
Generation of One-Dimensional Fibrous Polyethylene Nanocrystals in Epoxy Thermosets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Nanostructured Thermosets
3. Results and Discussion
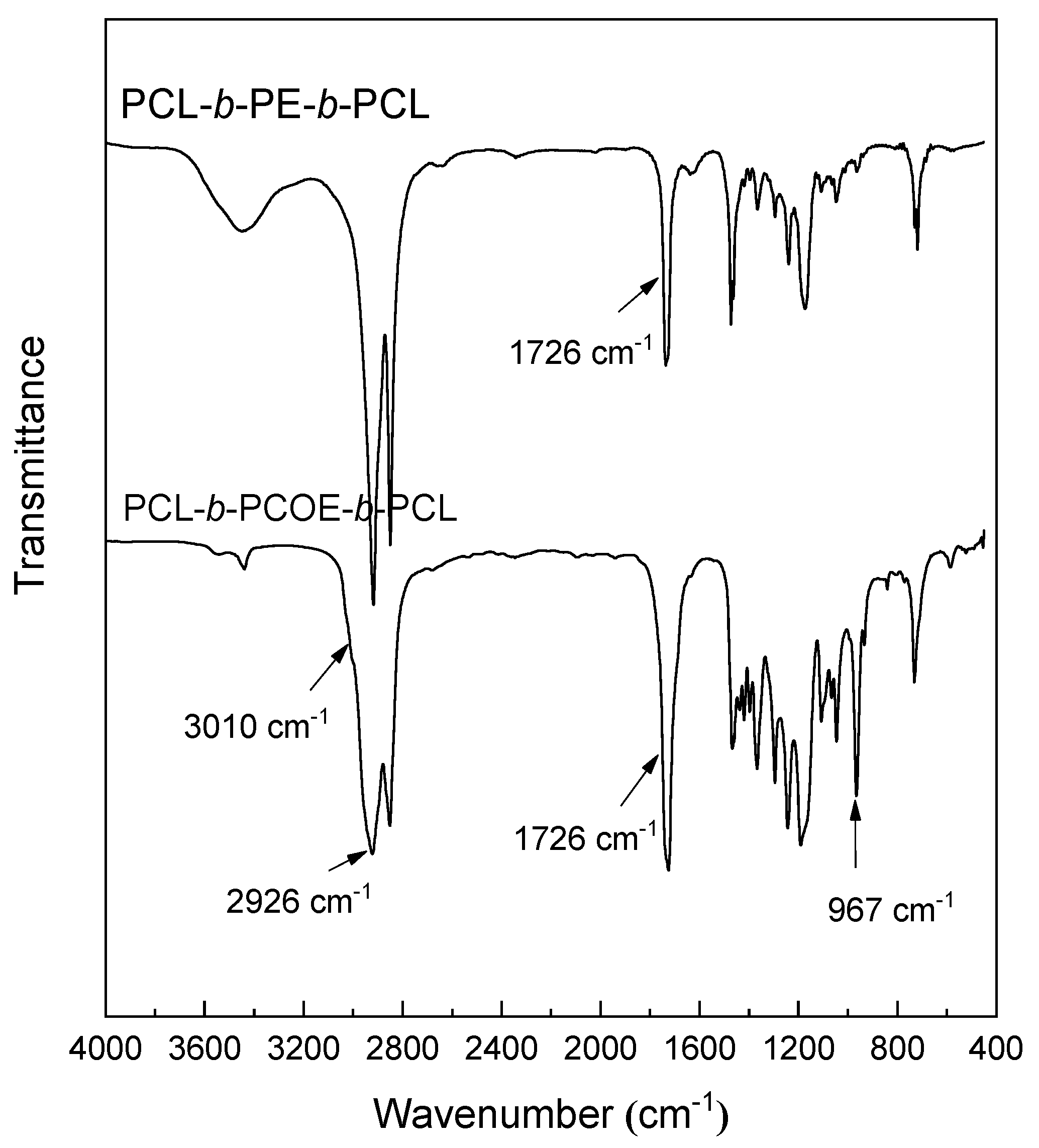
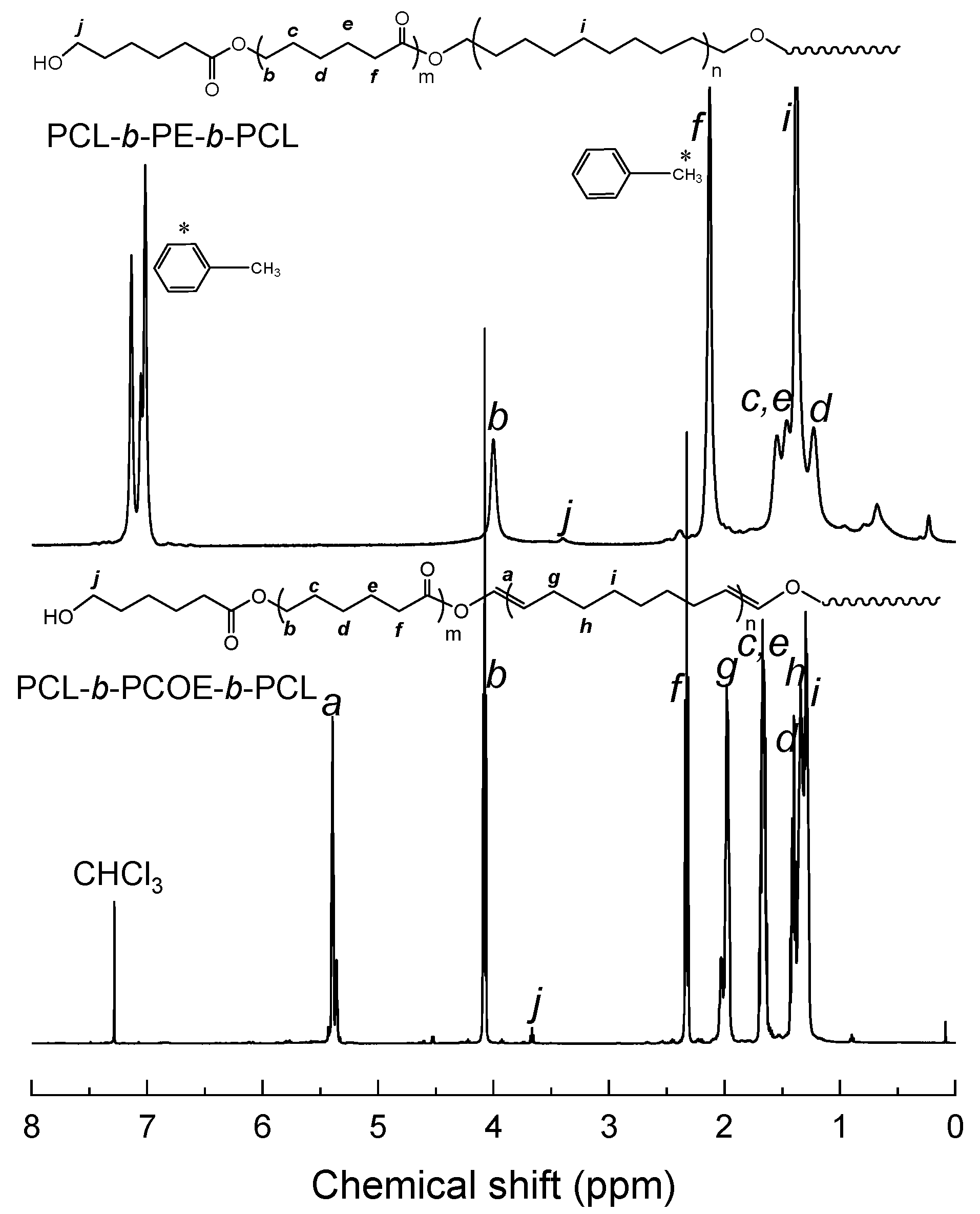
3.1. Synthesis of PCL-b-PE-b-PCL Triblock Copolymer
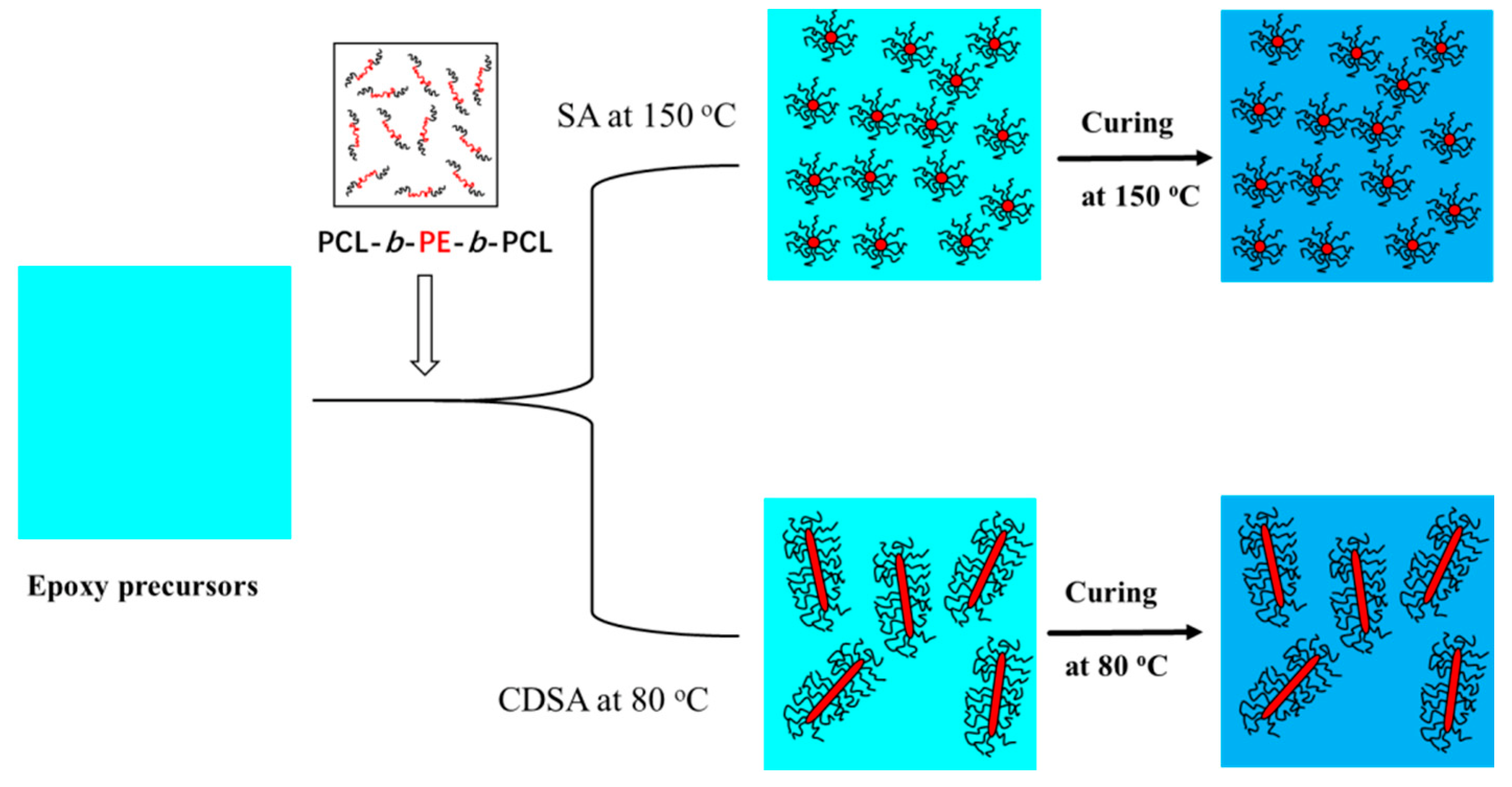
3.2. Preparation of Nanostructured Epoxy Thermosets
