
Comprehensive Analysis of Chromatin Accessibility and Transcriptional Landscape Identified BRCA1 Repression as a Potential Pathological Factor for Keloid


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subject
2.2. DF Extraction and Cell Culture
2.3. BRCA1 Knockdown
2.4. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.5. ATAC-Seq
2.6. Cleavage under Targets and Tagmentation Sequencing (CUT&Tag-Seq)
2.7. RNA-Seq
2.8. ATAC-Seq Analysis
2.9. CUT&Tag-Seq Analysis
2.10. RNA-Seq Analysis
3. Results
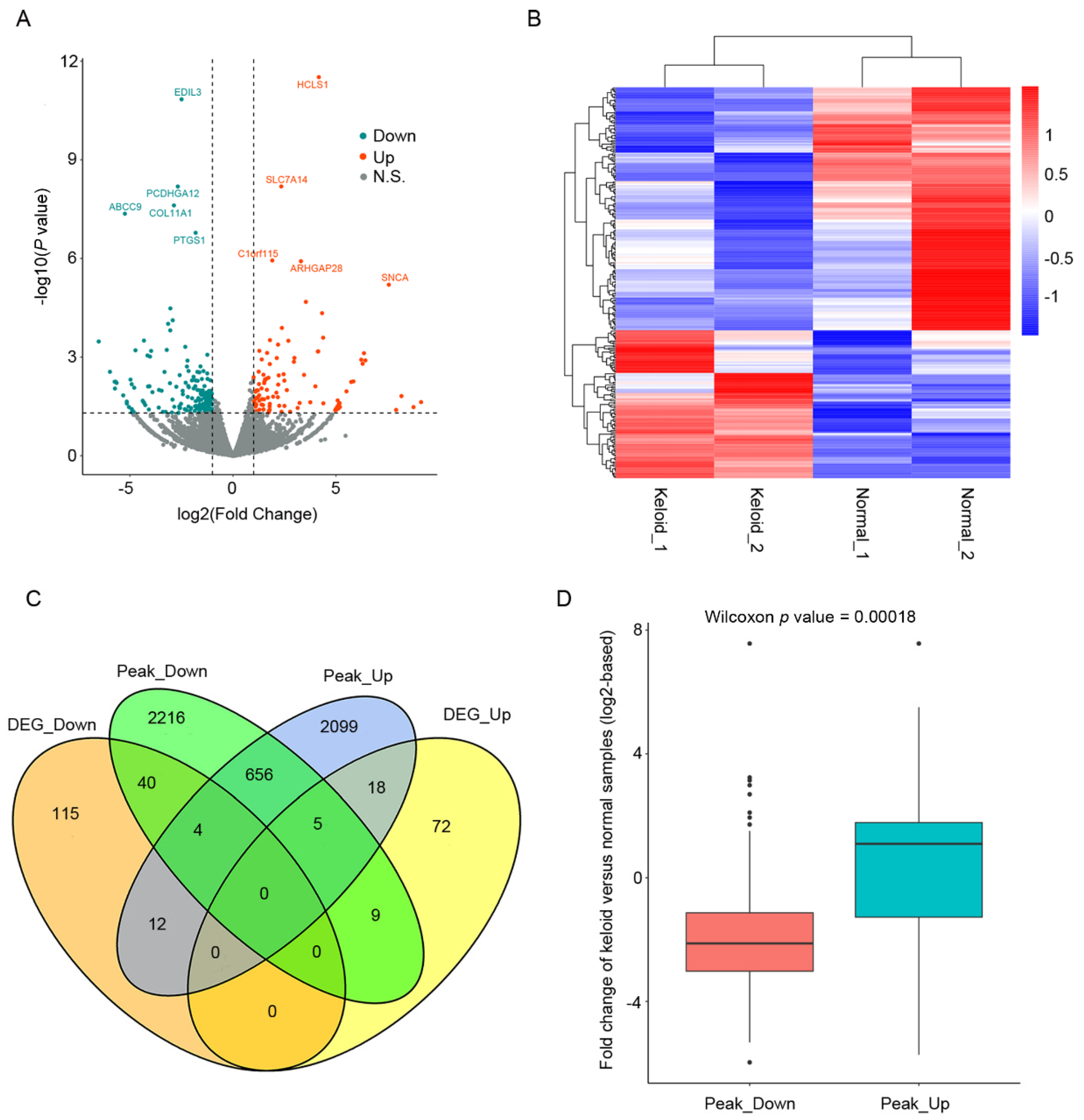
3.1. Keloid DFs Exhibited Extensive Chromatin State Changes
3.2. Chromatin State Transformation Is Related to Gene Expression Changes in Keloid Formation
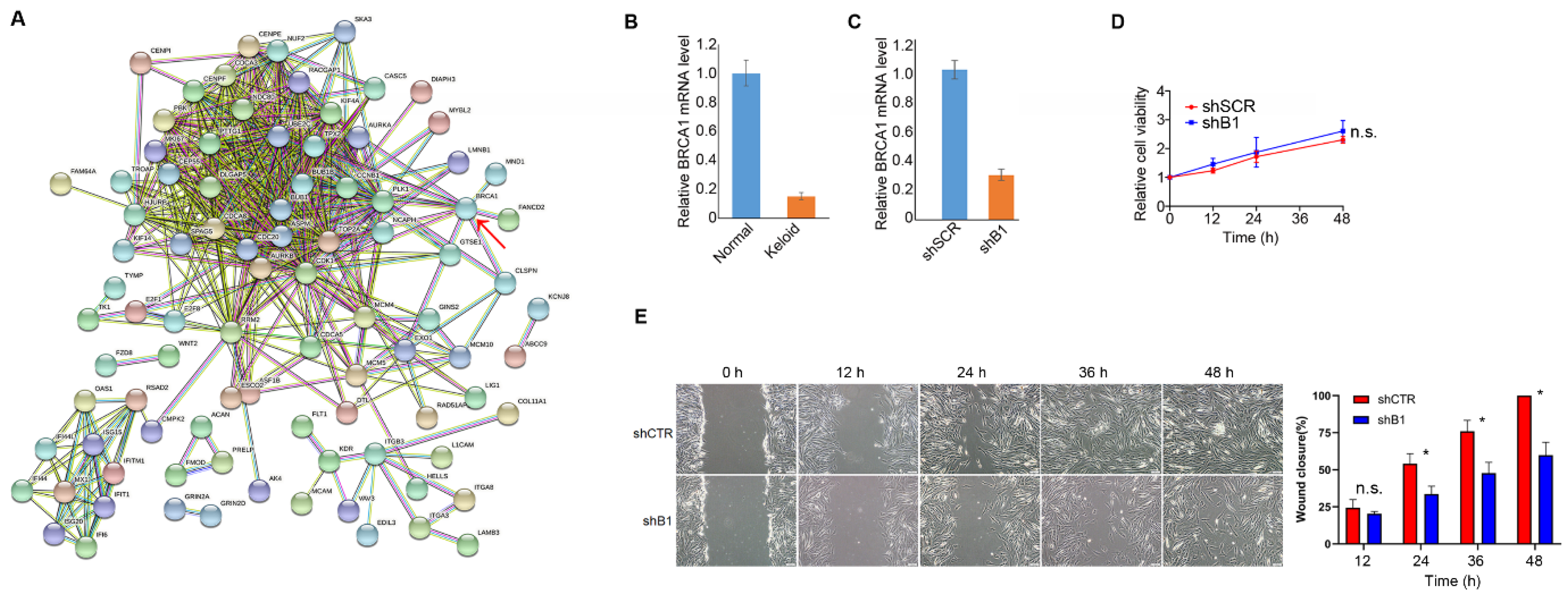
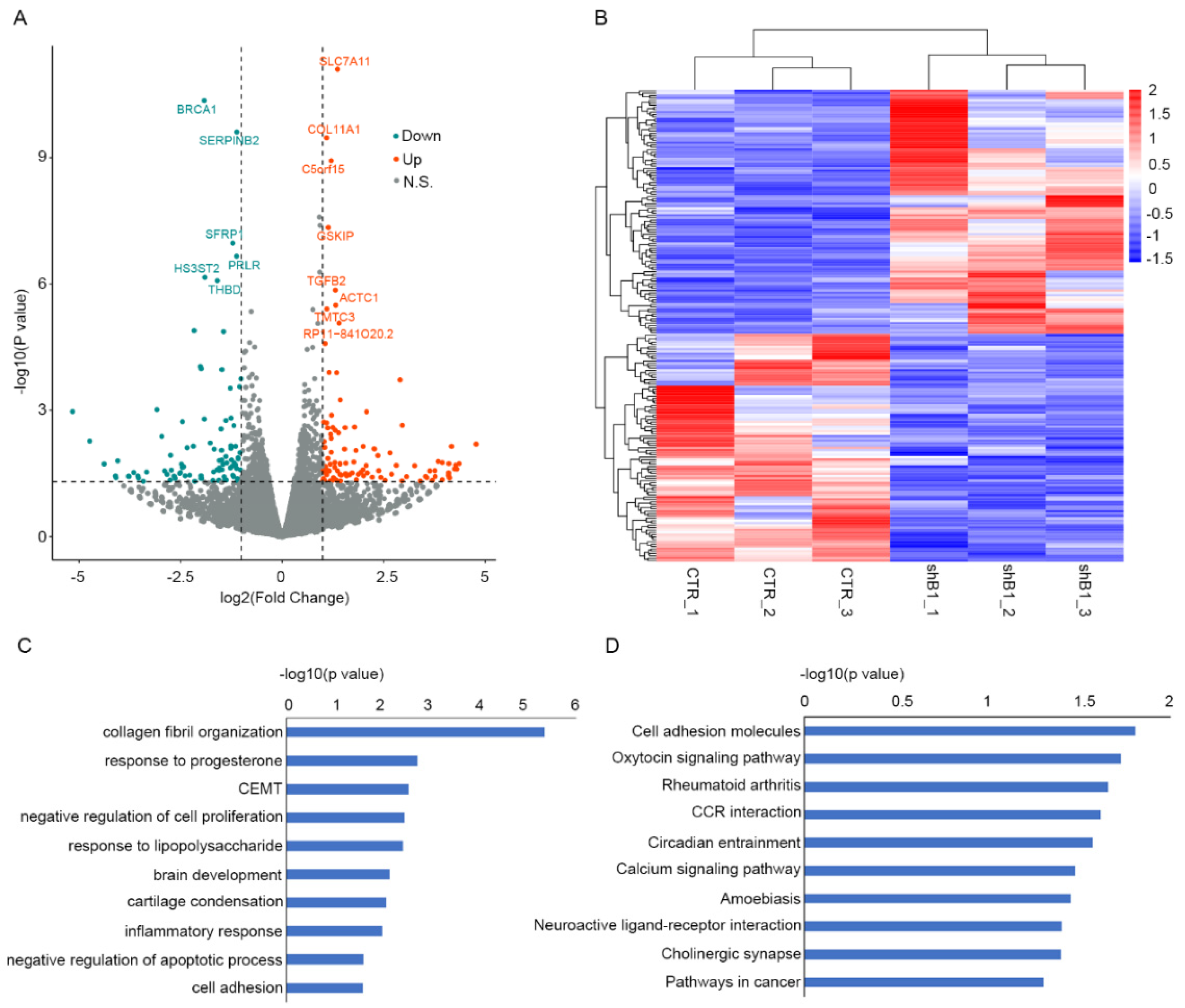
3.3. BRCA1 Suppression Is Potentially Associated with Keloid Pathogenesis
3.4. NPTX2 Is a Potential Target in Keloid Formation Induced by BRCA1 Suppression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, P.H.; Huang, B.S.; Horng, H.C.; Yeh, C.C.; Chen, Y.J. Wound healing. J. Chin. Med. Assoc. 2018, 81, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.X.; Wang, D.L.; Xu, G.C.; Deng, C.L.; Wei, Z.R. Advances in the research of effects of epithelial-mesenchymal interaction on wound healing and scar formation. Zhonghua Shao Shang Za Zhi 2020, 36, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Raquel Guillamat-Prats. The Role of MSC in Wound Healing, Scarring and Regeneration. Cells 2021, 10, 1729. [Google Scholar] [CrossRef]
- Russo, B.; Brembilla, N.C.; Chizzolini, C. Interplay Between Keratinocytes and Fibroblasts: A Systematic Review Providing a New Angle for Understanding Skin Fibrotic Disorders. Front. Immunol. 2020, 11, 648. [Google Scholar] [CrossRef]
- Naik, P.P. Novel targets and therapies for keloid. Clin. Exp. Dermatol. 2022, 47, 507–515. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X.F.; Wang, Z.C.; Lou, D.; Fang, Q.Q.; Hu, Y.Y.; Zhao, W.Y.; Zhang, L.Y.; Wu, L.H.; Tan, W.Q. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation. Biomed. Pharmacother. 2020, 129, 110287. [Google Scholar] [CrossRef]
