The University of Texas at Austin: Austin, TX, USA, 2006. [Google Scholar]
- Han, L.-H.; Suri, S.; Schmidt, C.E.; Chen, S. Fabrication of three-dimensional scaffolds for heterogeneous tissue engineering. Biomed. Microdevices 2010, 12, 721–725. [Google Scholar] [CrossRef]
- Wang, Q.; Jackson, J.A.; Ge, Q.; Hopkins, J.B.; Spadaccini, C.M.; Fang, N.X. Lightweight Mechanical Metamaterials with Tunable Negative Thermal Expansion. Phys. Rev. Lett. 2016, 117, 175901. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Hirata, H.; Furukawa, T.; Maruo, S. Multi-material microstereolithography using a palette with multicolor photocurable resins. Opt. Mater. Express 2020, 10, 2522. [Google Scholar] [CrossRef]
- Han, D.; Yang, C.; Fang, N.X.; Lee, H. Rapid multi-material 3D printing with projection micro-stereolithography using dynamic fluidic control. Addit. Manuf. 2019, 27, 606–615. [Google Scholar] [CrossRef]
- Mayer, F.; Richter, S.; Westhauser, J.; Blasco, E.; Barner-Kowollik, C.; Wegener, M. Multimaterial 3D laser microprinting using an integrated microfluidic system. Sci. Adv. 2019, 5, eaau9160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamont, A.C.; Restaino, M.A.; Kim, M.J.; Sochol, R.D. A facile multi-material direct laser writing strategy. Lab Chip 2019, 19, 2340–2345. [Google Scholar] [CrossRef]
- Mao, H.; Jia, W.; Leung, Y.-S.; Jin, J.; Chen, Y. Multi-material stereolithography using curing-on-demand printheads. Rapid Prototyp. J. 2021, 27, 861–871. [Google Scholar] [CrossRef]
- Lundeen, J.S.; Sutherland, B.; Patel, A.; Stewart, C.; Bamber, C. Direct measurement of the quantum wavefunction. Nature 2011, 474, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Makey, G.; Yavuz, Ö.; Kesim, D.K.; Turnalı, A.; Elahi, P.; Ilday, S.; Tokel, O.; Ilday, F.Ö. Breaking crosstalk limits to dynamic holography using orthogonality of high-dimensional random vectors. Nat. Photonics 2019, 13, 251–256. [Google Scholar] [CrossRef]
- Barany, G.; Merrifield, R.B. A new amino protecting group removable by reduction. Chemistry of the dithiasuccinoyl (Dts) function. J. Am. Chem. Soc. 1977, 99, 7363–7365. [Google Scholar] [CrossRef]
- Wong, C.-H.; Zimmerman, S.C. Orthogonality in organic, polymer, and supramolecular chemistry: From Merrifield to click chemistry. Chem. Commun. 2013, 49, 1679–1695. [Google Scholar] [CrossRef]
- Wong, C.-H.; Ye, X.-S.; Zhang, Z. Assembly of Oligosaccharide Libraries with a Designed Building Block and an Efficient Orthogonal Protection−Deprotection Strategy. J. Am. Chem. Soc. 1998, 120, 7137–7138. [Google Scholar] [CrossRef]
- Corrigan, N.; Boyer, C. 100th Anniversary of Macromolecular Science Viewpoint: Photochemical Reaction Orthogonality in Modern Macromolecular Science. ACS Macro Lett. 2019, 8, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Gräfe, D.; Wickberg, A.; Zieger, M.M.; Wegener, M.; Blasco, E.; Barner-Kowollik, C. Adding chemically selective subtraction to multi-material 3D additive manufacturing. Nat. Commun. 2018, 9, 2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibfarth, F.A.; Mattson, K.M.; Fors, B.P.; Collins, H.A.; Hawker, C.J. External regulation of controlled polymerizations. Angew. Chem. Int. Ed. 2013, 52, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Teator, A.J.; Lastovickova, D.N.; Bielawski, C.W. Switchable Polymerization Catalysts. Chem. Rev. 2016, 116, 1969–1992. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Festphasen-Synthese (Nobel-Vortrag). Angew. Chem. 1985, 97, 801–812. [Google Scholar] [CrossRef]
- Carmean, R.N.; Figg, C.A.; Becker, T.E.; Sumerlin, B.S. Closed-System One-Pot Block Copolymerization by Temperature-Modulated Monomer Segregation. Angew. Chem. 2016, 128, 8766–8771. [Google Scholar] [CrossRef]
- Corrigan, N.; Ciftci, M.; Jung, K.; Boyer, C. Mediating Reaction Orthogonality in Polymer and Materials Science. Angew. Chem. Int. Ed. 2021, 60, 1748–1781. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Gibson, M.I.; Klok, H.-A. Synthese funktioneller Polymere durch polymeranaloge Reaktionen. Angew. Chem. 2009, 121, 50–60. [Google Scholar] [CrossRef]
- Liu, S.; Dicker, K.T.; Jia, X. Modular and orthogonal synthesis of hybrid polymers and networks. Chem. Commun. 2015, 51, 5218–5237. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.J.; Boydston, A.J. Multimaterial actinic spatial control 3D and 4D printing. Nat. Commun. 2019, 10, 791. [Google Scholar] [CrossRef]
- Kuang, X.; Wu, J.; Chen, K.; Zhao, Z.; Ding, Z.; Hu, F.; Fang, D.; Qi, H.J. Grayscale digital light processing 3D printing for highly functionally graded materials. Sci. Adv. 2019, 5, eaav5790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, G.I.; Schwartz, J.J.; Zhang, D.; Weiss, B.M.; Ganter, M.A.; Storti, D.W.; Boydston, A.J. Production of Materials with Spatially-Controlled Cross-Link Density via Vat Photopolymerization. ACS Appl. Mater. Interfaces 2016, 8, 29037–29043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, J.; Mu, X.; Chen, H.; Qi, H.J.; Fang, D. Origami by frontal photopolymerization. Sci. Adv. 2017, 3, e1602326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wu, J.; Mu, X.; Chen, H.; Qi, H.J.; Fang, D. Desolvation Induced Origami of Photocurable Polymers by Digit Light Processing. Macromol. Rapid Commun. 2017, 38, 1600625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhao, Z.; Kuang, X.; Hamel, C.M.; Fang, D.; Qi, H.J. Reversible shape change structures by grayscale pattern 4D printing. Multifunct. Mater. 2018, 1, 15002. [Google Scholar] [CrossRef]
- Wang, W.; Hutchinson, R.A. Free-Radical Acrylic Polymerization Kinetics at Elevated Temperatures. Chem. Eng. Technol. 2010, 33, 1745–1753. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, X. Multi-material Additive Manufacturing of Metamaterials with Giant, Tailorable Negative Poisson’s Ratios. Sci. Rep. 2018, 8, 9139. [Google Scholar] [CrossRef]
- Wu, X.; Lian, Q.; Li, D.; Jin, Z. Biphasic osteochondral scaffold fabrication using multi-material mask projection stereolithography. Rapid Prototyp. J. 2019, 25, 277–288. [Google Scholar] [CrossRef]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.-K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Orellano, I.; Lam, T.; Noichl, B.; Geiger, M.-A.; Amler, A.-K.; Kreuder, A.-E.; Palmer, C.; Duda, G.; Lauster, R.; et al. Vascular bioprinting with enzymatically degradable bioinks via multi-material projection-based stereolithography. Acta Biomater. 2020, 117, 121–132. [Google Scholar] [CrossRef]
- Miri, A.K.; Nieto, D.; Iglesias, L.; Goodarzi Hosseinabadi, H.; Maharjan, S.; Ruiz-Esparza, G.U.; Khoshakhlagh, P.; Manbachi, A.; Dokmeci, M.R.; Chen, S.; et al. Microfluidics-Enabled Multimaterial Maskless Stereolithographic Bioprinting. Adv. Mater. 2018, 30, e1800242. [Google Scholar] [CrossRef] [PubMed]
- Robles-Martinez, P.; Xu, X.; Trenfield, S.J.; Awad, A.; Goyanes, A.; Telford, R.; Basit, A.W.; Gaisford, S. 3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method. Pharmaceutics 2019, 11, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Robles-Martinez, P.; Madla, C.M.; Joubert, F.; Goyanes, A.; Basit, A.W.; Gaisford, S. Stereolithography (SLA) 3D printing of an antihypertensive polyprintlet: Case study of an unexpected photopolymer-drug reaction. Addit. Manuf. 2020, 33, 101071. [Google Scholar] [CrossRef]
- Arcaute, K.; Zuverza, N.; Mann, B.; Wicker, R. Multi-Material Stereolithography: Spatially-Controlled Bioactive Poly(Ethylene Glycol) Scaffolds for Tissue Engineering. In 2007 International Solid Freeform Fabrication Symposium; The University of Texas at Austin: Austin, TX, USA, 2007. [Google Scholar]
- Mu, X.; Bertron, T.; Dunn, C.; Qiao, H.; Wu, J.; Zhao, Z.; Saldana, C.; Qi, H.J. Porous polymeric materials by 3D printing of photocurable resin. Mater. Horiz. 2017, 4, 442–449. [Google Scholar] [CrossRef]
- Arcaute, K.; Mann, B.; Wicker, R. Stereolithography of spatially controlled multi-material bioactive poly(ethylene glycol) scaffolds. Acta Biomater. 2010, 6, 1047–1054. [Google Scholar] [CrossRef]
- Raman, R.; Bhaduri, B.; Mir, M.; Shkumatov, A.; Lee, M.K.; Popescu, G.; Kong, H.; Bashir, R. High-Resolution Projection Microstereolithography for Patterning of Neovasculature. Adv. Healthc. Mater. 2016, 5, 610–619. [Google Scholar] [CrossRef]
- Arcaute, K.; Mann, B.K.; Wicker, R.B. Stereolithography of three-dimensional bioactive poly(ethylene glycol) constructs with encapsulated cells. Ann. Biomed. Eng. 2006, 34, 1429–1441. [Google Scholar] [CrossRef]
- Tiller, B.; Reid, A.; Zhu, B.; Guerreiro, J.; Domingo-Roca, R.; Curt Jackson, J.; Windmill, J. Piezoelectric microphone via a digital light processing 3D printing process. Mater. Des. 2019, 165, 107593. [Google Scholar] [CrossRef]
- Chan, V.; Zorlutuna, P.; Jeong, J.H.; Kong, H.; Bashir, R. Three-dimensional photopatterning of hydrogels using stereolithography for long-term cell encapsulation. Lab Chip 2010, 10, 2062–2070. [Google Scholar] [CrossRef]
- Chan, V.; Jeong, J.H.; Bajaj, P.; Collens, M.; Saif, T.; Kong, H.; Bashir, R. Multi-material bio-fabrication of hydrogel cantilevers and actuators with stereolithography. Lab Chip 2012, 12, 88–98. [Google Scholar] [CrossRef]
- Mandon, C.A.; Blum, L.J.; Marquette, C.A. Adding Biomolecular Recognition Capability to 3D Printed Objects. Anal. Chem. 2016, 88, 10767–10772. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Mapili, G.; Suhali, G.; Chen, S.; Roy, K. A digital micro-mirror device-based system for the microfabrication of complex, spatially patterned tissue engineering scaffolds. J. Biomed. Mater. Res. A 2006, 77, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zorlutuna, P.; Jeong, J.H.; Kong, H.; Bashir, R. Stereolithography-Based Hydrogel Microenvironments to Examine Cellular Interactions. Adv. Funct. Mater. 2011, 21, 3642–3651. [Google Scholar] [CrossRef]
- Grigoryan, B.; Sazer, D.W.; Avila, A.; Albritton, J.L.; Padhye, A.; Ta, A.H.; Greenfield, P.T.; Gibbons, D.L.; Miller, J.S. Development, characterization, and applications of multi-material stereolithography bioprinting. Sci. Rep. 2021, 11, 3171. [Google Scholar] [CrossRef]
- Patel, G.C.; Parmar, V.K.; Patel, P.S. Stimuli-responsive polymers for ocular therapy. In Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications. Volume 2, Advanced Nanocarriers for Therapeutics; Salam Hamdy Makhlouf, A., Abu-Thabit, N.Y., Eds.; Woodhead Publishing: Cambridge, MA, USA, 2019; pp. 463–489. ISBN 9780081019955. [Google Scholar]
- Thrasher, C.J.; Schwartz, J.J.; Boydston, A.J. Modular Elastomer Photoresins for Digital Light Processing Additive Manufacturing. ACS Appl. Mater. Interfaces 2017, 9, 39708–39716. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lu, Z.; Chester, S.A.; Lee, H. Micro 3D Printing of a Temperature-Responsive Hydrogel Using Projection Micro-Stereolithography. Sci. Rep. 2018, 8, 1963. [Google Scholar] [CrossRef]
- Odent, J.; Vanderstappen, S.; Toncheva, A.; Pichon, E.; Wallin, T.J.; Wang, K.; Shepherd, R.F.; Dubois, P.; Raquez, J.-M. Hierarchical chemomechanical encoding of multi-responsive hydrogel actuators via 3D printing. J. Mater. Chem. A 2019, 7, 15395–15403. [Google Scholar] [CrossRef]
- Alabiso, W.; Schlögl, S. The Impact of Vitrimers on the Industry of the Future: Chemistry, Properties and Sustainable Forward-Looking Applications. Polymers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Zhang, B.; Kowsari, K.; Serjouei, A.; Dunn, M.L.; Ge, Q. Reprocessable thermosets for sustainable three-dimensional printing. Nat. Commun. 2018, 9, 1831. [Google Scholar] [CrossRef] [Green Version]
- Giebler, M.; Alabiso, W.; Wieser, V.; Radl, S.; Schlögl, S. Photopatternable and Rewritable Epoxy-Anhydride Vitrimers. Macromol. Rapid Commun. 2021, 42, e2000466. [Google Scholar] [CrossRef]
- Denissen, W.; Droesbeke, M.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers. Nat. Commun. 2017, 8, 14857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically activated, catalyst-free polyhydroxyurethane vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef] [PubMed]
- Bähr, F.; Westkämper, E. Correlations between Influencing Parameters and Quality Properties of Components Produced by Fused Deposition Modeling. Procedia CIRP 2018, 72, 1214–1219. [Google Scholar] [CrossRef]
- Kallel, A.; Koutiri, I.; Babaeitorkamani, E.; Khavandi, A.; Tamizifar, M.; Shirinbayan, M.; Tcharkhtchi, A. Study of Bonding Formation between the Filaments of PLA in FFF Process. Int. Polym. Process. 2019, 34, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Bellehumeur, C.; Li, L.; Sun, Q.; Gu, P. Modeling of Bond Formation Between Polymer Filaments in the Fused Deposition Modeling Process. J. Manuf. Process. 2004, 6, 170–178. [Google Scholar] [CrossRef]
- Costa, S.F.; Duarte, F.M.; Covas, J.A. Estimation of filament temperature and adhesion development in fused deposition techniques. J. Mater. Process. Technol. 2017, 245, 167–179. [Google Scholar] [CrossRef]
- Yang, C.; Tian, X.; Li, D.; Cao, Y.; Zhao, F.; Shi, C. Influence of thermal processing conditions in 3D printing on the crystallinity and mechanical properties of PEEK material. J. Mater. Process. Technol. 2017, 248, 1–7. [Google Scholar] [CrossRef]
- Oropallo, W.; Piegl, L.A. Ten challenges in 3D printing. Eng. Comput. 2016, 32, 135–148. [Google Scholar] [CrossRef]
- Flores Ituarte, I.; Kretzschmar, N.; Chekurov, S.; Partanen, J.; Tuomi, J. Additive Manufacturing Validation Methods, Technology Transfer Based on Case Studies. In Additive Manufacturing—Developments in Training and Education; Springer: Cham, Switzerland, 2019; pp. 99–112. [Google Scholar]
- Formslab. FDM vs. SLA: Compare Filament and Resin 3D Printers. Available online: https://formlabs.com/blog/fdm-vs-sla-compare-types-of-3d-printers/ (accessed on 6 May 2022).
- Anycubic. Commercial 3D-Printers. Available online: https://www.anycubic.com/collections/new-arrival/products/photon-m3-max (accessed on 6 May 2022).
- Bialas, S.; Michalek, L.; Marschner, D.E.; Krappitz, T.; Wegener, M.; Blinco, J.; Blasco, E.; Frisch, H.; Barner-Kowollik, C. Access to Disparate Soft Matter Materials by Curing with Two Colors of Light. Adv. Mater. 2019, 31, e1807288. [Google Scholar] [CrossRef]
- Frey, H.; Johann, T. Celebrating 100 years of “polymer science”: Hermann Staudinger’s 1920 manifesto. Polym. Chem. 2020, 11, 8–14. [Google Scholar] [CrossRef]
- Liang, Y.; Kiick, K.L. Multifunctional lipid-coated polymer nanogels crosslinked by photo-triggered Michael-type addition. Polym. Chem. 2014, 5, 1728–1736. [Google Scholar] [CrossRef] [Green Version]
- Zieger, M.M.; Müller, P.; Blasco, E.; Petit, C.; Hahn, V.; Michalek, L.; Mutlu, H.; Wegener, M.; Barner-Kowollik, C. A Subtractive Photoresist Platform for Micro- and Macroscopic 3D Printed Structures. Adv. Funct. Mater. 2018, 28, 1801405. [Google Scholar] [CrossRef]
- Worrell, B.T.; McBride, M.K.; Lyon, G.B.; Cox, L.M.; Wang, C.; Mavila, S.; Lim, C.-H.; Coley, H.M.; Musgrave, C.B.; Ding, Y.; et al. Bistable and photoswitchable states of matter. Nat. Commun. 2018, 9, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xi, W.; Huang, S.; Long, K.; Bowman, C.N. Wavelength-Selective Sequential Polymer Network Formation Controlled with a Two-Color Responsive Initiation System. Macromolecules 2017, 50, 5652–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenor, S.R.; Shultz, A.R.; Love, B.J.; Long, T.E. Coumarins in polymers: From light harvesting to photo-cross-linkable tissue scaffolds. Chem. Rev. 2004, 104, 3059–3077. [Google Scholar] [CrossRef]
- Huyck, R.H.; Trenor, S.R.; Love, B.J.; Long, T.E. Photodimerization of Coumarin Functionalized Poly(alkyl Acrylate) and Poly(alkyl Methacrylate) Random Copolymers: Influence of Copolymer Composition on Photocrosslinking. J. Macromol. Sci. Part A 2007, 45, 9–15. [Google Scholar] [CrossRef]
- Furuta, T.; Wang, S.S.; Dantzker, J.L.; Dore, T.M.; Bybee, W.J.; Callaway, E.M.; Denk, W.; Tsien, R.Y. Brominated 7-hydroxycoumarin-4-ylmethyls: Photolabile protecting groups with biologically useful cross-sections for two photon photolysis. Proc. Natl. Acad. Sci. USA 1999, 96, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Gnanaguru, K.; Ramasubbu, N.; Venkatesan, K.; Ramamurthy, V. A study on the photochemical dimerization of coumarins in the solid state. J. Org. Chem. 1985, 50, 2337–2346. [Google Scholar] [CrossRef]
- Cazin, I.; Rossegger, E.; La Guedes de Cruz, G.; Griesser, T.; Schlögl, S. Recent Advances in Functional Polymers Containing Coumarin Chromophores. Polymers 2020, 13, 56. [Google Scholar] [CrossRef]
- Govindarajan, S.R.; Jain, T.; Choi, J.-W.; Joy, A.; Isayeva, I.; Vorvolakos, K. A hydrophilic coumarin-based polyester for ambient-temperature initiator-free 3D printing: Chemistry, rheology and interface formation. Polymer 2018, 152, 9–17. [Google Scholar] [CrossRef]
- Truong, V.X.; Li, F.; Forsythe, J.S. Versatile Bioorthogonal Hydrogel Platform by Catalyst-Free Visible Light Initiated Photodimerization of Anthracene. ACS Macro Lett. 2017, 6, 657–662. [Google Scholar] [CrossRef]
- Claus, T.K.; Telitel, S.; Welle, A.; Bastmeyer, M.; Vogt, A.P.; Delaittre, G.; Barner-Kowollik, C. Light-driven reversible surface functionalization with anthracenes: Visible light writing and mild UV erasing. Chem. Commun. 2017, 53, 1599–1602. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Desvergne, J.-P.; Castellan, A.; Lapouyade, R. Photodimerization of anthracenes in fluid solution: Structural aspects. Chem. Soc. Rev. 2000, 29, 43–55. [Google Scholar] [CrossRef]
- Kaiser, S.; Radl, S.V.; Manhart, J.; Ayalur-Karunakaran, S.; Griesser, T.; Moser, A.; Ganser, C.; Teichert, C.; Kern, W.; Schlögl, S. Switching “on” and “off” the adhesion in stimuli-responsive elastomers. Soft Matter 2018, 14, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Manhart, J.; Ayalur-Karunakaran, S.; Radl, S.; Oesterreicher, A.; Moser, A.; Ganser, C.; Teichert, C.; Pinter, G.; Kern, W.; Griesser, T.; et al. Design and application of photo-reversible elastomer networks by using the [4πs+4πs] cycloaddition reaction of pendant anthracene groups. Polymer 2016, 102, 10–20. [Google Scholar] [CrossRef]
- Gernhardt, M.; Frisch, H.; Welle, A.; Jones, R.; Wegener, M.; Blasco, E.; Barner-Kowollik, C. Multi-material 3D microstructures with photochemically adaptive mechanical properties. J. Mater. Chem. C 2020, 8, 10993–11000. [Google Scholar] [CrossRef]
- Romano, A.; Roppolo, I.; Rossegger, E.; Schlögl, S.; Sangermano, M. Recent Trends in Applying Rrtho-Nitrobenzyl Esters for the Design of Photo-Responsive Polymer Networks. Materials 2020, 13, 2777. [Google Scholar] [CrossRef]
- Batchelor, R.; Messer, T.; Hippler, M.; Wegener, M.; Barner-Kowollik, C.; Blasco, E. Two in One: Light as a Tool for 3D Printing and Erasing at the Microscale. Adv. Mater. 2019, 31, e1904085. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Tong, X.; Morris, D.; Zhao, Y. Toward Photocontrolled Release Using Light-Dissociable Block Copolymer Micelles. Macromolecules 2006, 39, 4633–4640. [Google Scholar] [CrossRef]
- Johnson, J.A.; Lu, Y.Y.; Burts, A.O.; Lim, Y.-H.; Finn, M.G.; Koberstein, J.T.; Turro, N.J.; Tirrell, D.A.; Grubbs, R.H. Core-clickable PEG-branch-azide bivalent-bottle-brush polymers by ROMP: Grafting-through and clicking-to. J. Am. Chem. Soc. 2011, 133, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Rossegger, E.; Hennen, D.; Griesser, T.; Roppolo, I.; Schlögl, S. Directed motion of water droplets on multi-gradient photopolymer surfaces. Polym. Chem. 2019, 10, 1882–1893. [Google Scholar] [CrossRef]
- Rossegger, E.; Nees, D.; Turisser, S.; Radl, S.; Griesser, T.; Schlögl, S. Photo-switching of surface wettability on micropatterned photopolymers for fast transport of water droplets over a long-distance. Polym. Chem. 2020, 11, 3125–3135. [Google Scholar] [CrossRef]
- Radl, S.V.; Schipfer, C.; Kaiser, S.; Moser, A.; Kaynak, B.; Kern, W.; Schlögl, S. Photo-responsive thiol–ene networks for the design of switchable polymer patterns. Polym. Chem. 2017, 8, 1562–1572. [Google Scholar] [CrossRef]
- Romano, A.; Angelini, A.; Rossegger, E.; Palmara, G.; Castellino, M.; Frascella, F.; Chiappone, A.; Chiadò, A.; Sangermano, M.; Schlögl, S.; et al. Laser-Triggered Writing and Biofunctionalization of Thiol-Ene Networks. Macromol. Rapid Commun. 2020, 41, e2000084. [Google Scholar] [CrossRef] [PubMed]
- Krappitz, T.; Feist, F.; Lamparth, I.; Moszner, N.; John, H.; Blinco, J.P.; Dargaville, T.R.; Barner-Kowollik, C. Polymer networks based on photo-caged diene dimerization. Mater. Horiz. 2019, 6, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Marschner, D.E.; Frisch, H.; Offenloch, J.T.; Tuten, B.T.; Becer, C.R.; Walther, A.; Goldmann, A.S.; Tzvetkova, P.; Barner-Kowollik, C. Visible Light [2 + 2] Cycloadditions for Reversible Polymer Ligation. Macromolecules 2018, 51, 3802–3807. [Google Scholar] [CrossRef]
- Alves, J.; Krappitz, T.; Feist, F.; Blinco, J.P.; Barner-Kowollik, C. Combining Photodeprotection and Ligation into a Dual-Color Gated Reaction System. Chemistry 2020, 26, 16985–16989. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

