
Microstructure Analysis of Drawing Effect and Mechanical Properties of Polyacrylonitrile Precursor Fiber According to Molecular Weight


Abstract
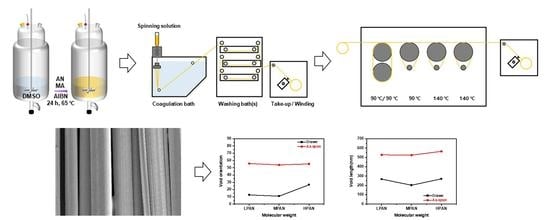
:
1. Introduction
2. Materials and Methods
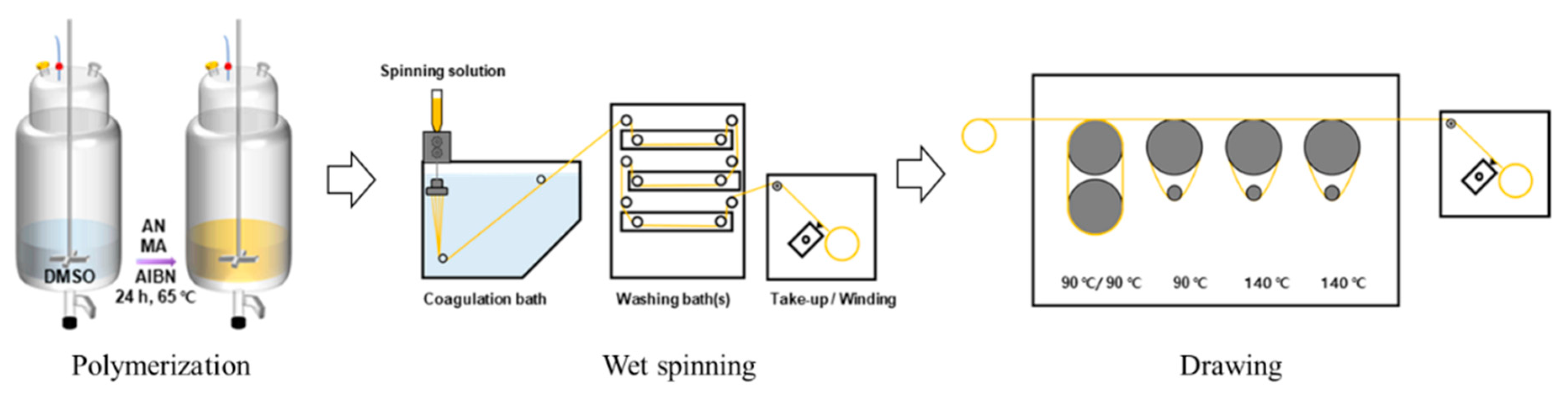
2.1. Materials and Polymerization
2.2. Fiber Spinning and Drawing
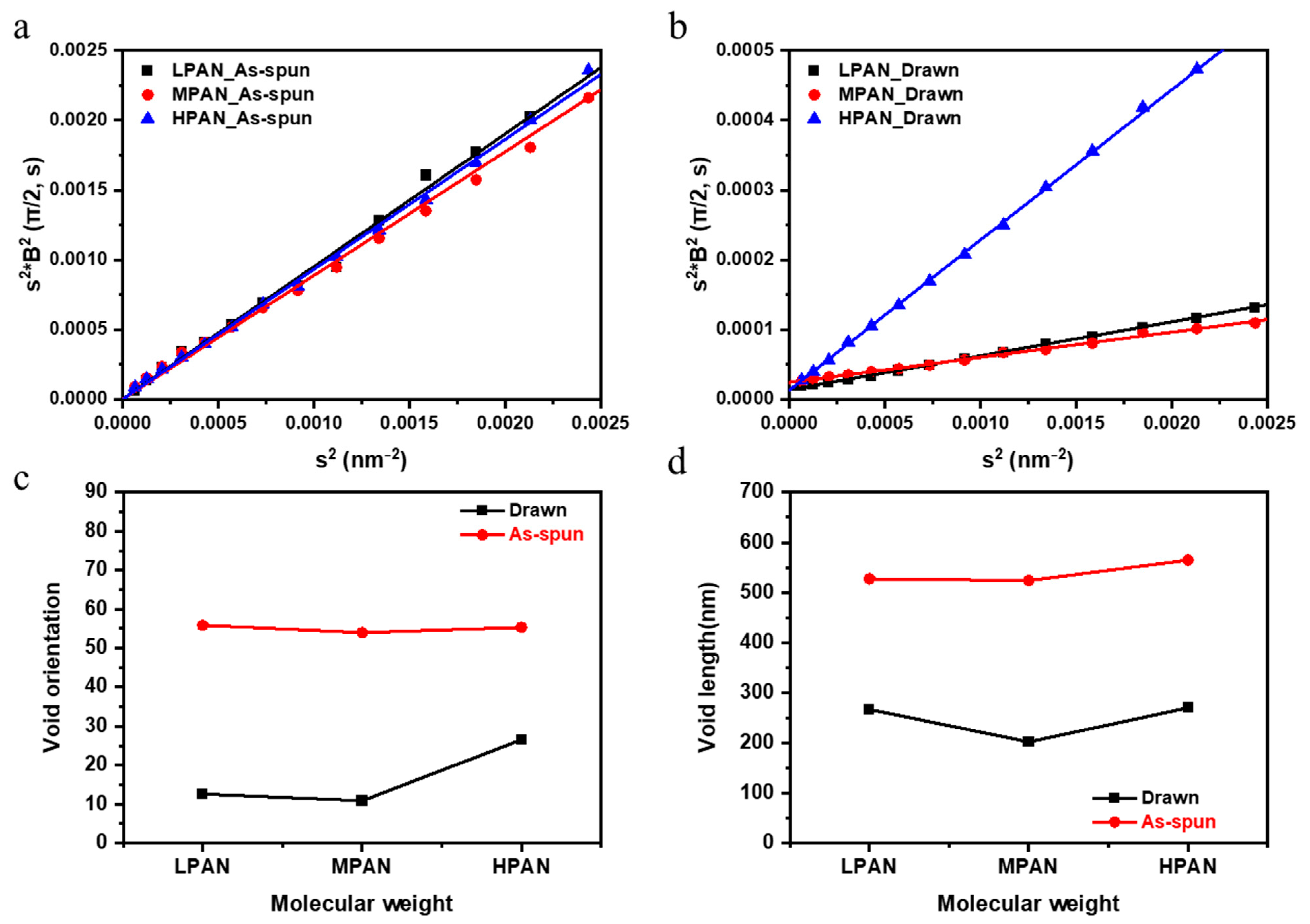
2.3. Characterization
3. Results and Discussion
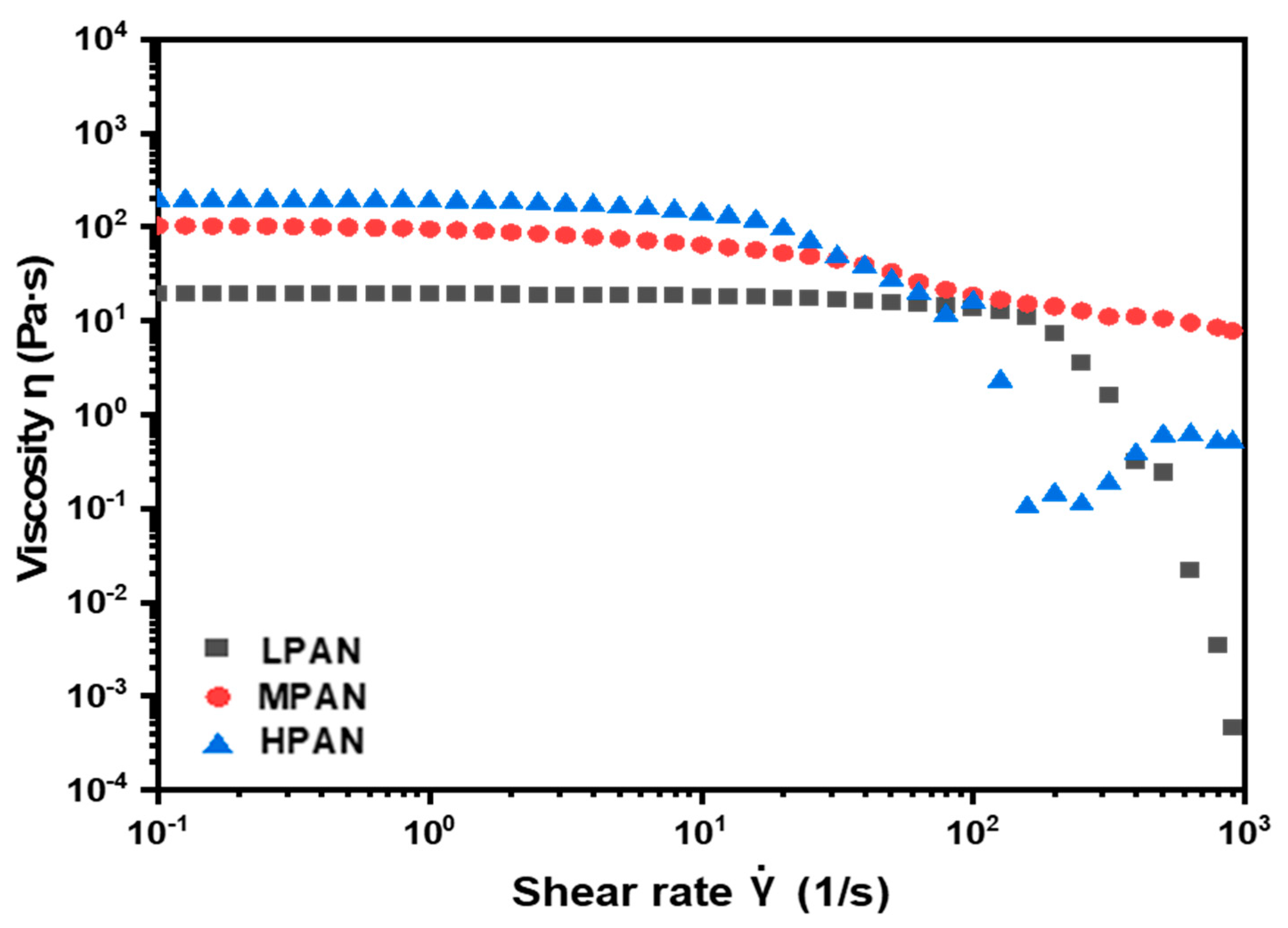
3.1. Polymerization
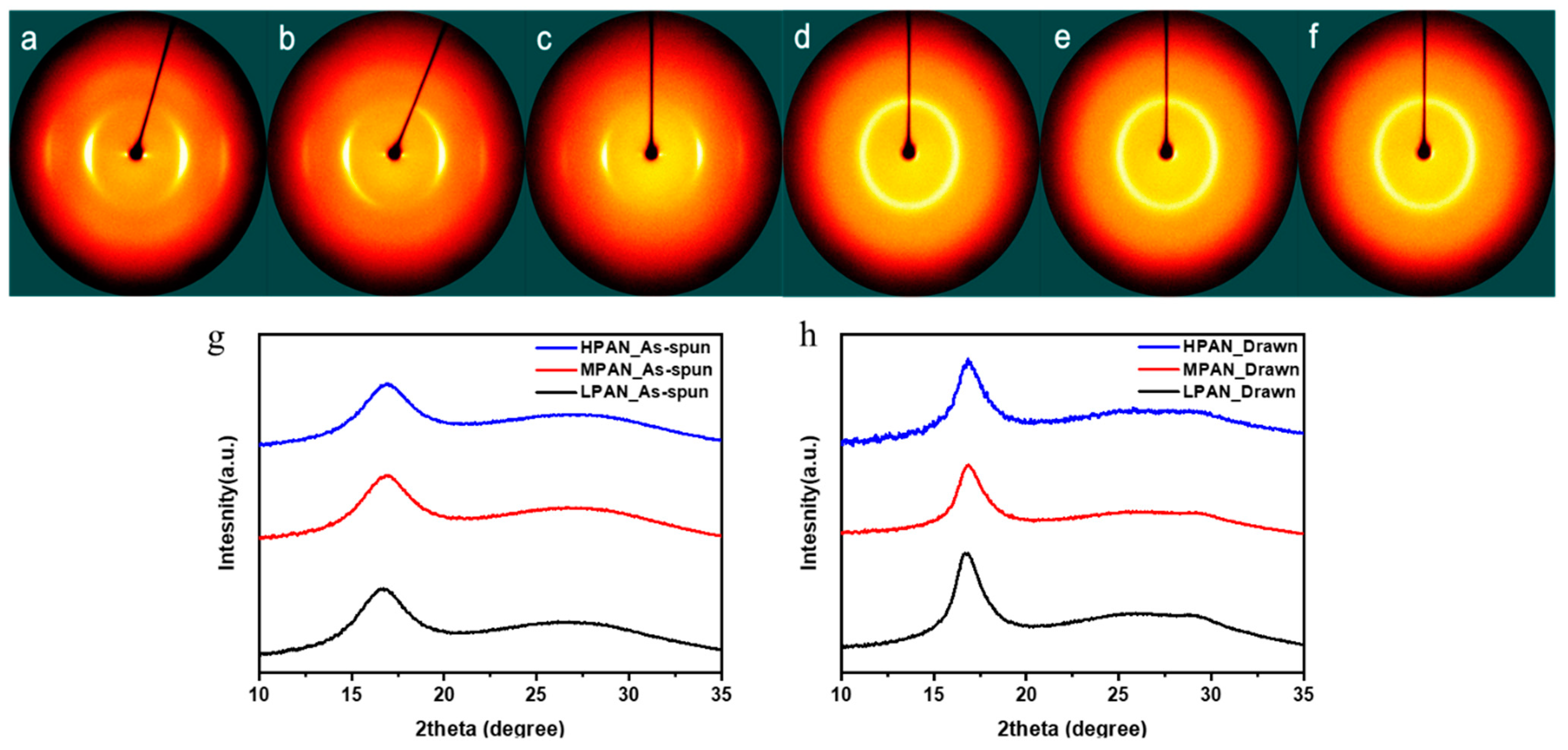
3.2. Microstructures
3.3. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gupta, A.; Harrison, I.R. New aspects in the oxidative stabilization of PAN-based carbon fibers. Carbon 1996, 34, 1427–1445. [Google Scholar] [CrossRef]
- Mikolajczyk, T.; Krucińska, I. Correlation Between Copolymer Characteristics, Conditions of Fiber Formation, and Mechanical Properties of PAN Carbon Fiber Precursor: Part I: Effect of Copolymer Intrinsic Viscosity Distribution on Mechanical Properties. Text. Res. J. 1989, 59, 536–540. [Google Scholar] [CrossRef]
- Tsai, J.-S.; Lin, C.-H. The effect of molecular weight on the cross section and properties of polyacrylonitrile precursor and resulting carbon fiber. J. Appl. Polym. Sci. 1991, 42, 3045–3050. [Google Scholar] [CrossRef]
- Huang, Y.; Young, R.J. Effect of fibre microstructure upon the modulus of PAN- and pitch-based carbon fibres. Carbon 1995, 33, 97–107. [Google Scholar] [CrossRef]
- Edie, D.D. The effect of processing on the structure and properties of carbon fibers. Carbon 1998, 36, 345–362. [Google Scholar] [CrossRef]