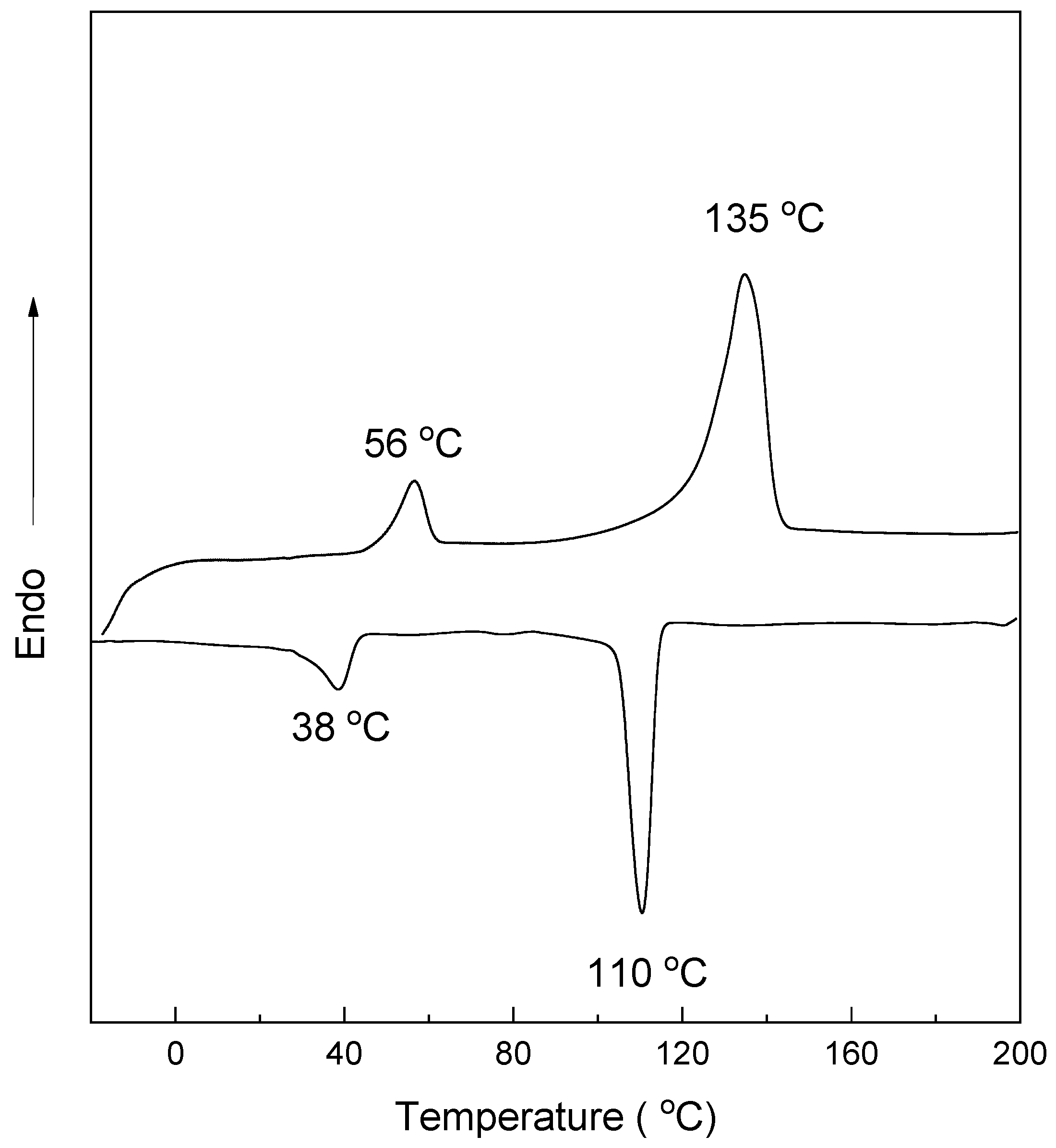
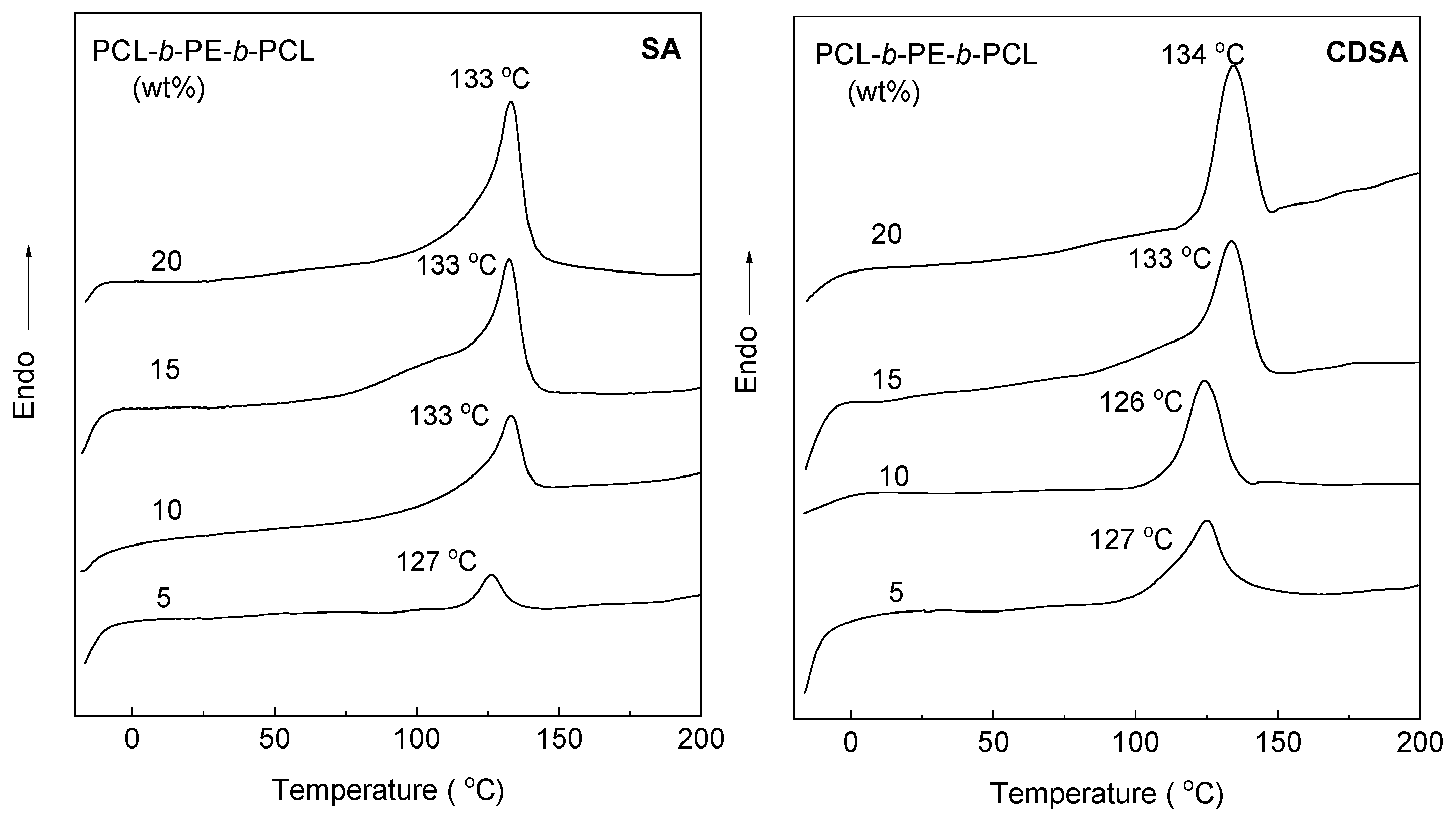
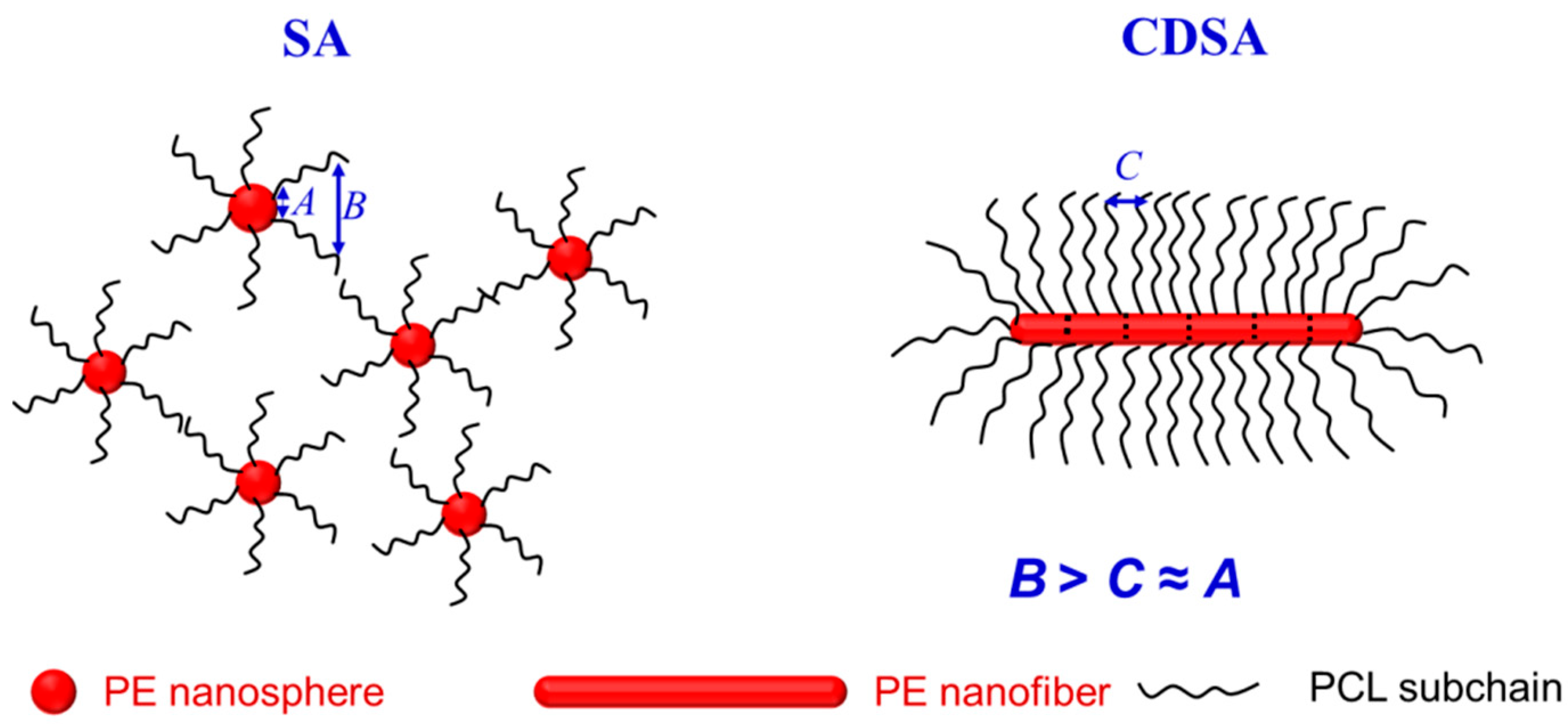
3.3. Crystallization and Melting Behavior
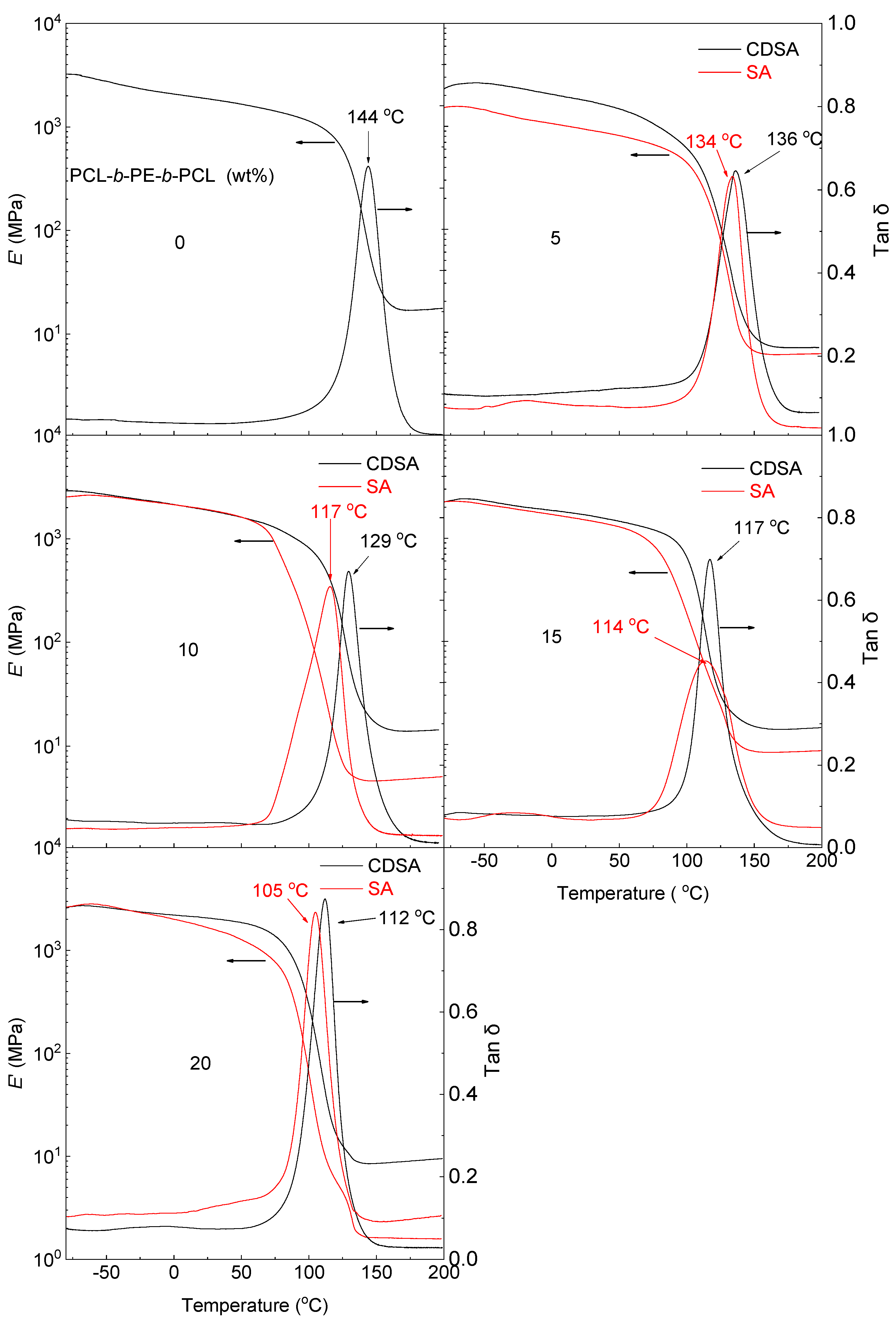
3.4. Dynamic Mechanical Thermal Properties
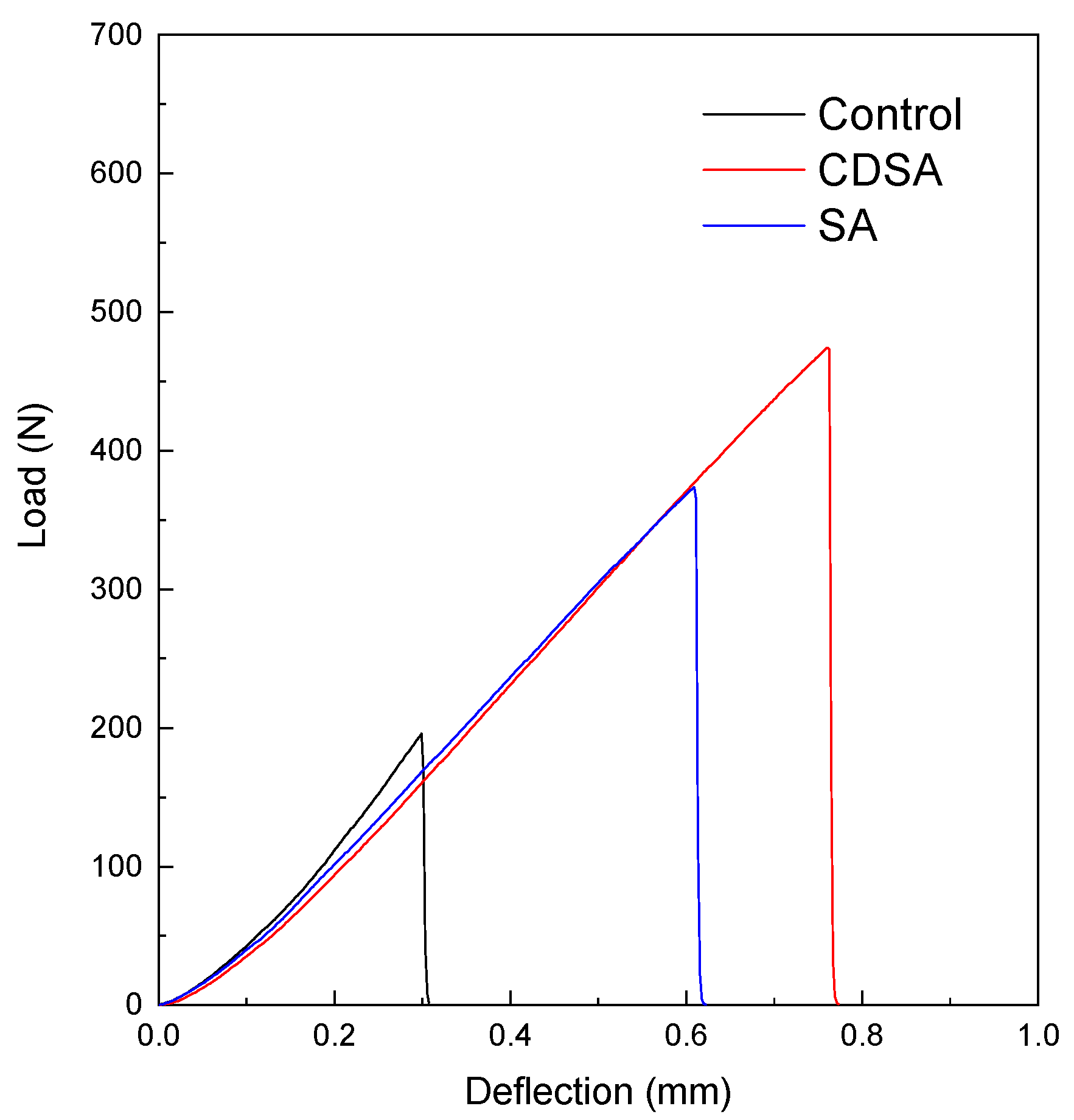
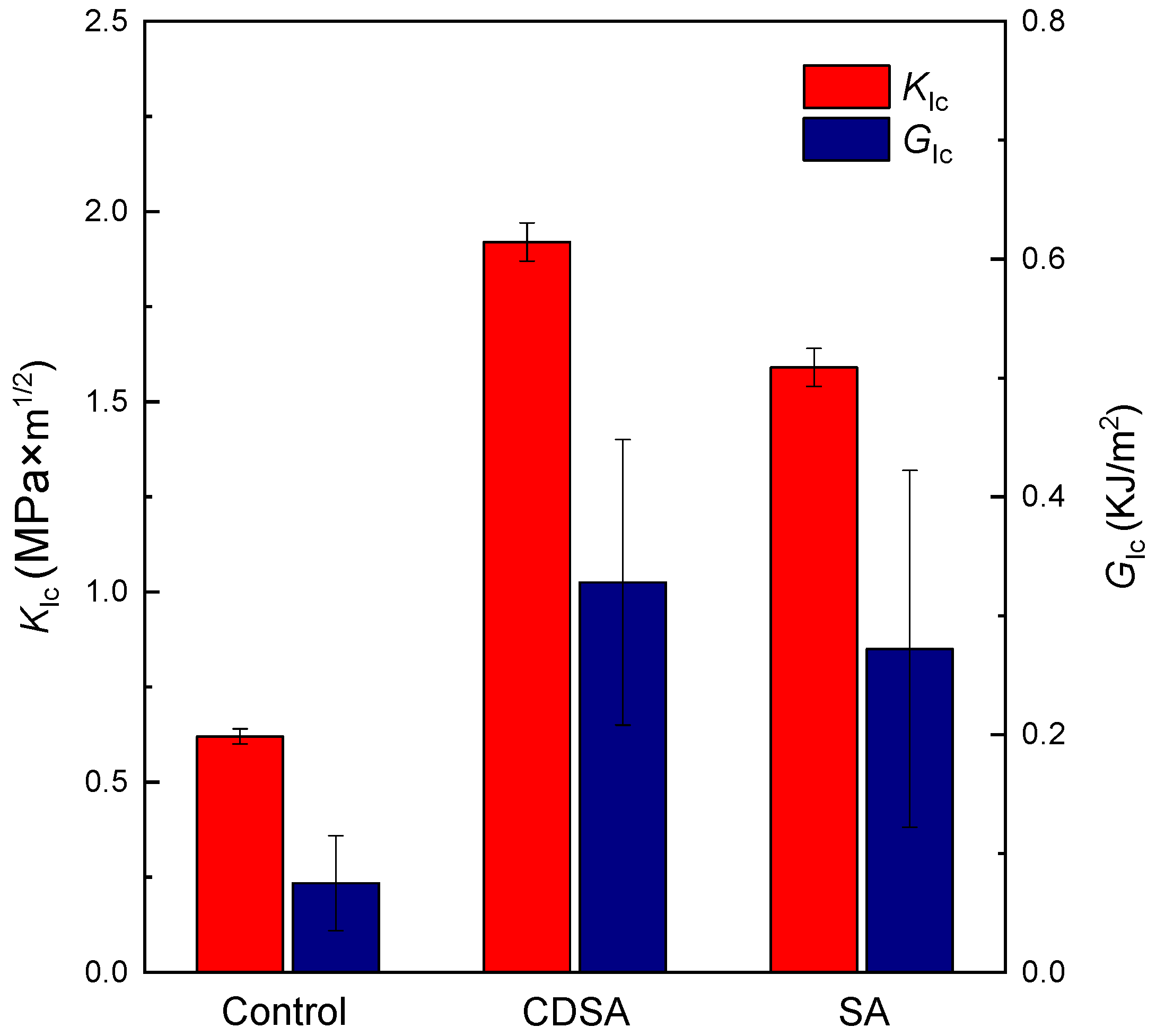
3.5. Toughening with PE Nanocrystals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hillmyer, M.A.; Lipic, P.M.; Hajduk, D.A.; Almdal, K.; Bates, F.S. Self-assembly