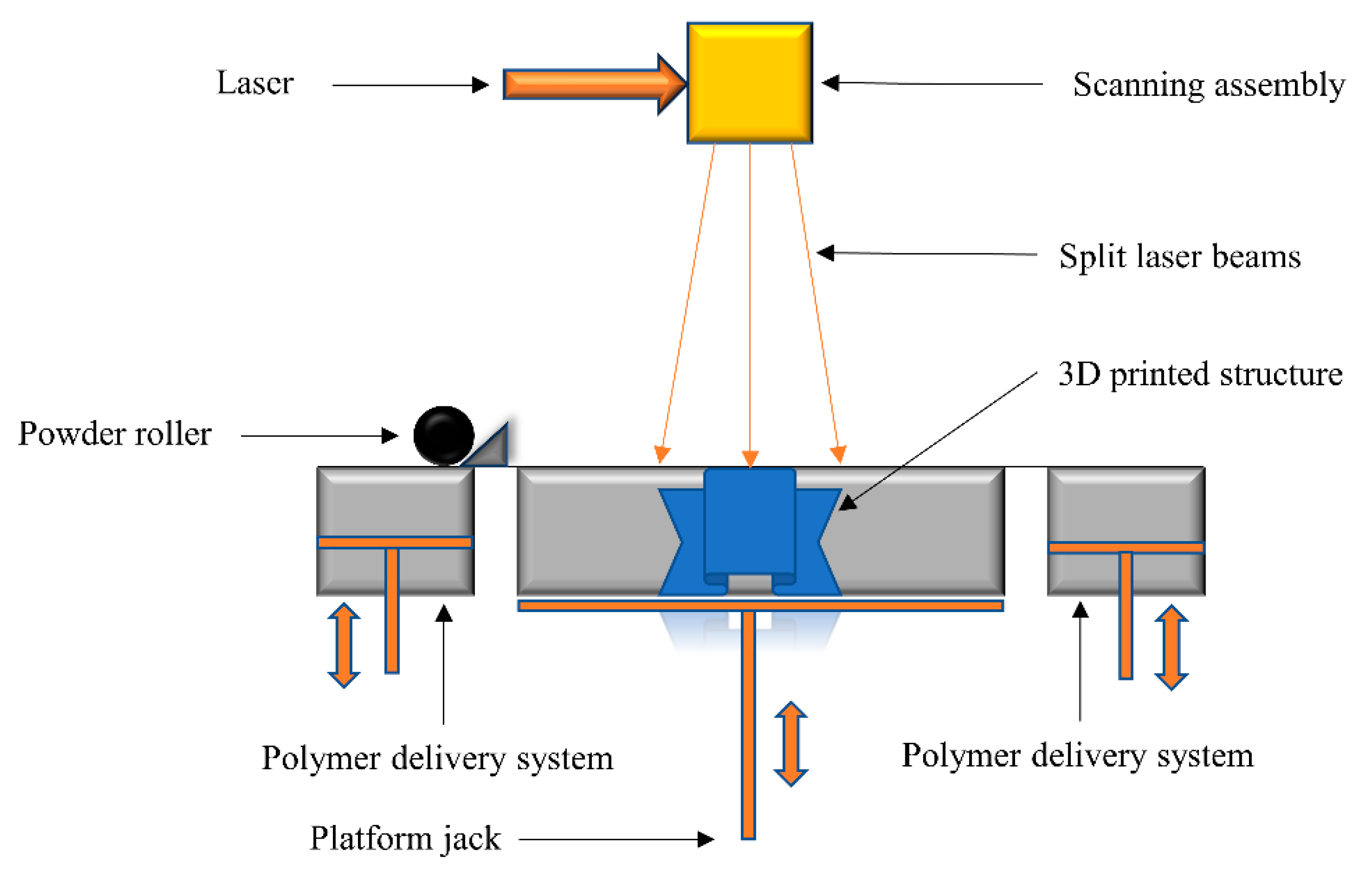

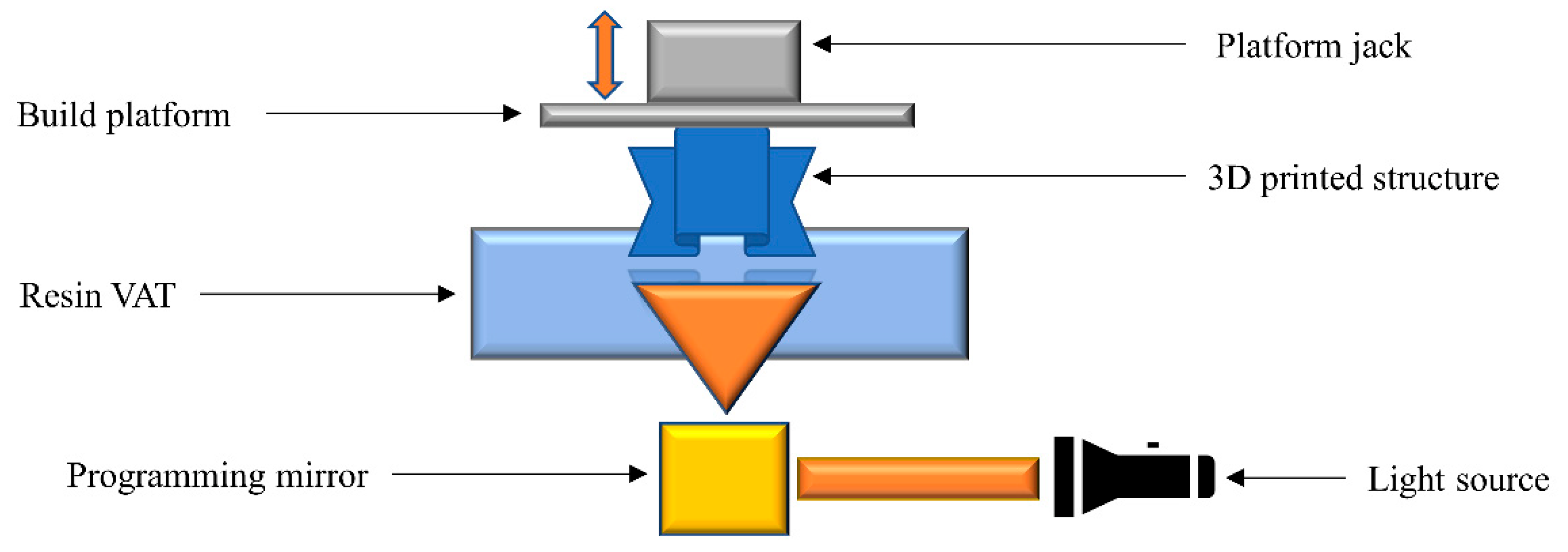
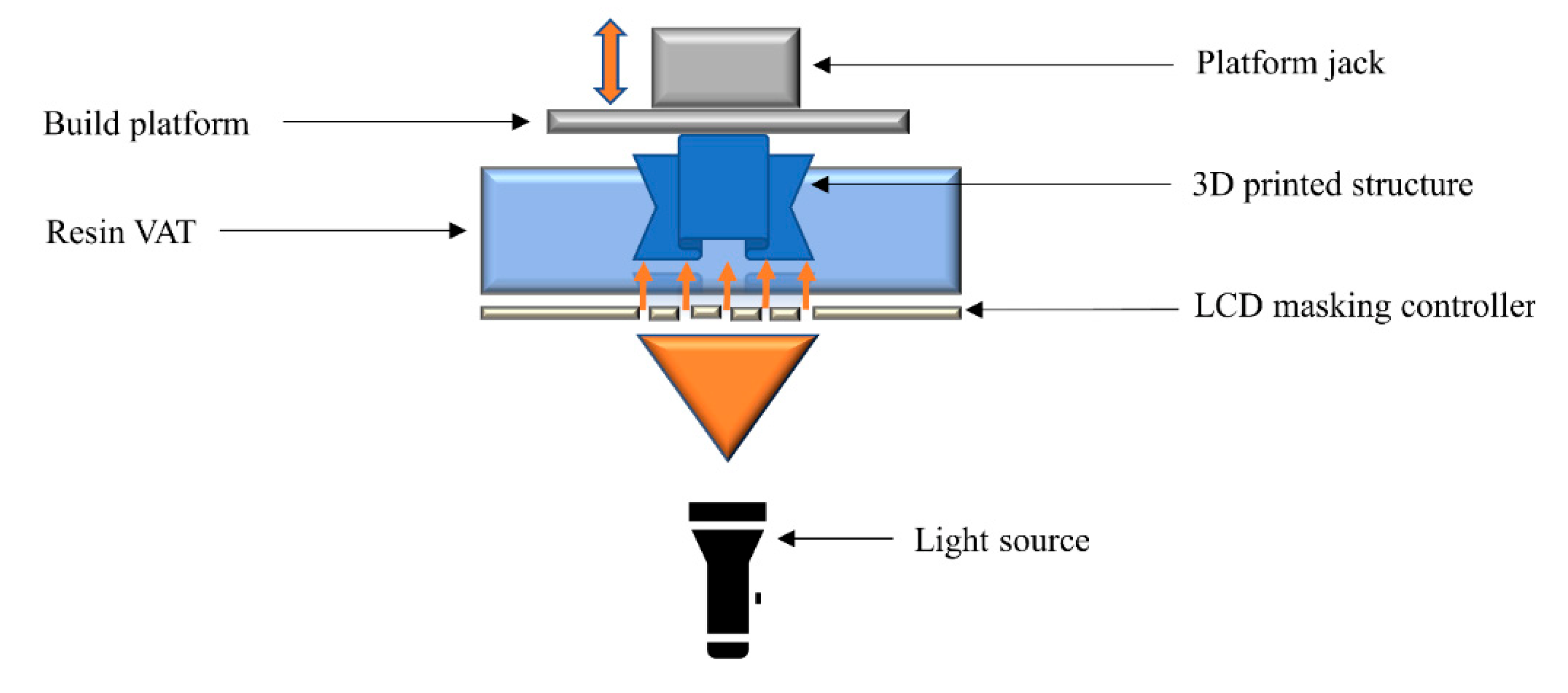
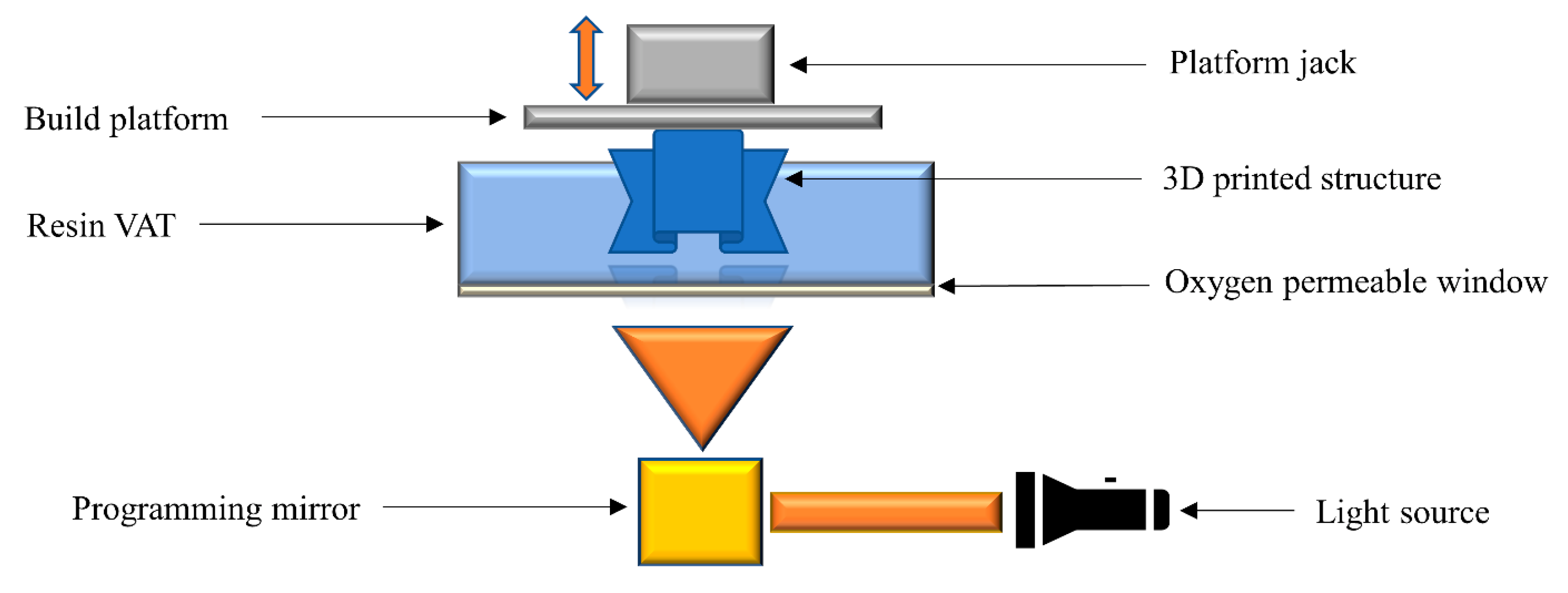
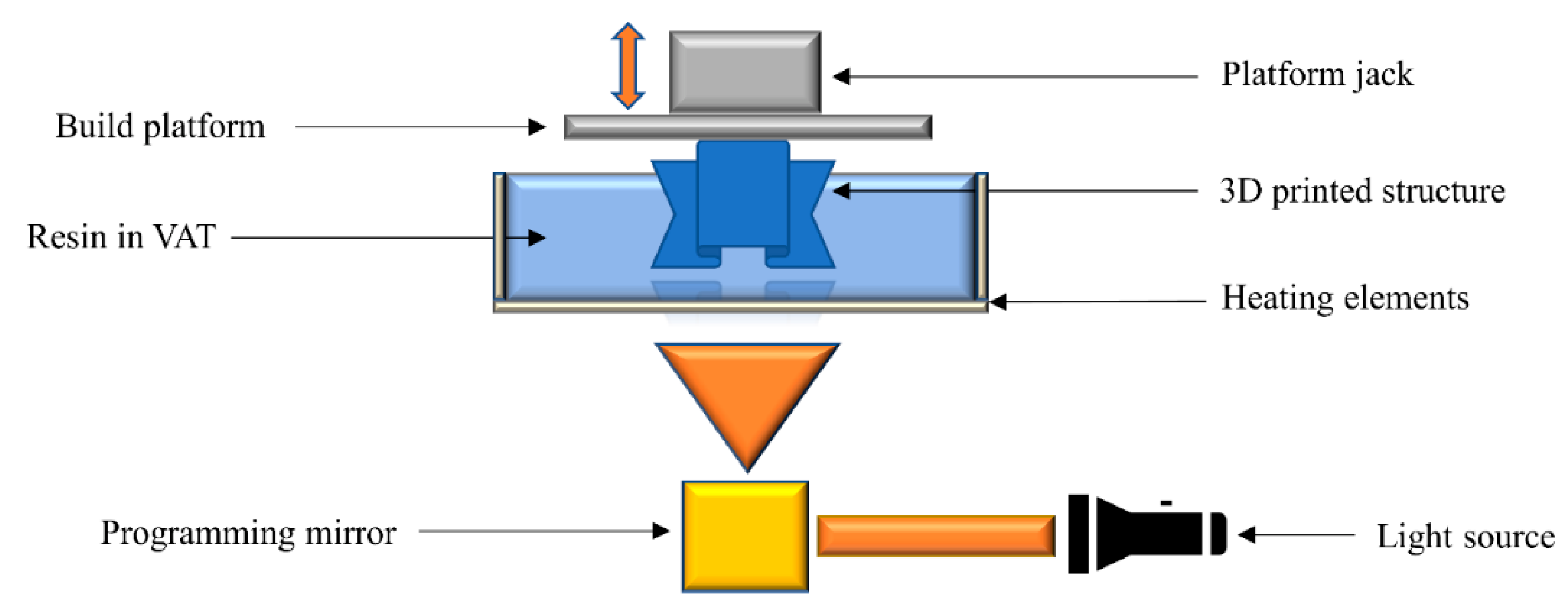
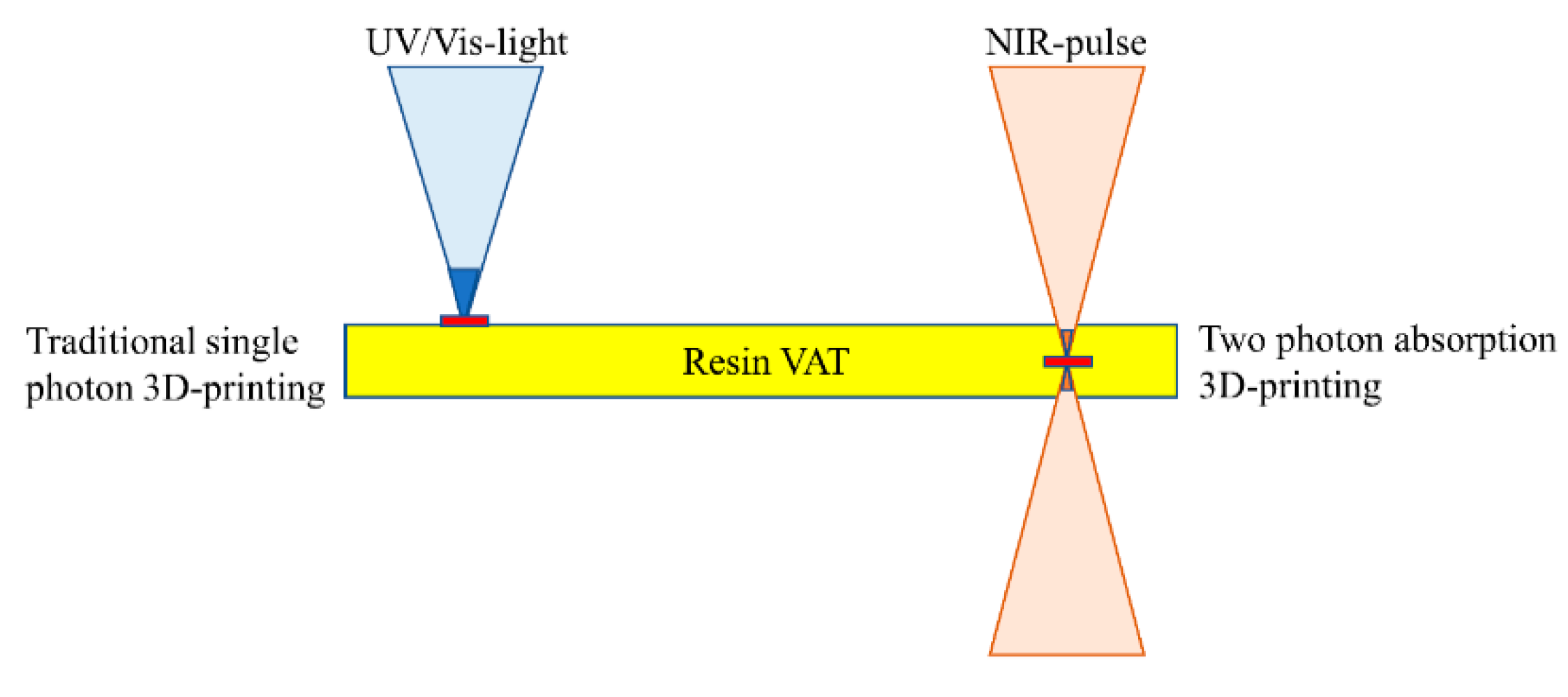