- Glass, D.A., 2nd. Current Understanding of the Genetic Causes of Keloid Formation. J. Investig. Dermatol. Symp. Proc. 2017, 18, S50–S53. [Google Scholar] [CrossRef]
- Marneros, A.G.; Norris, J.E.; Watanabe, S.; Reichenberger, E.; Olsen, B.R. Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J. Investig. Dermatol. 2004, 122, 1126–1132. [Google Scholar] [CrossRef]
- Yan, X.; Gao, J.H.; Chen, Y.; Song, M.; Liu, X.J. Preliminary linkage analysis and mapping of keloid susceptibility locus in a Chinese pedigree. Zhonghua Zheng Xing Wai Ke Za Zhi 2007, 23, 32–35. [Google Scholar]
- Stevenson, A.W.; Deng, Z.; Allahham, A.; Prele, C.M.; Wood, F.M.; Fear, M.W. The epigenetics of keloids. Exp. Dermatol. 2021, 30, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S. Pioneer Transcription Factors Initiating Gene Network Changes. Annu. Rev. Genet. 2020, 54, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Gorisch, S.M.; Wachsmuth, M.; Toth, K.F.; Lichter, P.; Rippe, K. Histone acetylation increases chromatin accessibility. J. Cell Sci. 2005, 118, 5825–5834. [Google Scholar] [CrossRef] [PubMed]
- Adachi, A.; Senmatsu, S.; Asada, R.; Abe, T.; Hoffman, C.S.; Ohta, K.; Hirota, K. Interplay between chromatin modulators and histone acetylation regulates the formation of accessible chromatin in the upstream regulatory region of fission yeast fbp1. Genes Genet. Syst. 2018, 92, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef]
- Loh, C.; Park, S.H.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-Induced inflammatory genes escape repression in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214. [Google Scholar] [CrossRef]
- Tang, L.; Wang, M.; Shen, C.; Wen, L.; Li, M.; Wang, D.; Zheng, X.; Sheng, Y.; Wu, W.; Zhang, C.; et al. Assay for Transposase-Accessible Chromatin Using Sequencing Analysis Reveals a Widespread Increase in Chromatin Accessibility in Psoriasis. J. Investig. Dermatol. 2021, 141, 1745–1753. [Google Scholar] [CrossRef]