and polymerization of epoxy resin-amphiphilic block copolymer nanocomposites. J. Am. Chem. Soc. 1997, 119, 2749–2750. [Google Scholar] [CrossRef]
- Meng, F.; Zheng, S.; Zhang, W.; Li, H.; Liang, Q. Nanostructured thermosetting blends of epoxy resin and amphiphilic poly(ε-caprolactone)-block- polybutadiene-block-poly(ε-caprolactone) triblock copolymer. Macromolecules 2006, 39, 711–719. [Google Scholar] [CrossRef]
- Zheng, S. Natured epoxyes by the use of block copolymers. In Epoxy Polymers: New Materials and Innovations; Pascault, J.P., Williams, R.J.J., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; p. 79. [Google Scholar]
- Li, L.; Zheng, S. Mechanical Properties of Epoxy/Block Copolymer Blends. In Handbook of Epoxy Blends; Parameswaranpillai, J., Hameed, N., Pionteck, J., Woo, E., Eds.; Springer: Cham, Switzerland, 2015; pp. 1–29. [Google Scholar]
- Williams, R.J.J.; Rozenberg, B.A.; Pascault, J.P. Reaction-induced phase separation in modified thermosetting polymers. Polym. Anal. Polym. Phys. 1997, 128, 95–156. [Google Scholar]
- Verchere, D.; Pascault, J.P.; Sautereau, H.; Moschiar, S.M.; Riccardi, C.C.; Williams, R.J.J. Rubber-modified epoxies. II. Influence of the cure schedule and rubber concentration on the generated