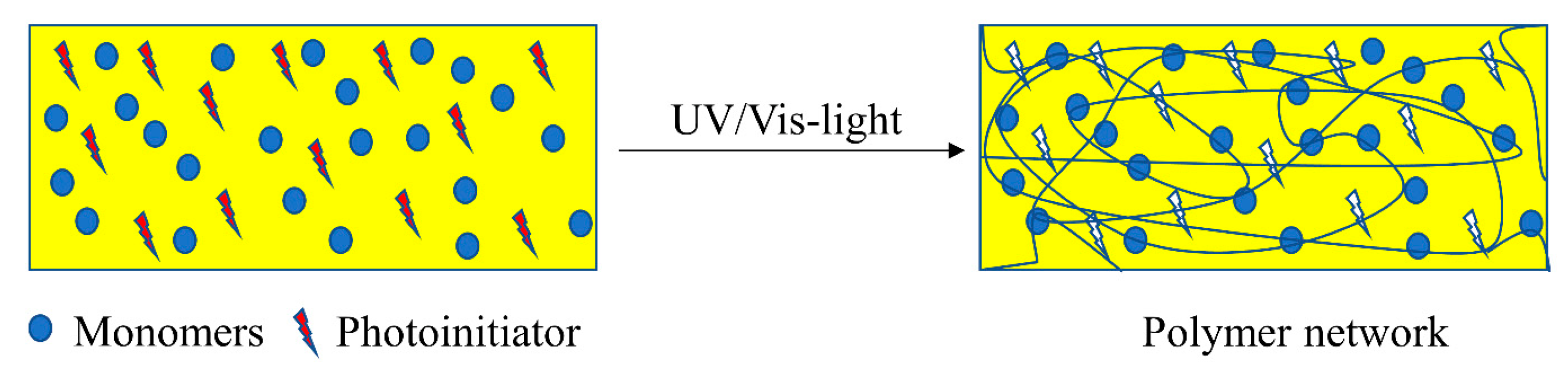
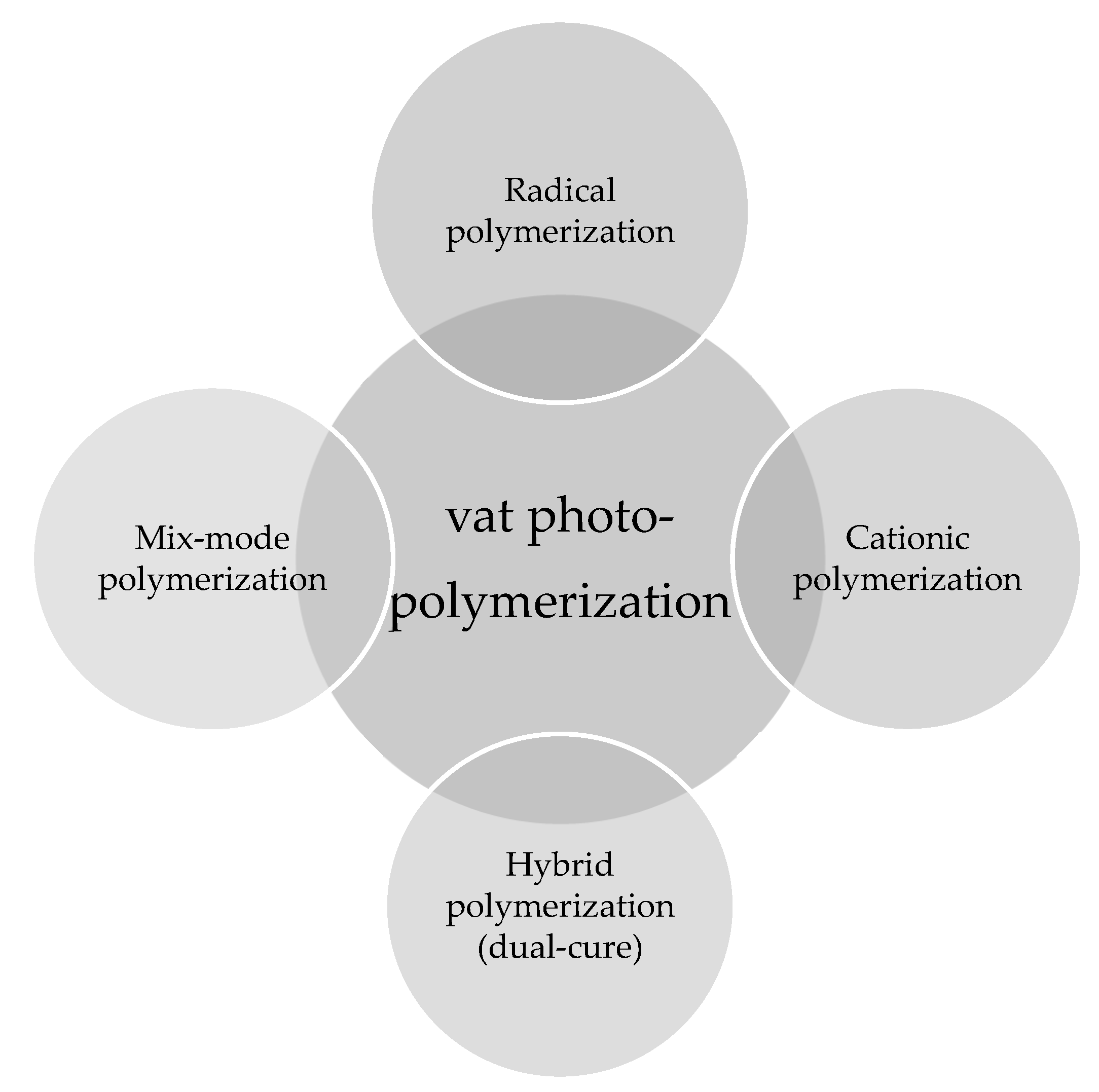
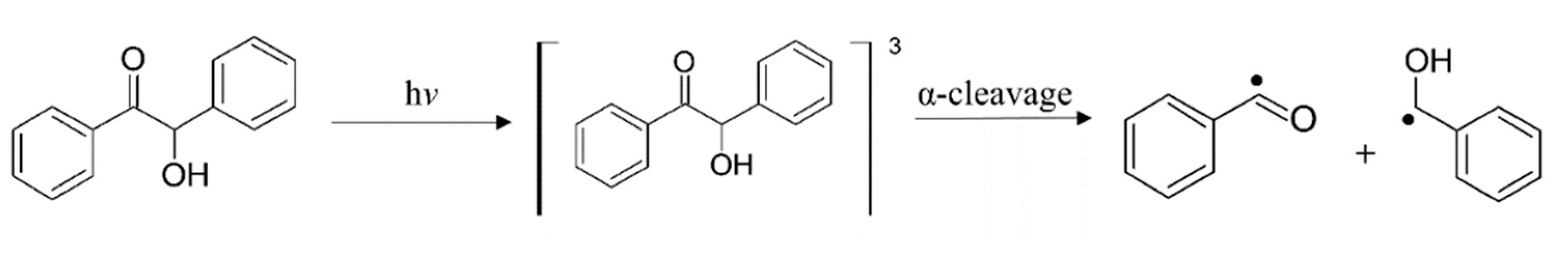
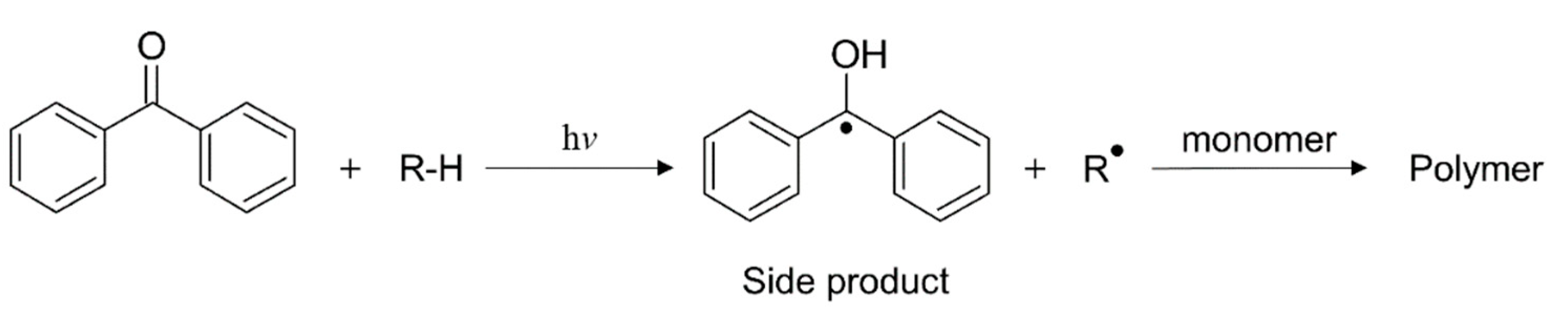
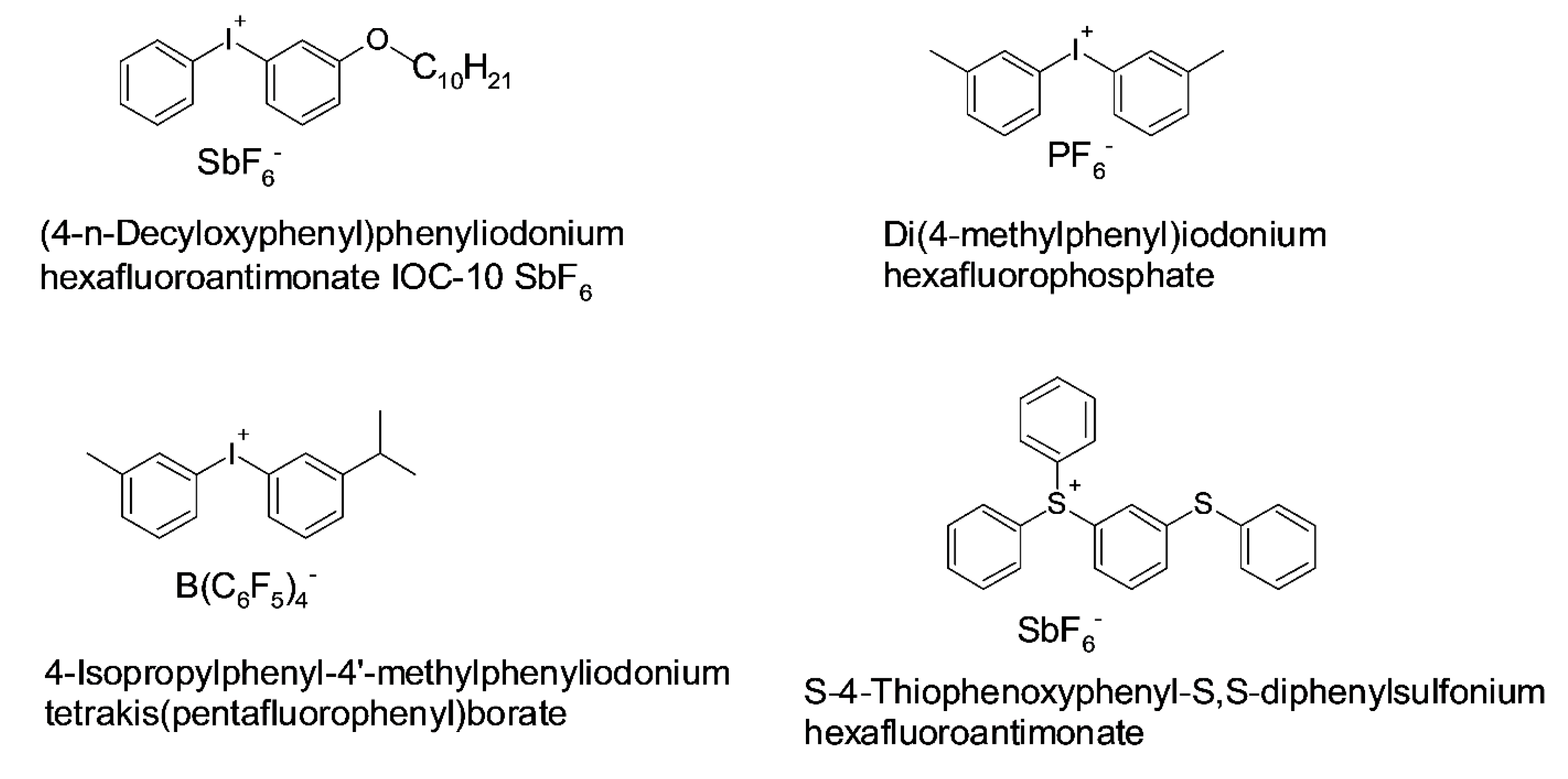
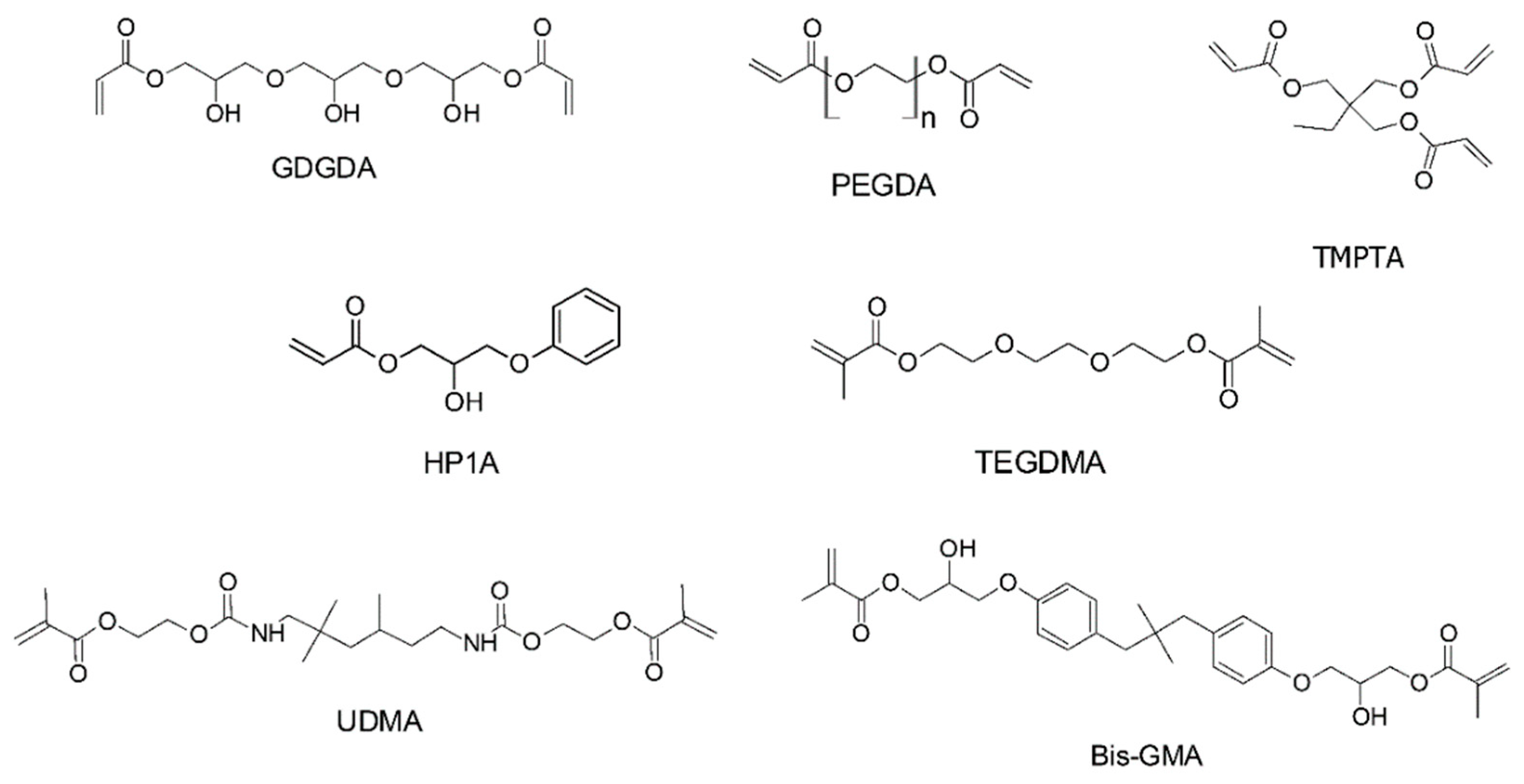
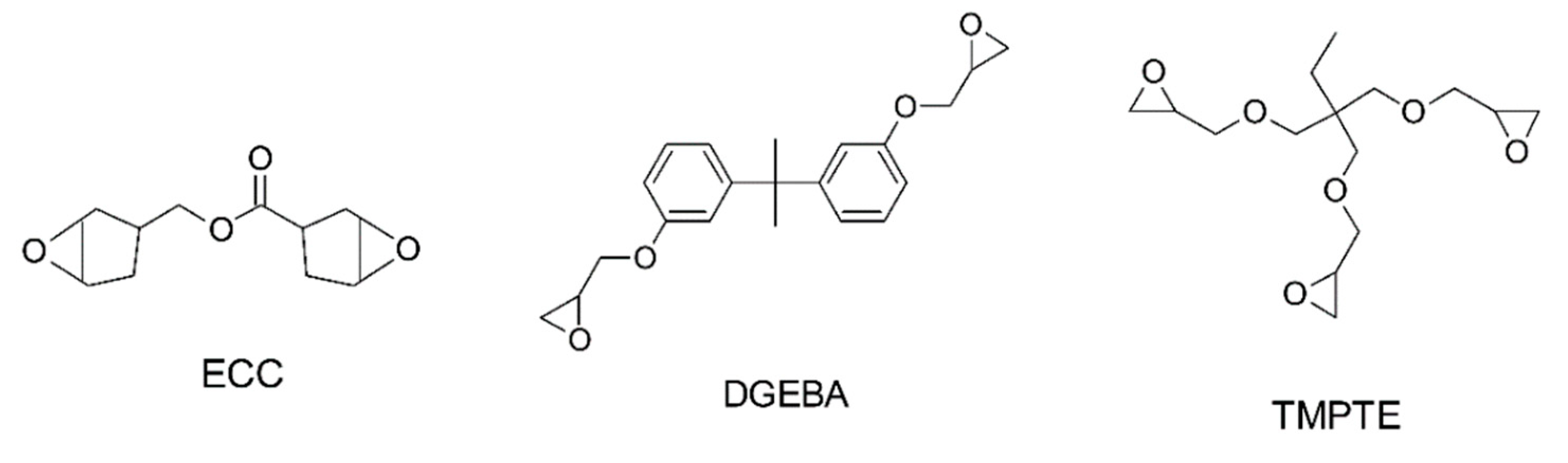
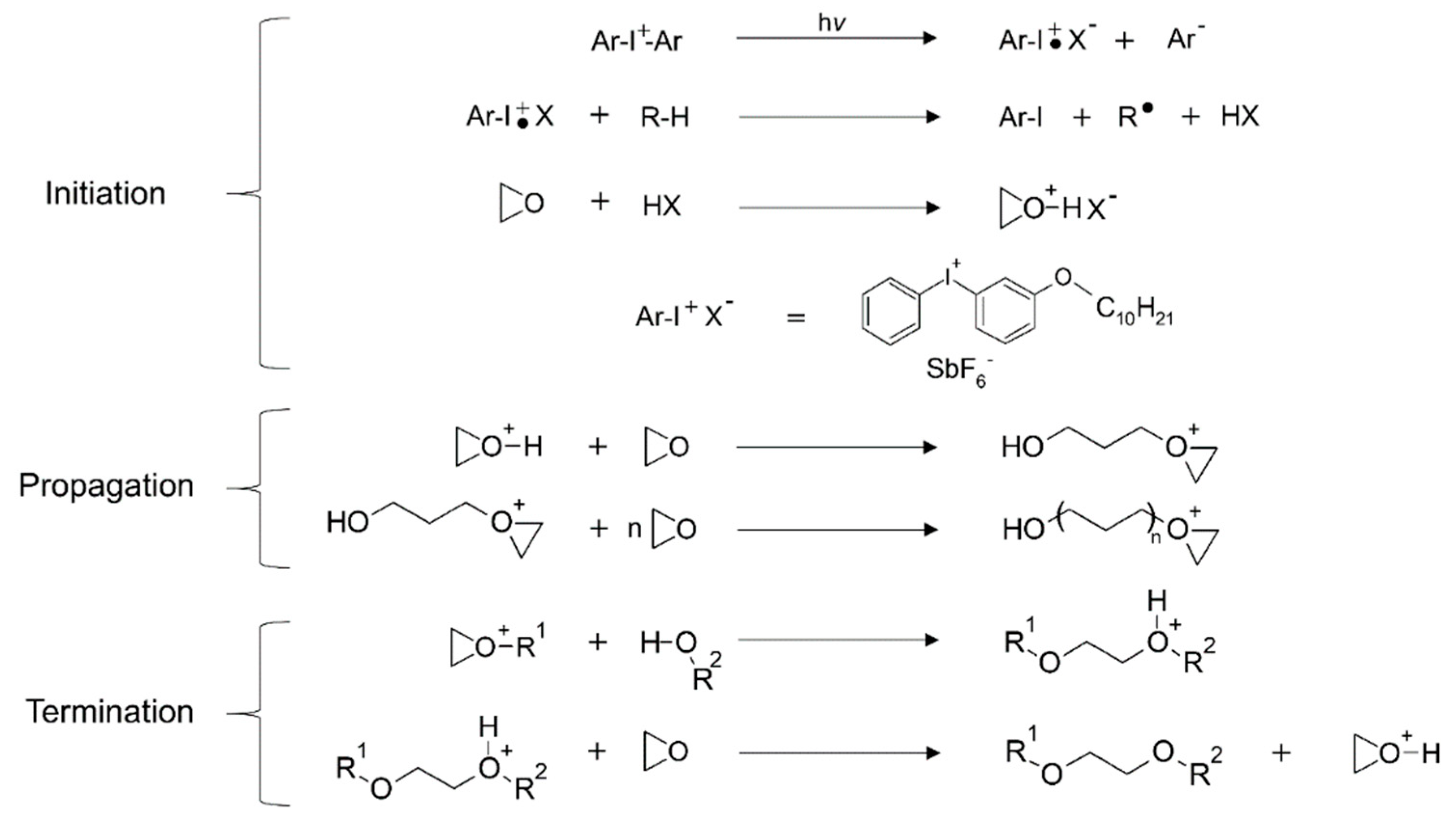