- Bonnomet, A.; Terryn, C.; Cutrona, J.; Jonquet, A.; Birembaut, P.; Zahm, J.M. Analysis of cell dispersion and migration by video-microscopy. Methods Enzymol. 2012, 505, 233–254. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhang, Y.; Yuan, G.C.; Orkin, S.H.; Waxman, D.J. MAnorm: A robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012, 13, R16. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Meers, M.P.; Tenenbaum, D.; Henikoff, S. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin 2019, 12, 42. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, C.; Song, K.; Qi, Z.; Huang, W.; Wang, Y. lncRNA-H19/miR-29a axis affected the viability and apoptosis of keloid fibroblasts through acting upon COL1A1 signaling. J. Cell. Biochem. 2020, 121, 4364–4376. [Google Scholar] [CrossRef]
- Bozic, M.; Caus, M.; Rodrigues-Diez, R.R.; Pedraza, N.; Ruiz-Ortega, M.; Gari, E.; Gallel, P.; Panades, M.J.; Martinez, A.; Fernandez, E.; et al. Protective role of renal proximal tubular α-synuclein in the pathogenesis of kidney fibrosis. Nat. Commun. 2020, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, R.F.; Paul, M.A.; Valmaseda, A.; Francois, A.; Jabr, R.; Anjum, S.; Marber, M.S.; Budhram-Mahadeo, V.; Heads, R.J. Protein kinase Cepsilon-calcineurin cosignaling downstream of toll-like receptor 4 downregulates fibrosis and induces wound healing gene expression in cardiac myofibroblasts. Mol. Cell. Biol. 2014, 34, 574–594. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Nakashima, M.; Zembutsu, H.; Nakamura, Y. Possible involvement of NEDD4 in keloid formation; its critical role in fibroblast proliferation and collagen production. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tian, G.; Jiang, C.; Xue, H.; Kuerbanjiang, M.; Sun, L.; Gu, L.; Zhou, H.; Liu, Y.; Zhang, Z.; et al. NPTX2 promotes colorectal cancer growth and liver metastasis by the activation of the canonical Wnt/β-catenin pathway via FZD6. Cell Death Dis. 2019, 10, 217. [Google Scholar] [CrossRef]