- Chen, J.C.; Harrison, I.R. Modification of polyacrylonitrile (PAN) carbon fiber precursor via post-spinning plasticization and stretching in dimethyl formamide (DMF). Carbon 2002, 40, 25–45. [Google Scholar] [CrossRef]
- Huang, X. Fabrication and Properties of Carbon Fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef]
- Ahn, H.; Kim, Y.M.; Yang, H.-S.; Yeo, S.Y.; Yu, W.-R.; Lee, B.-S. Moisturized Polyacrylonitrile Copolymer for Stronger Precursor Fibers. ACS Appl. Polym. Mater. 2021, 3, 6285–6293. [Google Scholar] [CrossRef]
- Zhang, H.; Quan, L.; Gao, A.; Tong, Y.; Shi, F.; Xu, L. Thermal Analysis and Crystal Structure of Poly(Acrylonitrile-Co-Itaconic Acid) Copolymers Synthesized in Water. Polymers 2020, 12, 221. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.; Yeo, S.Y.; Lee, B.-S. Designing Materials and Processes for Strong Polyacrylonitrile Precursor Fibers. Polymers 2021, 13, 2863. [Google Scholar] [CrossRef]
- Arbab, S.; Noorpanah, P.; Mohammadi, N.; Zeinolebadi, A. Exploring the effects of non-solvent concentration, jet-stretching and hot-drawing on microstructure formation of poly(acrylonitrile) fibers during wet-spinning. J. Polym. Res. 2011, 18, 1343–1351. [Google Scholar] [CrossRef]
- Oroumei, A.; Naebe, M. Mechanical property optimization of wet-spun lignin/polyacrylonitrile carbon fiber precursor by response surface methodology. Fibers Polym. 2017, 18, 2079–2093. [Google Scholar] [CrossRef]
- An, M.; Xu, H.; Lv, Y.; Gu, Q.; Tian, F.; Wang, Z. An in situ small-angle X-ray scattering study of the structural effects of temperature and draw ratio of the hot-drawing process on ultra-high molecular weight polyethylene fibers. RSC Adv. 2016, 6, 51125–51134. [Google Scholar] [CrossRef]