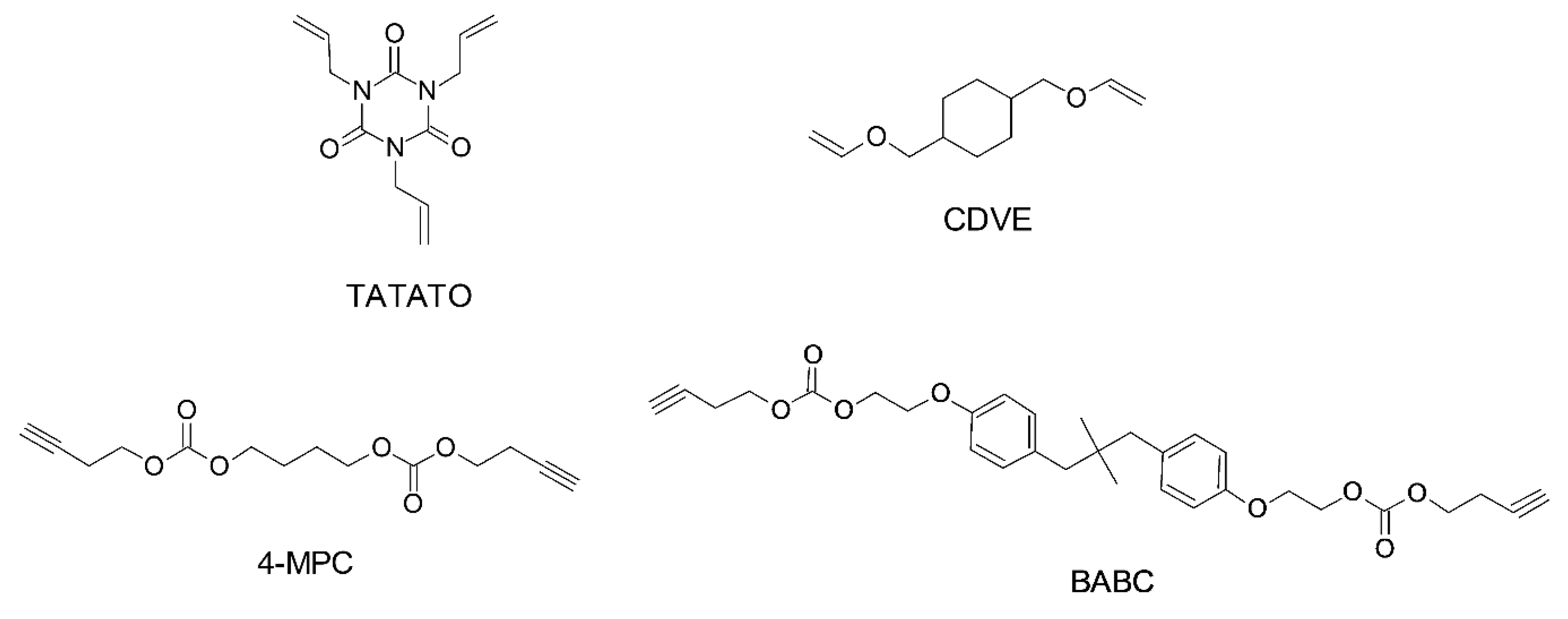

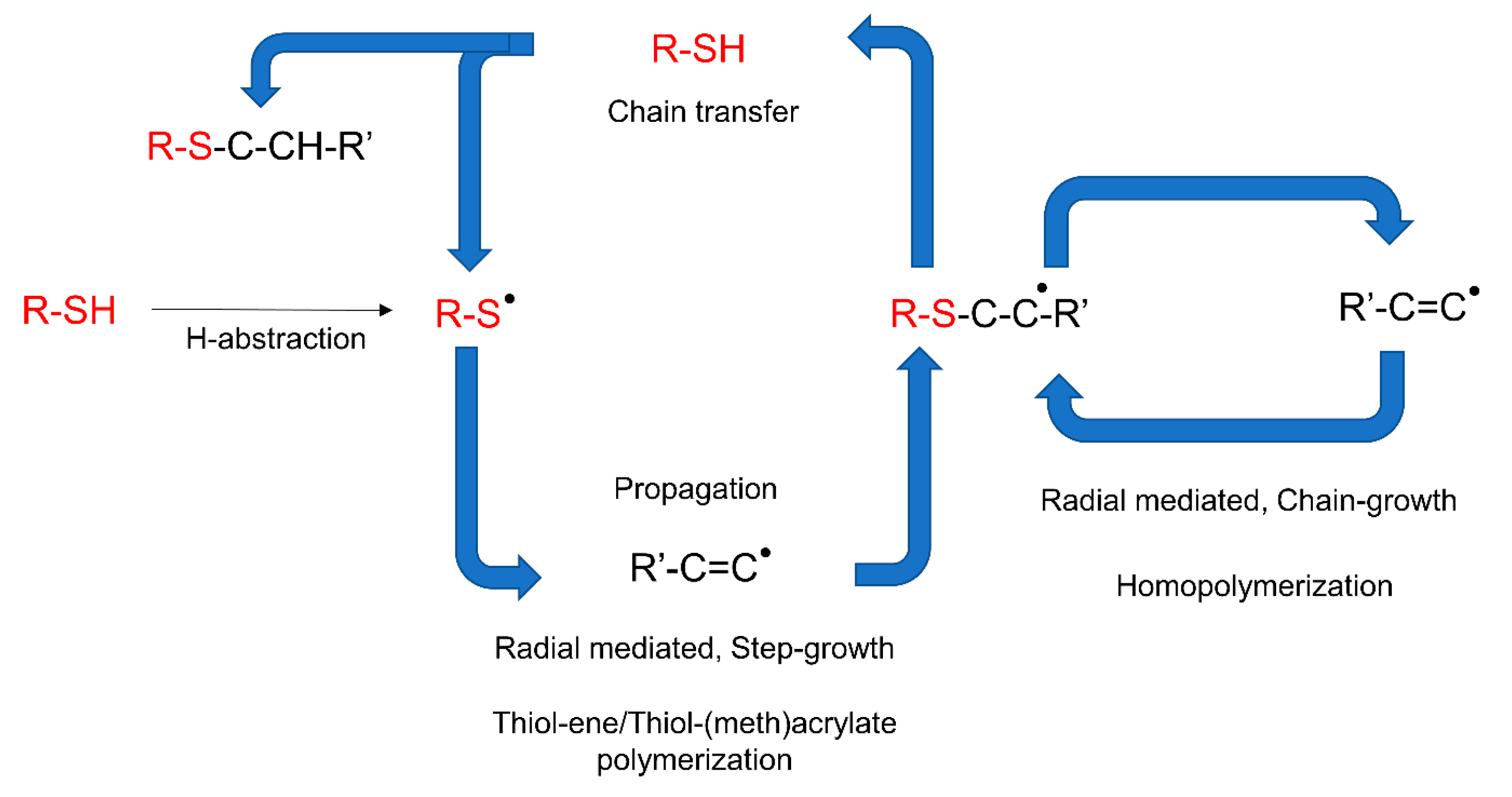
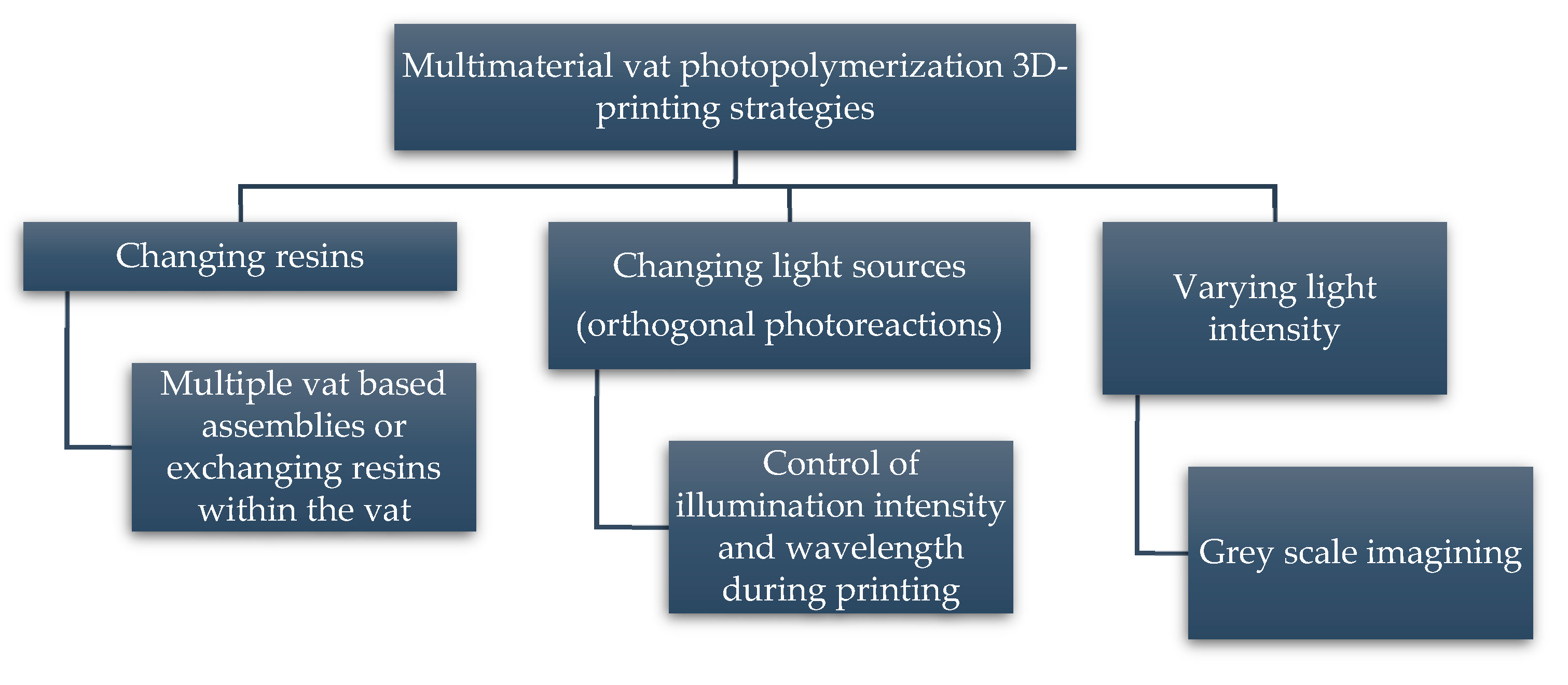