- Xu, G.; Fan, L.; Zhao, S.; OuYang, C. Neuronal pentraxin II (NPTX2) hypermethylation promotes cell proliferation but inhibits cell cycle arrest and apoptosis in gastric cancer cells by suppressing the p53 signaling pathway. Bioengineered 2021, 12, 1311–1323. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-Wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Chiang, H.C.; Hsieh, Y.P.; Lu, C.; Park, B.H.; Jatoi, I.; Jin, V.X.; Hu, Y.; Li, R. BRCA1 mutations attenuate super-enhancer function and chromatin looping in haploinsufficient human breast epithelial cells. Breast Cancer Res. 2019, 21, 51. [Google Scholar] [CrossRef]
- Shaker, S.A.; Ayuob, N.N.; Hajrah, N.H. Cell talk: A phenomenon observed in the keloid scar by immunohistochemical study. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 153–159. [Google Scholar] [CrossRef]
- Alexanian, M.; Przytycki, P.F.; Micheletti, R.; Padmanabhan, A.; Ye, L.; Travers, J.G.; Gonzalez-Teran, B.; Silva, A.C.; Duan, Q.; Ranade, S.S.; et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595, 438–443. [Google Scholar] [CrossRef]
- Tampe, B.; Zeisberg, M. Chromatin dynamics at the core of kidney fibrosis. Matrix Biol. 2018, 68–69, 194–229. [Google Scholar] [CrossRef]
- Beisaw, A.; Kuenne, C.; Guenther, S.; Dallmann, J.; Wu, C.C.; Bentsen, M.; Looso, M.; Stainier, D.Y.R. AP-1 Contributes to Chromatin Accessibility to Promote Sarcomere Disassembly and Cardiomyocyte Protrusion During Zebrafish Heart Regeneration. Circ. Res. 2020, 126, 1760–1778. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Feng, S.; Duttke, S.H.; Potok, M.E.; Zhang, Y.; Gallego-Bartolome, J.; Liu, W.; Jacobsen, S.E. DNA methylation-linked chromatin accessibility affects genomic architecture in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023347118. [Google Scholar] [CrossRef]