- Morris, E.A.; Weisenberger, M.C.; Bradley, S.B.; Abdallah, M.G.; Mecham, S.J.; Pisipati, P.; McGrath, J.E. Synthesis, spinning, and properties of very high molecular weight poly(acrylonitrile-co-methyl acrylate) for high performance precursors for carbon fiber. Polymer 2014, 55, 6471–6482. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-S.; Kim, Y.-M.; Choi, H.; Jang, J.; Youk, J.H.; Lee, B.-S.; Yu, W.-R. Electrochemical wet-spinning process for fabricating strong PAN fibers via an in situ induced plasticizing effect. Polymer 2020, 202, 122641. [Google Scholar] [CrossRef]
- Cho, D.W.; Hong, S.C. Synergistic effect of comonomers on the thermal oxidative stabilization of polyacrylonitrile copolymers for carbon materials. Polym. Degrad. Stab. 2019, 161, 191–197. [Google Scholar] [CrossRef]
- Hou, C.; Ying, L. Stability of acrylonitrile/methyl vinyl ketone copolymer solutions. J. Appl. Polym. Sci. 2006, 100, 3377–3381. [Google Scholar] [CrossRef]
- Ju, A.; Guang, S.; Xu, H. Effect of comonomer structure on the stabilization and spinnability of polyacrylonitrile copolymers. Carbon 2013, 54, 323–335. [Google Scholar] [CrossRef]
- Al Faruque, M.A.; Remadevi, R.; Razal, J.M.; Naebe, M. Impact of the wet spinning parameters on the alpaca-based polyacrylonitrile composite fibers: Morphology and enhanced mechanical properties study. J. Appl. Polym. Sci. 2020, 137, 49264. [Google Scholar] [CrossRef]