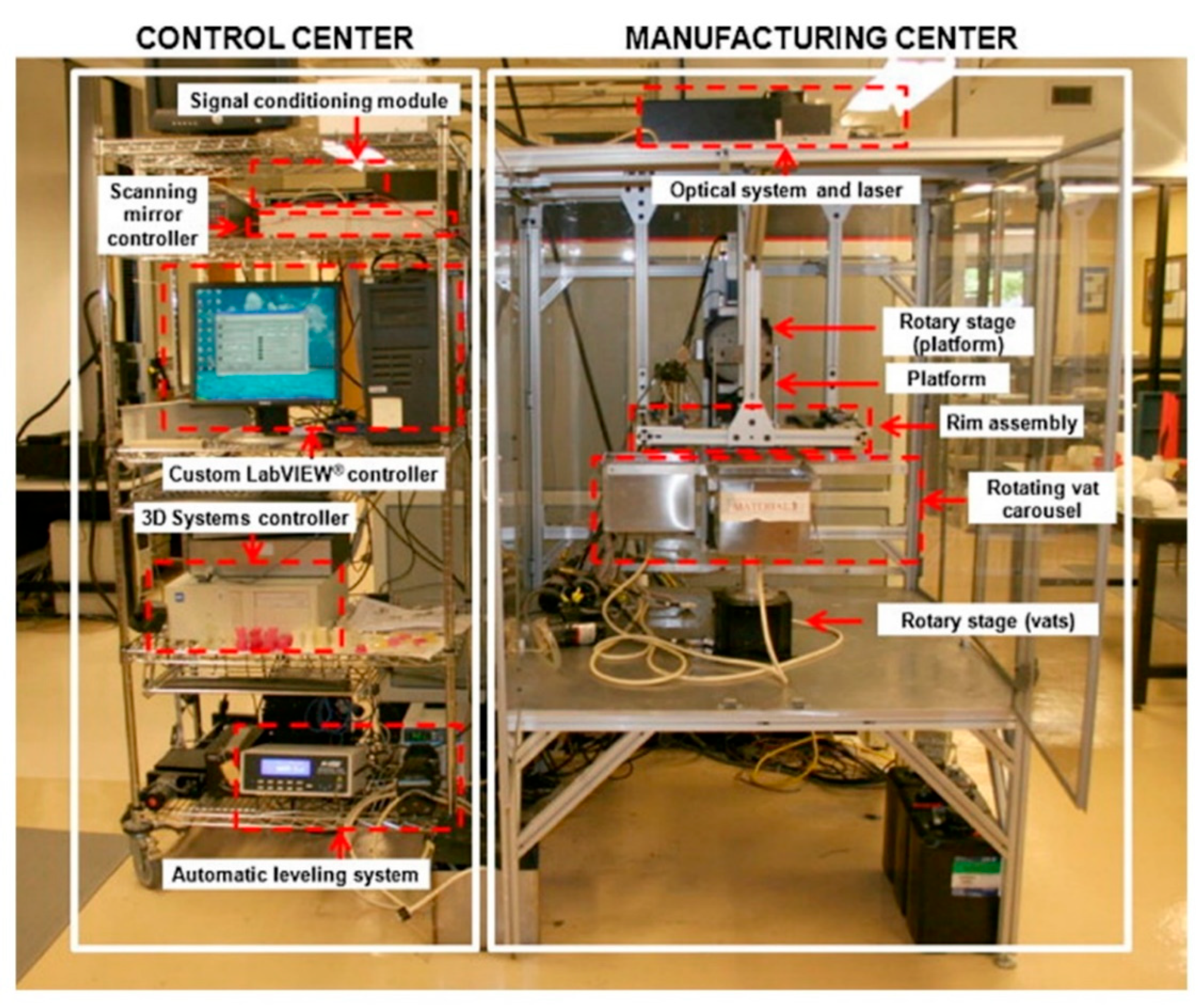

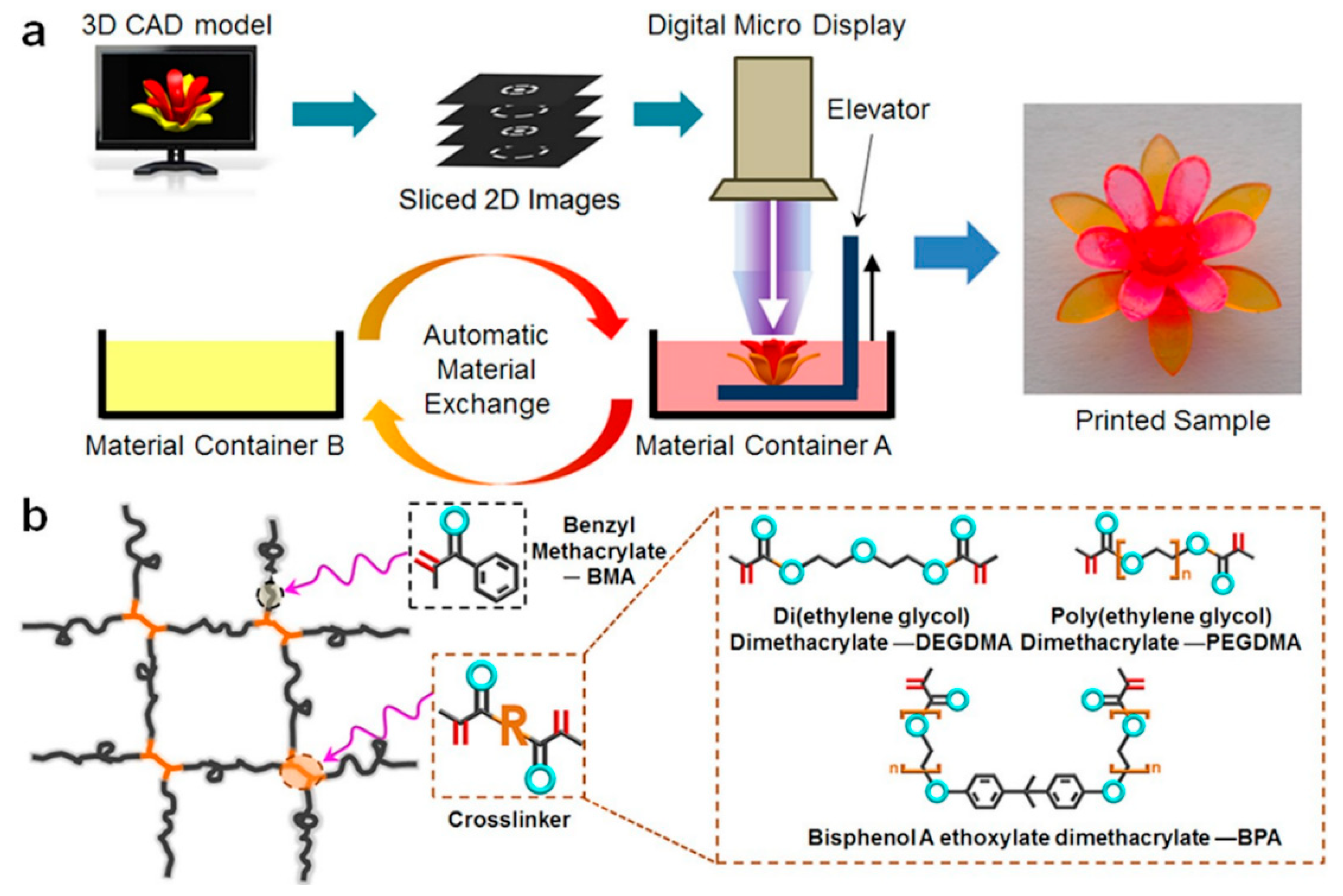
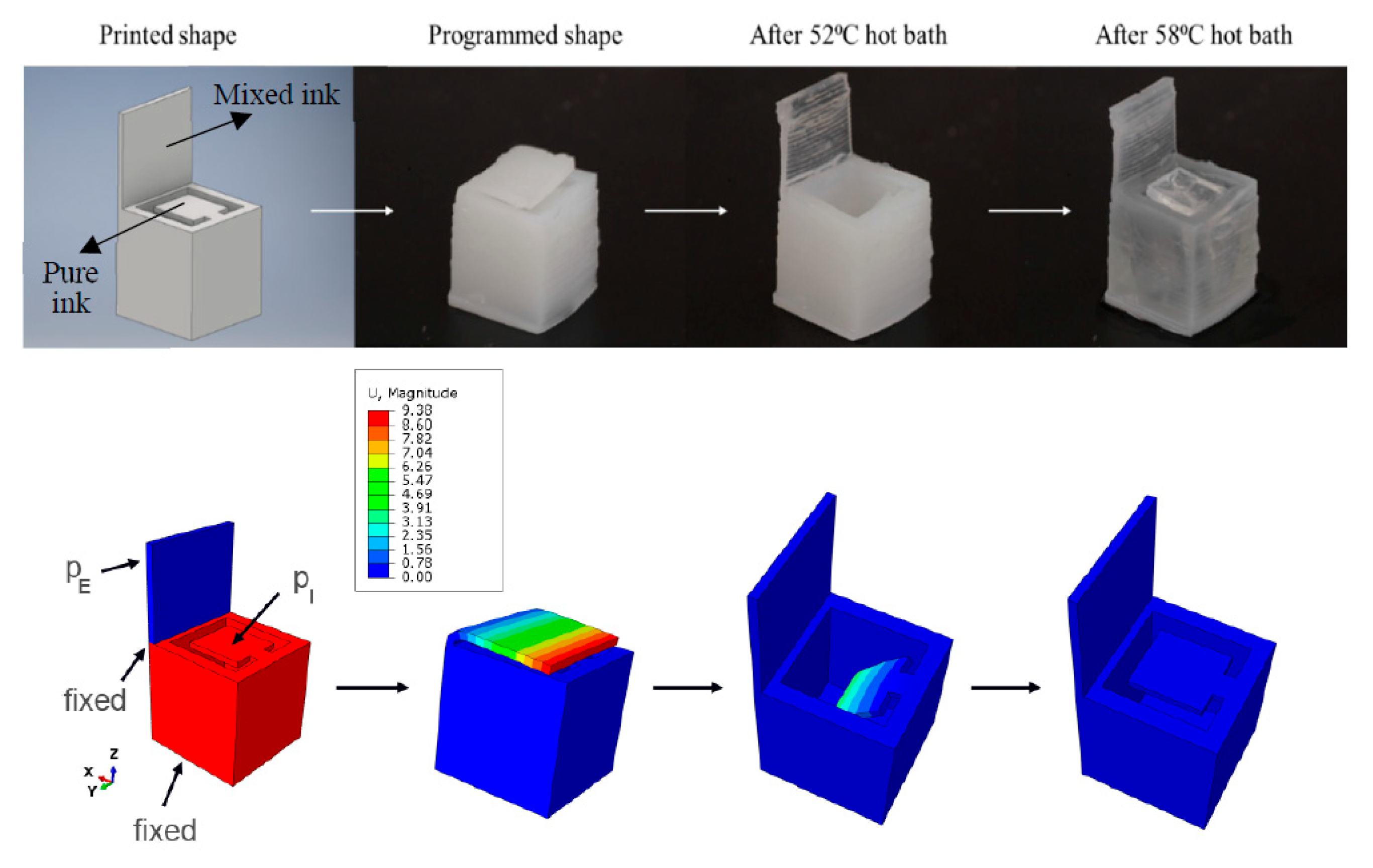
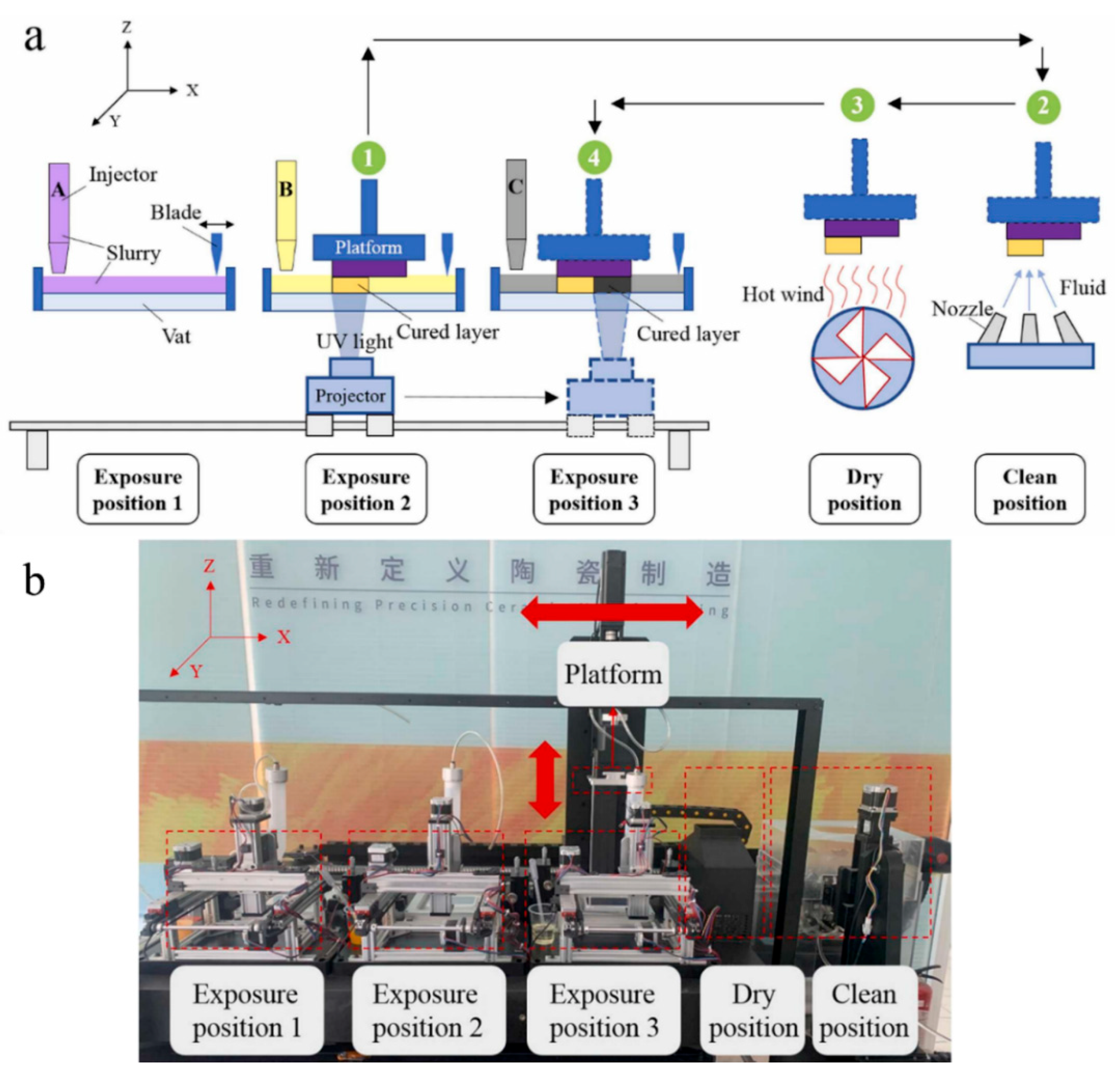