- Schick, S.; Grosche, S.; Kohl, K.E.; Drpic, D.; Jaeger, M.G.; Marella, N.C.; Imrichova, H.; Lin, J.G.; Hofstatter, G.; Schuster, M.; et al. Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet. 2021, 53, 269–278. [Google Scholar] [CrossRef]
- Zhang, G.; Guan, Q.; Chen, G.; Qian, F.; Liang, J. DNA methylation of the CDC2L1 gene promoter region decreases the expression of the CDK11p58 protein and reduces apoptosis in keloid fibroblasts. Arch. Dermatol. Res. 2018, 310, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Paul, S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front. Biosci. 2014, 19, 605–618. [Google Scholar] [CrossRef]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Botuyan, M.V.; Zhao, D.; Cui, G.; Mer, E.; Mer, G. Mechanisms of BRCA1-BARD1 nucleosome recognition and ubiquitylation. Nature 2021, 596, 438–443. [Google Scholar] [CrossRef]
- Feng, J.; Xue, S.; Pang, Q.; Rang, Z.; Cui, F. miR-141-3p inhibits fibroblast proliferation and migration by targeting GAB1 in keloids. Biochem. Biophys. Res. Commun. 2017, 490, 302–308. [Google Scholar] [CrossRef]
- Chen, J.; Liu, K.; Liu, Y.; Wang, X.; Zhang, Z. Targeting mTORC1/2 with OSI-027 inhibits proliferation and migration of keloid keratinocytes. Exp. Dermatol. 2019, 28, 270–275. [Google Scholar] [CrossRef]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Addis, R.; Cruciani, S.; Santaniello, S.; Bellu, E.; Sarais, G.; Ventura, C.; Maioli, M.; Pintore, G. Fibroblast Proliferation and Migration in Wound Healing by Phytochemicals: Evidence for a Novel Synergic Outcome. Int. J. Med. Sci. 2020, 17, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Gouzos, M.; Ramezanpour, M.; Bassiouni, A.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Antibiotics Affect ROS Production and Fibroblast Migration in an In-vitro Model of Sinonasal Wound Healing. Front. Cell. Infect. Microbiol. 