- Wu, Q.-Y.; Chen, X.-N.; Wan, L.-S.; Xu, Z.-K. Interactions between Polyacrylonitrile and Solvents: Density Functional Theory Study and Two-Dimensional Infrared Correlation Analysis. J. Phys. Chem. B 2012, 116, 8321–8330. [Google Scholar] [CrossRef]
- Newcomb, B.A.; Gulgunje, P.V.; Liu, Y.; Gupta, K.; Kamath, M.G.; Pramanik, C.; Ghoshal, S.; Chae, H.G.; Kumar, S. Polyacrylonitrile solution homogeneity study by dynamic shear rheology and the effect on the carbon fiber tensile strength. Polym. Eng. Sci. 2016, 56, 361–370. [Google Scholar] [CrossRef]
- Ahn, H.; Wee, J.-H.; Kim, Y.M.; Yu, W.-R.; Yeo, S.-Y. Microstructure and Mechanical Properties of Polyacrylonitrile Precursor Fiber with Dry and Wet Drawing Process. Polymers 2021, 13, 1613. [Google Scholar] [CrossRef]
- Kaur, J.; Millington, K.; Smith, S. Producing high-quality precursor polymer and fibers to achieve theoretical strength in carbon fibers: A review. J. Appl. Polym. Sci. 2016, 133, 43963. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, C.; Bai, Y.; Wang, Y.; Xu, Y. Influence of Precursor Properties on the Thermal Stabilization of Polyacrylonitrile Fibers. Polym. Bull. 2006, 57, 757–763. [Google Scholar] [CrossRef]
- Lu, J.; Li, W.; Kang, H.; Feng, L.; Xu, J.; Liu, R. Microstructure and properties of polyacrylonitrile based carbon fibers. Polym. Test. 2020, 81, 106267. [Google Scholar] [CrossRef]
- Mengfan, W.; Yingge, X.; Weiyu, C.; Na, J.; Wang, C.; Lianghua, X. SAXS and WAXD study of periodical structure for polyacrylonitrile fiber during coagulation. Polym. Adv. Technol. 2015, 26, 136–141. [Google Scholar] [CrossRef]