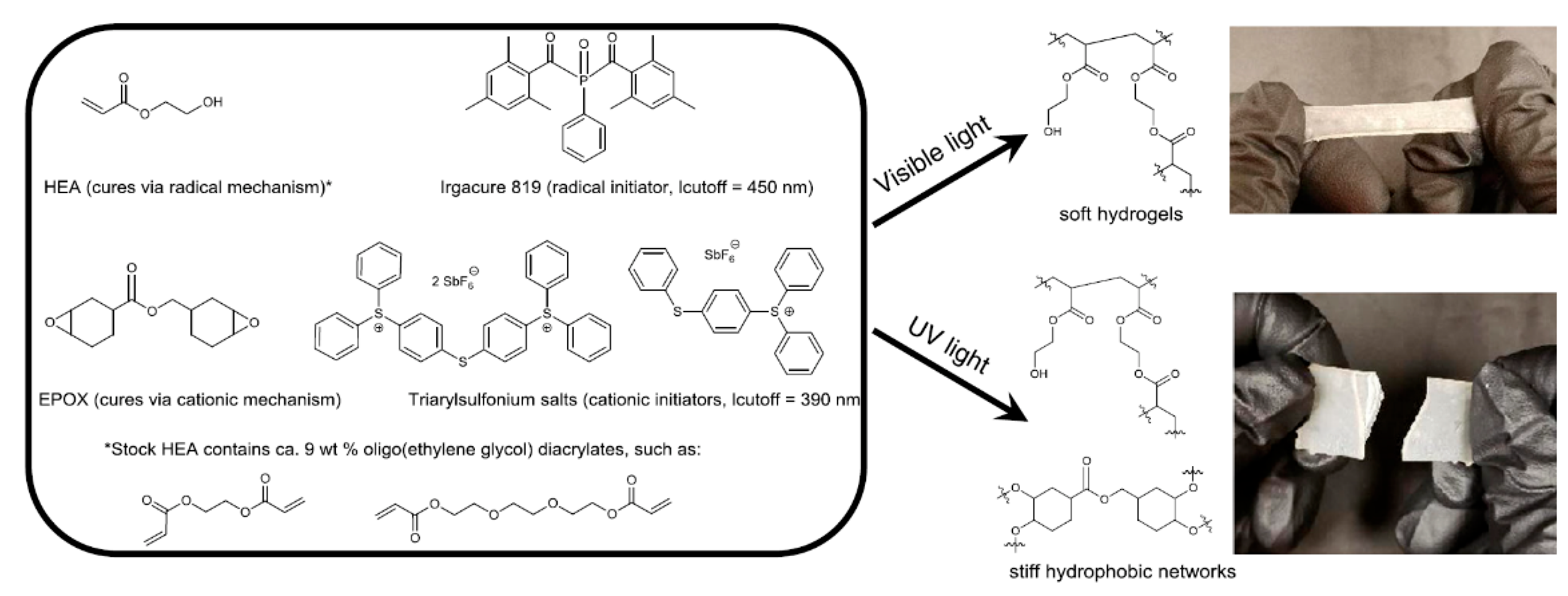
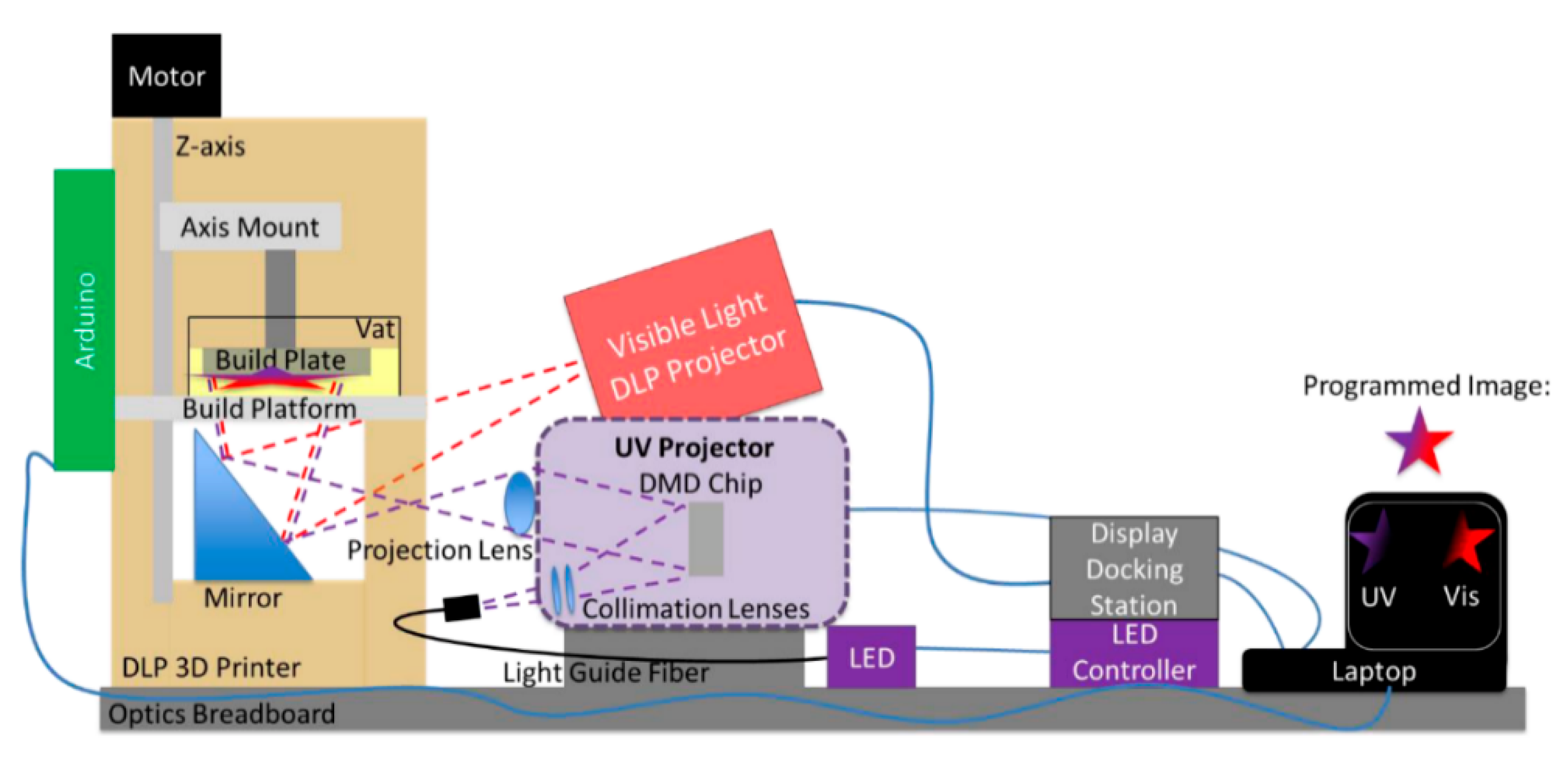
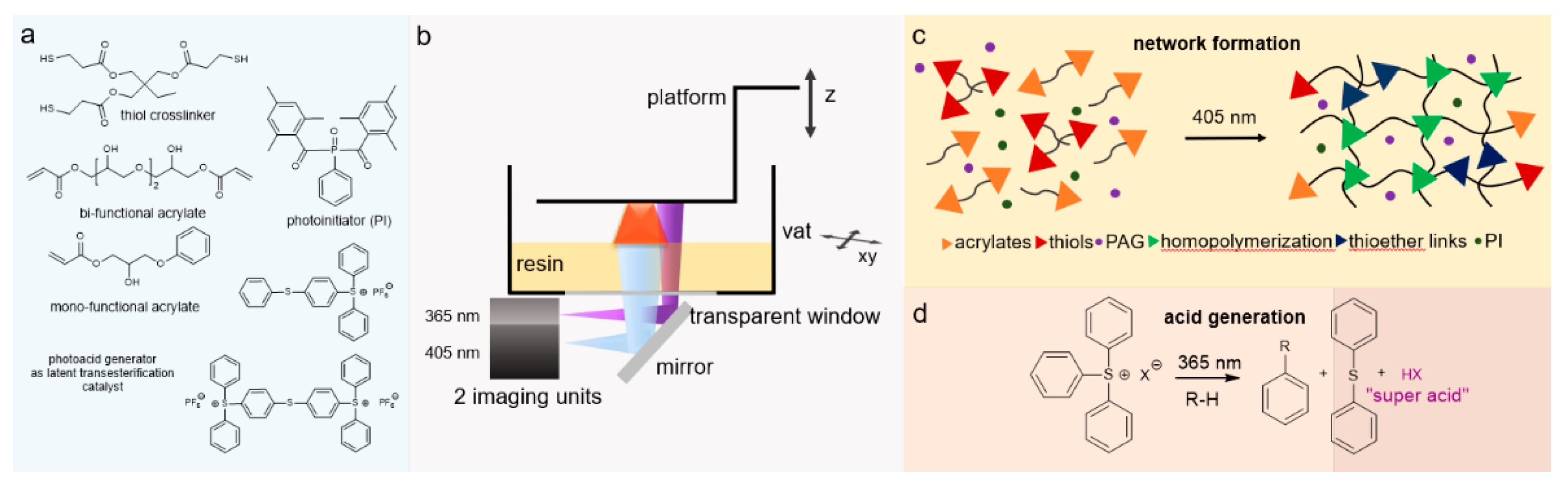
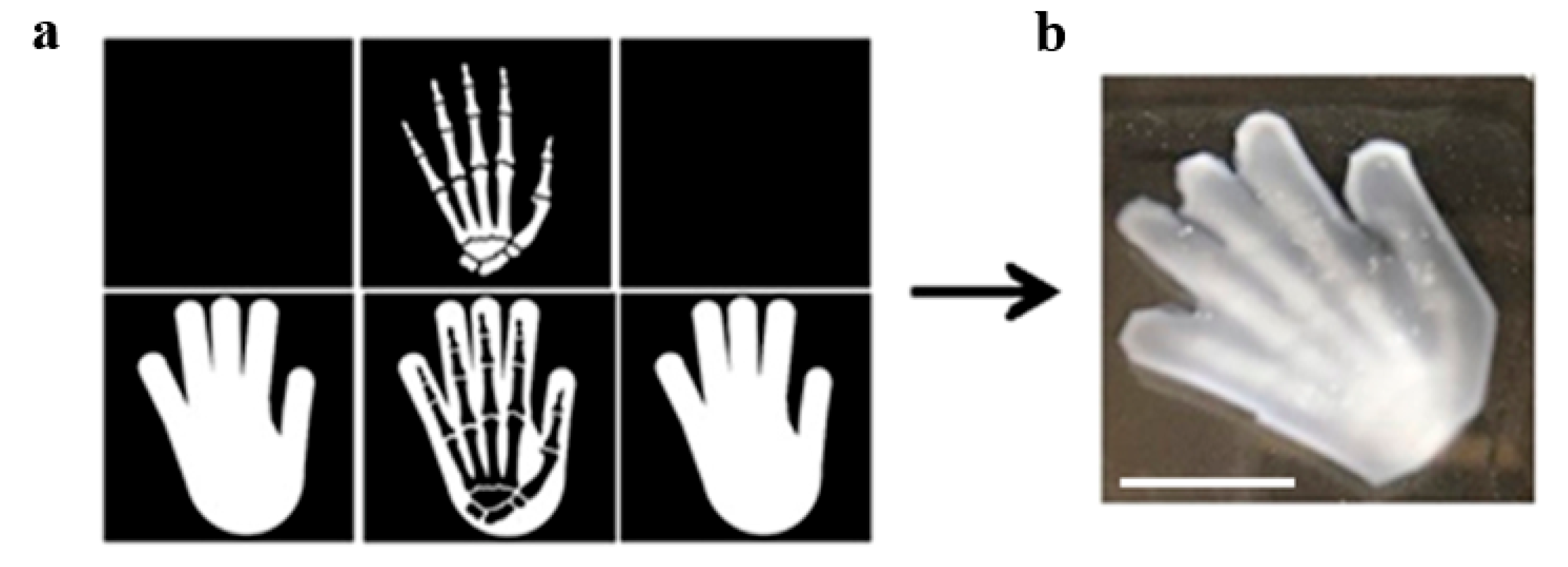

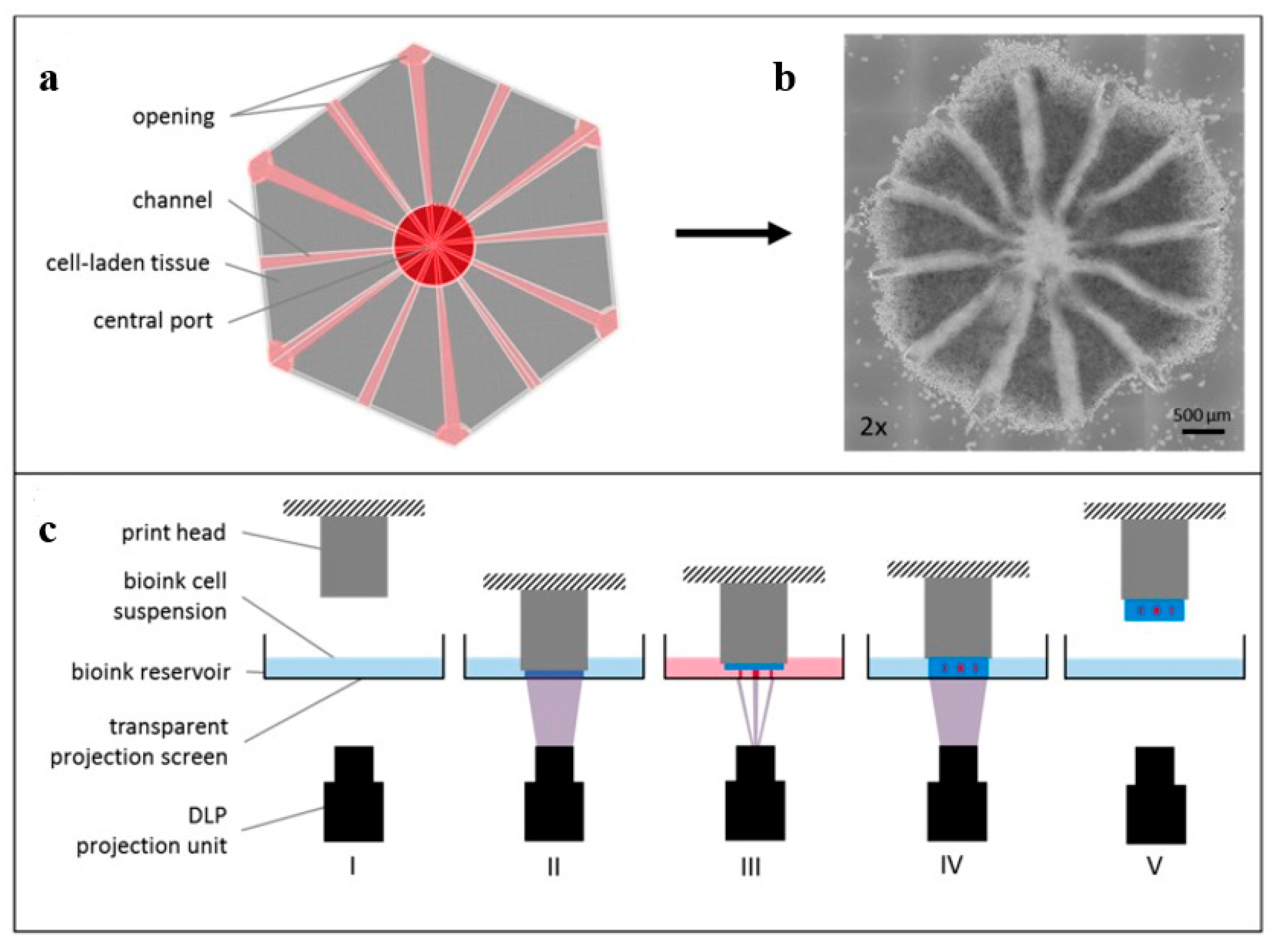
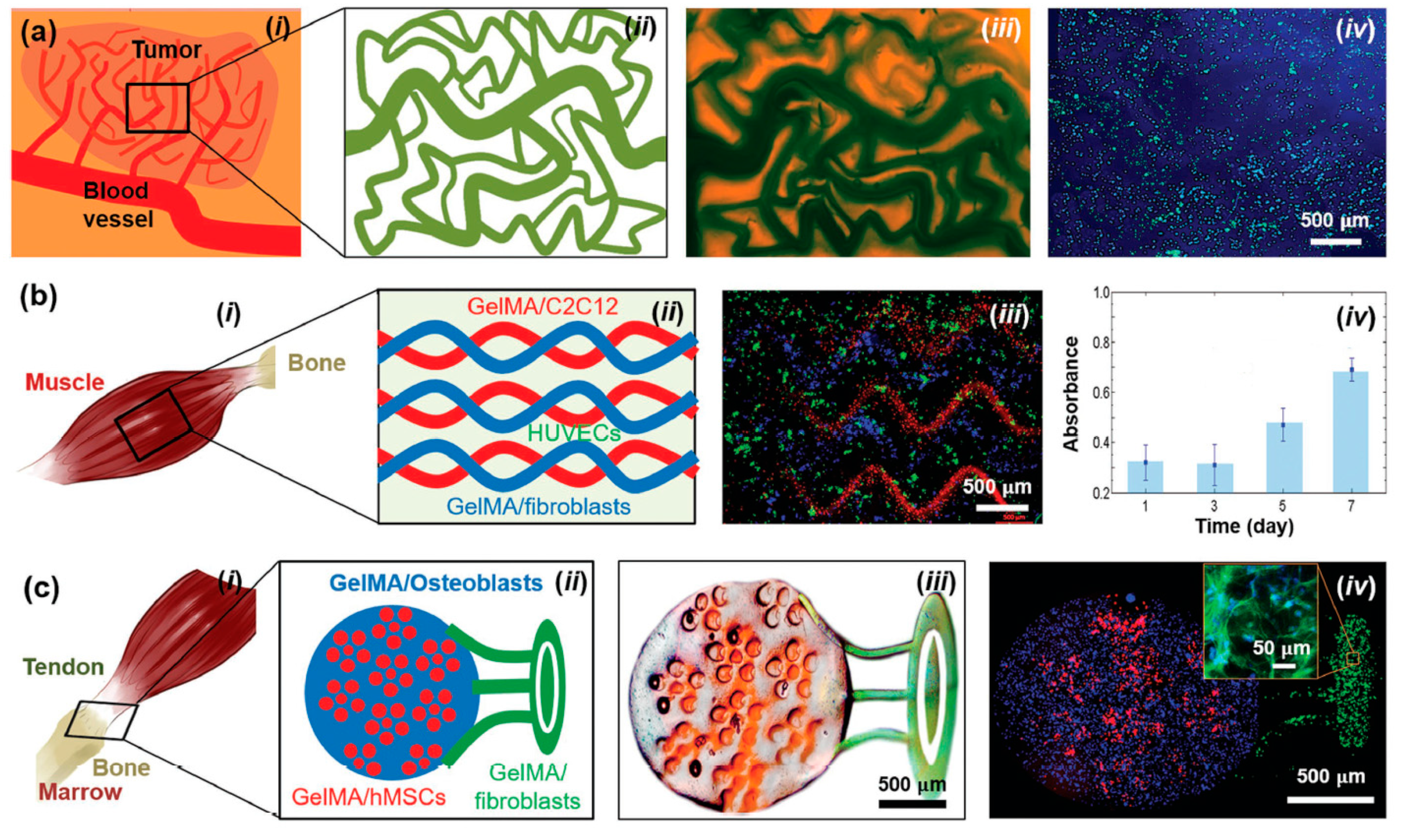