2020, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Swanson, A.; Willette, A.A.; Alzheimer’s Disease Neuroimaging Initiative. Neuronal Pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer’s disease spectrum. Brain Behav. Immun. 2016, 58, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Belbin, O.; Xiao, M.F.; Xu, D.; Carmona-Iragui, M.; Pegueroles, J.; Benejam, B.; Videla, L.; Fernandez, S.; Barroeta, I.; Nunez-Llaves, R.; et al. Cerebrospinal fluid profile of NPTX2 supports role of Alzheimer’s disease-related inhibitory circuit dysfunction in adults with Down syndrome. Mol. Neurodegener. 2020, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Lu, Y.; Li, X.; Xia, L.; Wen, H.; Feng, Z.; Ju, X.; Chen, X.; Wu, X. Overexpression of NPTX2 Promotes Malignant Phenotype of Epithelial Ovarian Carcinoma via IL6-JAK2/STAT3 Signaling Pathway Under Hypoxia. Front. Oncol. 2021, 11, 643986. [Google Scholar] [CrossRef]
- Yue, L.; Lin, H.; Yuan, S.; Wu, L.; Chen, G.; Wang, J.; Feng, J. miR-1251-5p Overexpression Inhibits Proliferation, Migration, and Immune Escape in Clear Cell Renal Cell Carcinoma by Targeting NPTX2. J. Oncol. 2022, 2022, 3058588. [Google Scholar] [CrossRef]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, K.; Yang, J.; Yao, Q.; Xu, Y.; Peng, Y.; Liu, X. Comprehensive Analysis of Chromatin Accessibility and Transcriptional Landscape Identified BRCA1 Repression as a Potential Pathological Factor for Keloid. Polymers 2022, 14, 3391. https://doi.org/10.3390/polym14163391
Xie K, Yang J, Yao Q, Xu Y, Peng Y, Liu X. Comprehensive Analysis of Chromatin Accessibility and Transcriptional Landscape Identified BRCA1 Repression as a Potential Pathological Factor for Keloid. Polymers. 2022; 14(16):3391. https://doi.org/10.3390/polym14163391
Chicago/Turabian StyleXie, Kuixia, Jingrun Yang, Qianqian Yao, Yang Xu, Yonglin Peng, and Xinhua Liu. 2022. "Comprehensive Analysis of Chromatin Accessibility and Transcriptional Landscape Identified BRCA1 Repression as a Potential Pathological Factor for Keloid" Polymers 14, no. 16: 3391. https://doi.org/10.3390/polym14163391