- Li, X.-Y.; Tian, F.; Gao, X.-P.; Bian, F.-G.; Li, X.-H.; Wang, J. WAXD/SAXS study and 2D fitting (SAXS) of the microstructural evolution of PAN-based carbon fibers during the pre-oxidation and carbonization process. New Carbon Mater. 2017, 32, 130–136. [Google Scholar] [CrossRef]
- Tang, H.; Meng, F.; Liu, Y.; Jin, S.; Wang, X.; Gao, Z.; Che, X. Investigation of Voids in Polyacrylonitrile Fibers by USAXS and SAXS. Chem. Res. Chin. Univ. 2019, 35, 1070–1075. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, X.; Liu, J.; Liang, J.; Wang, X. Structure and tensile properties of carbon fibers based on electron-beam irradiated polyacrylonitrile fibers. J. Mater. Sci. 2020, 55, 4962–4969. [Google Scholar] [CrossRef]
- Gong, Y.; Du, R.; Mo, G.; Xing, X.; Lü, C.-X.; Wu, Z. In-situ microstructural changes of polyacrylonitrile based fibers with stretching deformation. Polymer 2014, 55, 4270–4280. [Google Scholar] [CrossRef]
- Liu, L.; Wu, F.; Yao, H.; Shi, J.; Chen, L.; Xu, Z.; Deng, H. Investigation of surface properties of pristine and γ-irradiated PAN-based carbon fibers: Effects of fiber instinct structure and radiation medium. Appl. Surf. Sci. 2015, 337, 241–248. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P (AN-MA) | NMR Conversion Ratio (mol %) | GPC | ||
|---|---|---|---|---|
| AN | MA | Mw | PDI | |
| LPAN | 97 | 3 | 191 k | 1.93 |
| MPAN | 97.7 | 2.3 | 221 k | 1.86 |
| HPAN | 97 | 3 | 337 k | 1.91 |
| Mw | Crystallinity (%) | Preferred Orientation (%) | Crystal Size (nm) | |
|---|---|---|---|---|
| LPAN | As-spun | 58.87 | 28.48 | 28.71 |
| Drawn | 59.73 | 83.56 | 49.17 | |
| MPAN | As-spun | 59.14 | 38.54 | 28.66 |