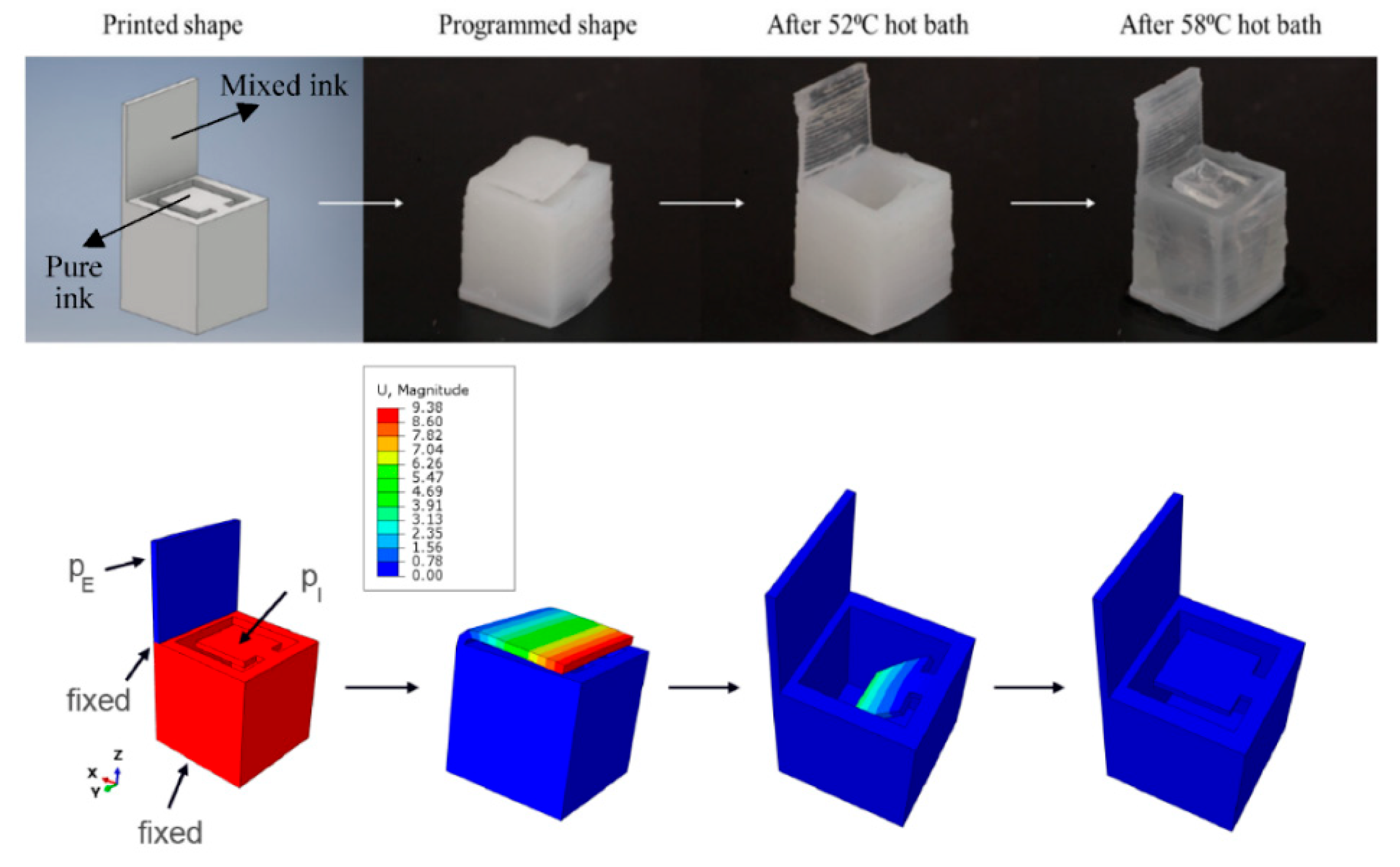

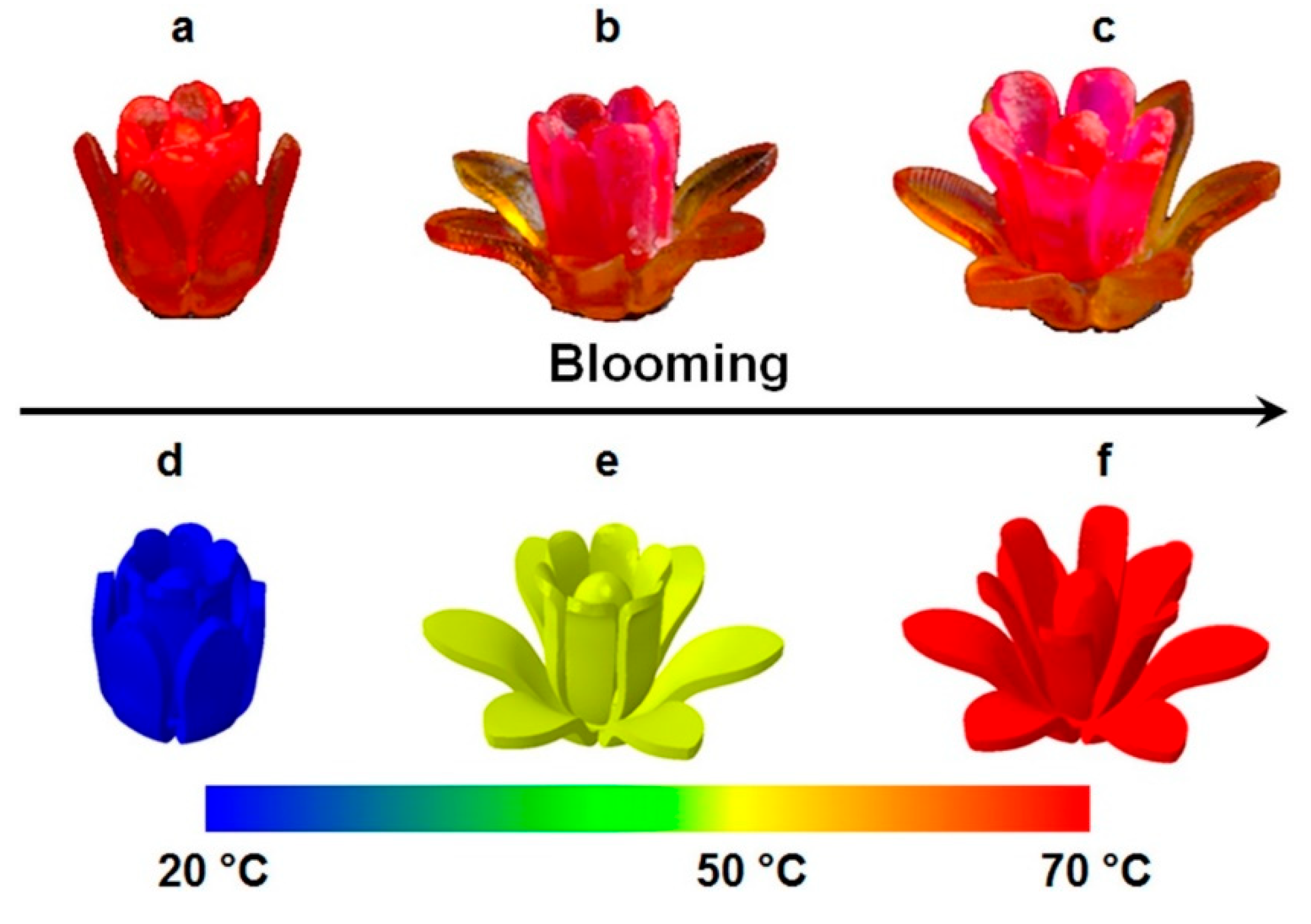

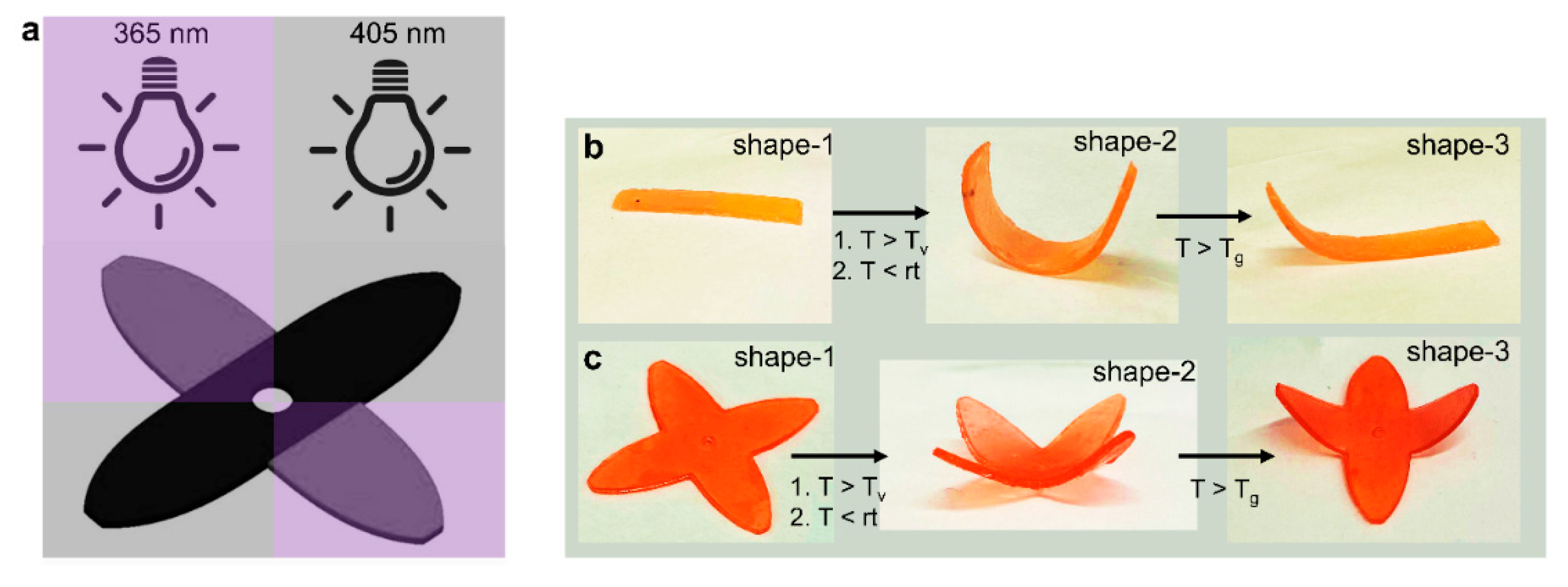

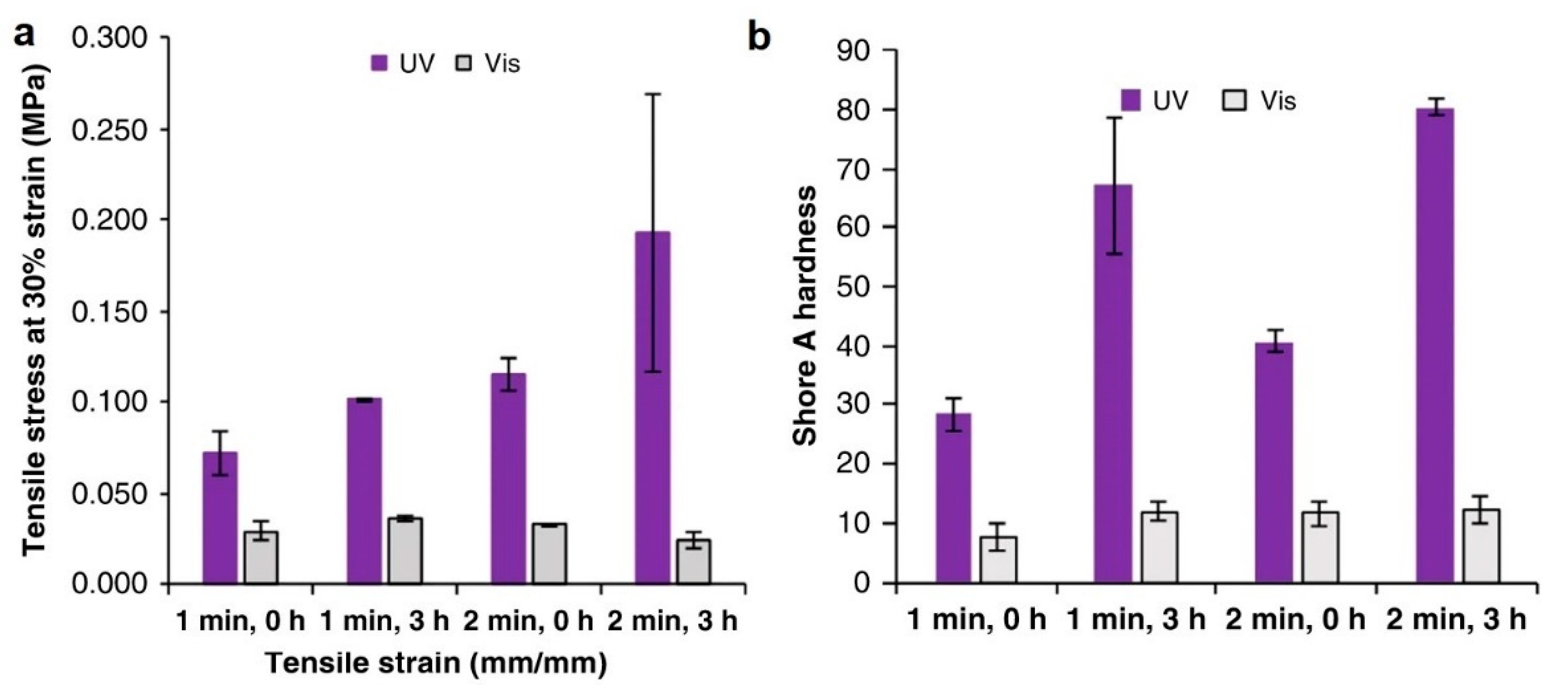
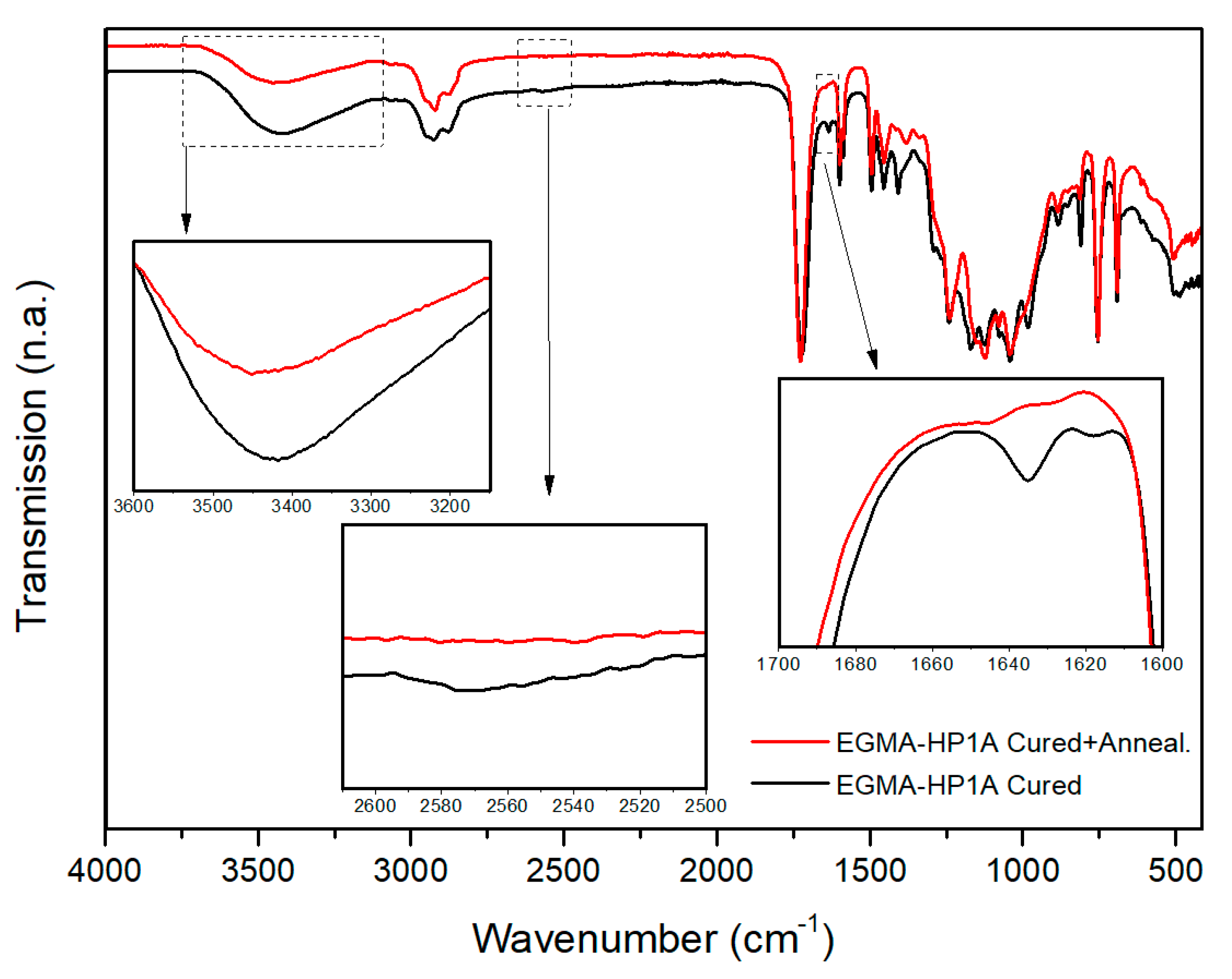