| Drawn | 61.63 | 80.45 | 50.74 | |
| HPAN | As-spun | 56.02 | 41.66 | 27.95 |
| Drawn | 58.67 | 84.83 | 48.55 | |
| Mw | Tensile Strength (MPa) | Elongation at Break (%) | |
|---|---|---|---|
| LPAN | As-spun | 102 ± 3 | 36.06 ± 4.29 |
| Drawn | 145 ± 4 | 11.85 ± 1.20 | |
| MPAN | As-spun | 159 ± 8 | 81.37 ± 6.59 |
| Drawn | 185 ± 22 | 13.79 ± 0.97 | |
| HPAN | As-spun | 126 ± 3 | 62.26 ± 9.36 |
| Drawn | 241 ± 7 | 12.49 ± 1.71 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, H.; Gwak, H.J.; Kim, Y.M.; Yu, W.-R.; Lee, W.J.; Yeo, S.Y. Microstructure Analysis of Drawing Effect and Mechanical Properties of Polyacrylonitrile Precursor Fiber According to Molecular Weight. Polymers 2022, 14, 2625. https://doi.org/10.3390/polym14132625
Ahn H, Gwak HJ, Kim YM, Yu W-R, Lee WJ, Yeo SY. Microstructure Analysis of Drawing Effect and Mechanical Properties of Polyacrylonitrile Precursor Fiber According to Molecular Weight. Polymers. 2022; 14(13):2625. https://doi.org/10.3390/polym14132625
Chicago/Turabian StyleAhn, Hyunchul, Hyeon Jung Gwak, Yong Min Kim, Woong-Ryeol Yu, Won Jun Lee, and Sang Young Yeo. 2022. "Microstructure Analysis of Drawing Effect and Mechanical Properties of Polyacrylonitrile Precursor Fiber According to Molecular Weight" Polymers 14, no. 13: 2625. https://doi.org/10.3390/polym14132625
APA StyleAhn, H., Gwak, H. J., Kim, Y. M., Yu, W. -R., Lee, W. J., & Yeo, S. Y. (2022). Microstructure Analysis of Drawing Effect and Mechanical Properties of Polyacrylonitrile Precursor Fiber According to Molecular Weight. Polymers, 14(13), 2625. https://doi.org/10.3390/polym14132625