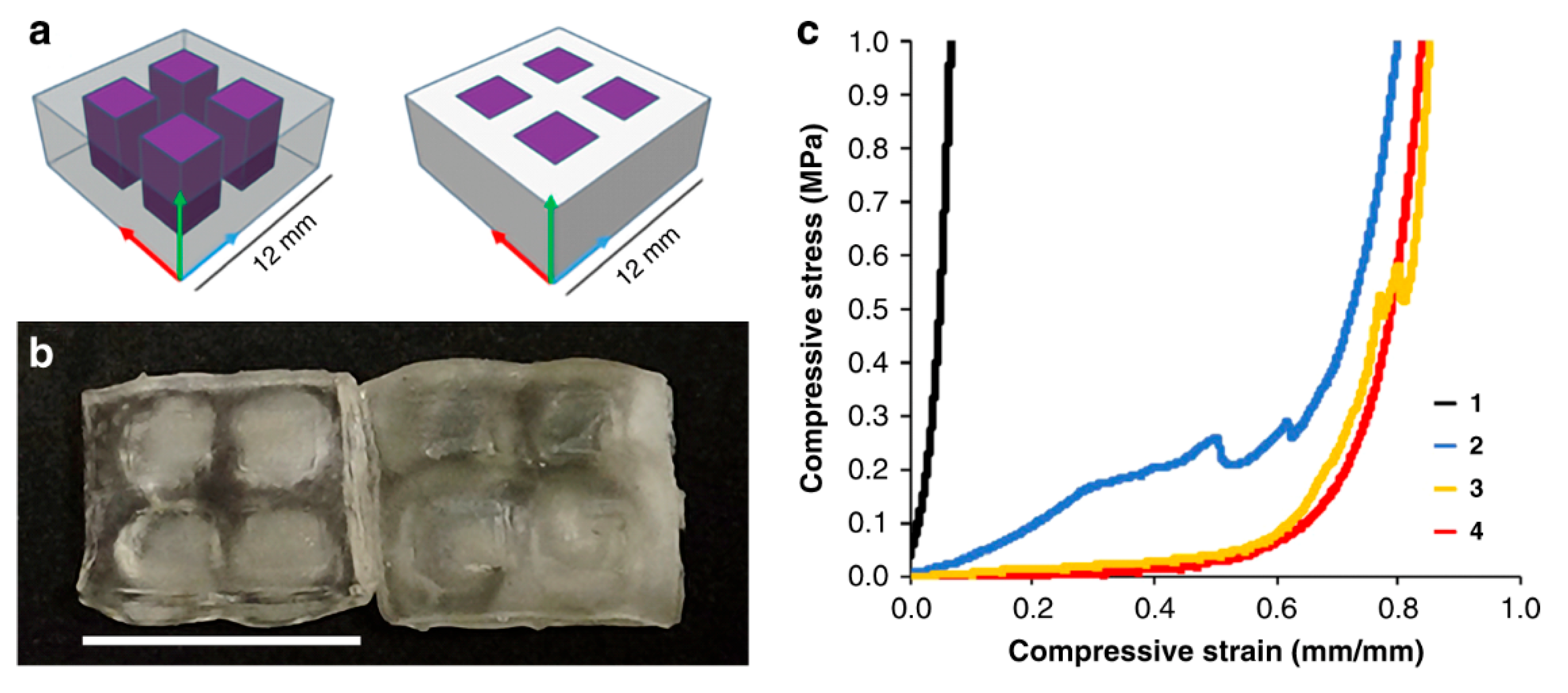


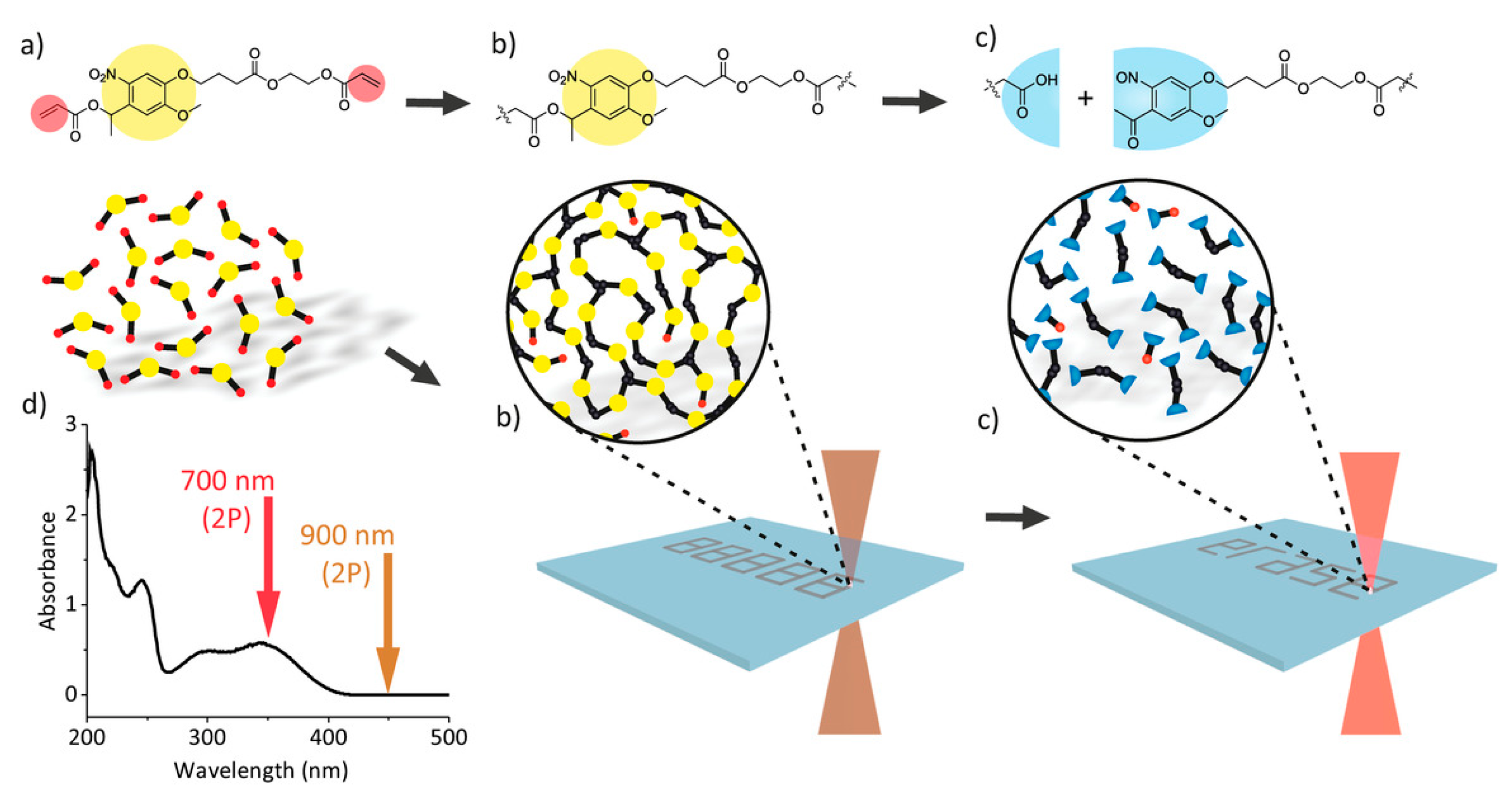

| Category | Material | Young’s Modulus (Pa) |
|---|---|---|
| Soft | Brain tissues | 103 |
| Alginate hydrogel fat | 104 | |
| Silicone elastomer | 105 | |
| Polydimethylsiloxane | 0.8 × 106 | |
| Polycaprolactone | 0.2 × 107 | |
| Biological skin | 0.5 × 108 | |
| Rubber | 0.8 × 108 | |
| Polyethylene (low density) | 0.5 × 109 | |
| Rigid | Polylactic acid | 0.5 × 1010 |
| Nylon | 0.6 × 1010 | |
| Wood | 1010 | |
| Polymethylmethacrylate | 4.5 × 1010 | |
| Polyethylene terephthalate | 5.5 × 1010 | |
| Bone | 0.5 × 1011 | |
| Glass | 0.8 × 1011 | |
| Copper | 1011 | |
| Steel | 0.5 × 1012 | |
| Diamond | 1012 |
| Processing Strategy | Application Area | Resolution Achieved | Reference | |
|---|---|---|---|---|
| TPA Photo polymerization | Radical quencher | Microstructures | 100 nm | [124] |
| Activation beam | Microdevices | 40 nm | [125] | |
| Scanning speed manipulation | Micromachines | 25 nm | [126] | |
| Self-smoothing | Micro-optics | 20 nm | [127] |
| Suitable Materials | Resolution | Building Speed | Benefits | Limitations | Reference | |
|---|---|---|---|---|---|---|
| Fused filament fabrication (FFF) | Acrylonitrile butadiene styrene, nylon, polylactic acid, polyethylene terephthalate, polyvinylalcohol, high impact polystyrene, thermoplastic polyurethane, polycarbonate, polypropylene | 100 µm | 1–10 m/min | Ability to print large functional materials with standard thermoplastic polymers | Low resolution, nozzle choking, non-homogenous filament melting, material, anisotropy along z-axis | [1,62,68,75,132,133] |
| Direct ink writing (DIW) | Liquid polymer/melt/gel/paste | 1–100 µm | 3 m/min | Highest resolution among all extrusion processes | Higher cost, not suitable for complex geometries | [1,62,68,134,135,136] |
| Selective laser sintering (SLS) | Polylaurylamide, polyether ketone ketone, polyamide, polycaprolactone | 80–100 µm | 1.80–3.6 m/min | Ability to process standard plastics, good mechanical properties | Lower resolution, rough surface | [1,10,68,137,138,139] |
| Stereolithography (SLA) | Acrylate, methacrylate, epoxy, vinyl monomers | 5–50 µm | 0.25 mm/min | High resolution and accuracy | Limited availability of photopolymers, toxicity of monomers | [1,68,99,138,140,141,142,143,144] |
| Digital light processing (DLP) | Acrylate, methacrylate, epoxy, vinyl monomers | 5–50 µm | 0.4–2.5 mm/min | High resolution and accuracy, lower cost, higher printing speed compared to SLA | Limited availability of photopolymers, toxicity of monomers | [1,68,143] |
| Liquid crystalline display (LCD) | Acrylate, methacrylate, epoxy, vinyl monomers | <50 µm | 10 mm/min | High resolution and accuracy, lower cost | Limited availability of photopolymers, toxicity of monomers | [37,103,104,105] |
| Continuous liquid interface printing (CLIP) | Acrylate, methacrylate, vinyl monomers, epoxies | <100 µm | 8–16 mm/min | High printing speed | Anisotropy of printed structures | [106,109] |
| Two-photon absorption (TPA) | Acrylate, methacrylate, vinyl monomers, epoxies | <100 nm | 0.08–33 mm3/min | Excellent resolution | Expensive, time consuming, requires tedious control strategies (rastering) | [145,146] |
| Volumetric 3D printing | Acrylate, methacrylate, vinyl monomers, epoxies | Up to 80 µm | 10 mm/min | Fast printing speed. | High viscosity resin (>10 Pa·s) required. Costly technology, tedious resin formulation strategies, low absorption and high reactivity of monomers required. | [86,128,130,147] |
| Solution mask liquid lithography (SMaLL) | Acrylate, methacrylate, vinyl monomers, epoxies | Up to 100 µm | 8.33 mm/min | Large curing depth, no moving parts required, rapid curing rates | Additional photochromes and sensitizer required. Reaction strategies must be developed before printing. | [24] |
| Technology | Strategy | Reference |
|---|---|---|
| SLA | Carousel-like rotating disks including various resins | [297] |
| SLA | Carousel-based rotary vats | [298] |
| SLA | Multiple resin injections under the servo-stage on the building stage through an orifice | [299] |
| SLA | Resin droplet delivery via rotary wheel | [300] |
| DLP | Resin exchange in vat with intermittent cleaning | [301] |
| µSLA | Multiple-resin dynamic liquid control within an integrated fluidic cell. Pumps for drawing/withdrawing of materials. | [302] |
| SLA | Multiple resin supply via microchannels | [303,304] |
| SLA | Multiple resin injection and intermittent cleaning | [305] |
| Applications | Printing Materials | Methodology | Reference |
|---|---|---|---|
| Scaffolds for tissue engineering | PEGDA, PEGDMA | Resin exchange, SLA | [334] |
| Multilayer polypills | PEGDA + dissolved drug | Resin exchange, SLA | [332] |
| Multilayer polypills | PEGDA, PEGDMA + dissolved drug | Resin exchange, SLA | [333] |
| Tissue-porous scaffolds | PEGDA, Commercial photocurable resins, + Leachable salt particulates | DLP | [335] |
| Bioactive scaffolds | PEGDA, PEGDMA, + fluorescently labeled components | Resin exchange, SLA | [336] |
| Neovasculature | PEGDA, PEGDMA, + Murine cells | Resin exchange, SLA | [337] |
| Constructs with Encapsulated Cells | PEGDA + human dermal fibroblasts | Resin exchange, SLA | [338] |
| Piezoelectric acoustic sensor | PEGDA + barium titanate nanopowder + multi-walled carbon nanotubes | Resin exchange, DLP | [339] |
| Cell encapsulation | PEGDA + cells | Resin exchange, SLA | [340] |
| Multi-material cantilevers | poly(ethylene glycol) diacrylate (PEGDA) and acrylic-PEG-collagen (PC) | Resin exchange, SLA | [341] |
| Selective Porous Barriers | PEGDA: MW 258, MW 575, MW 700 | Resin exchange, SLA | [65] |
| Biological sensors | PEGDA, commercial resin, + biomolecules | Resin exchange, SLA | [342] |
| Tissue scaffolds | PEGDA+ fluorescently-labeled polystyrene microparticles | Resin exchange, SLA | [343] |
| Spatially-designed biological sensor | oxidized methacrylic alginate, poly(ethylene glycol) methyl ether methacrylate, PEGDA + cells | Resin exchange, SLA | [344] |
| Cells interactive sensors | Gelatin methacrylate, PEGDA, fluorescent dextrans, cells | Resin exchange, SLA | [345] |
| Applications | Stimuli | Printing Materials | Multimaterial Strategy | Reference |
|---|---|---|---|---|
| Hydrogel cantilevers and actuators | Chemical stimuli | Poly(ethylene glycol) diacrylate (PEGDA) and acrylic-PEG-collagen (PC) mixtures | Resin exchange in vat, SLA | [341] |
| Hydrogels | Thermal stimuli | Poly(N-isopropylacrylamide), N,N′-Methylene-bis(acrylamide) mixtures | Resin exchange in vat, SLA | [348] |
| Hinges, robotic arms, bars, and sheets | Thermal stimuli | Bisphenol A ethoxylate diacrylate (BPADA), glycidyl methacrylate (GMA), n-butyl acrylate (BA), a diamine cross-linker [poly(propylene glycol) bis(2-aminopropyl ether); D230], mixtures | Grayscale DLP 3D printing | [321] |
| Hydrogels | Osmotic pressure, temperature and pH | N-Isopropylacrylamide, 2-carboxyethylacrylate, N,N′-ethylenebisacrylamide | 1. Swelling rates via high surface area patterning, 2. Crosslinking density via photo-exposure, 3. Chemical composition via resin vat exchange | [349] |
| Material A | Material B | Multi-Material | |
|---|---|---|---|
| Modulus (MPa) | 16.5 | 0.92 | 1.84 |
| Failure strain | 5% | 99% | 46% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaukat, U.; Rossegger, E.; Schlögl, S. A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization. Polymers 2022, 14, 2449. https://doi.org/10.3390/polym14122449
Shaukat U, Rossegger E, Schlögl S. A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization. Polymers. 2022; 14(12):2449. https://doi.org/10.3390/polym14122449
Chicago/Turabian StyleShaukat, Usman, Elisabeth Rossegger, and Sandra Schlögl. 2022. "A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization" Polymers 14, no. 12: 2449. https://doi.org/10.3390/polym14122449
APA StyleShaukat, U., Rossegger, E., & Schlögl, S. (2022). A Review of Multi-Material 3D Printing of Functional Materials via Vat Photopolymerization. Polymers, 14(12), 2449. https://doi.org/10.3390/polym14122449