morphology. J. Appl. Polym. Sci. 1991, 42, 701–716. [Google Scholar] [CrossRef]
- Chen, D.; Pascault, J.P.; Sautereau, H.; Ruseckaite, R.A.; Williams, R.J.J. Rubber-modified epoxies: III. Influence of the rubber molecular weight on the phase separation process. Polym. Int. 1994, 33, 253–261. [Google Scholar] [CrossRef]
- Yamanaka, K.; Takagi, Y.; Inoue, T. Reaction-induced phase separation in rubber-modified epoxy resins. Polymer 1989, 30, 1839–1844. [Google Scholar] [CrossRef]
- Inoue, T. Reaction-induced phase decomposition in polymer blends. Prog. Polym. Sci. 1995, 20, 119–153. [Google Scholar] [CrossRef]
- Declet-Perez, C.; Francis, L.F.; Bates, F.S. Cavitation in block copolymer modified epoxy revealed by in situ small-angle X-ray scattering. ACS Macro Lett. 2013, 2, 939–943. [Google Scholar] [CrossRef]
- Dean, J.M.; Grubbs, R.B.; Saad, W.; Cook, R.F.; Bates, F.S. Mechanical properties of block copolymer vesicle and micelle modified epoxies. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 2444–2456. [Google Scholar] [CrossRef]
- Dean, J.M.; Lipic, P.M.; Grubbs, R.B.; Cook, R.F.; Bates, F.S. Micellar structure and mechanical properties of block copolymer-modified epoxies. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 2996–3010. [Google Scholar] [CrossRef]
- Declet-Perez, C.; Francis, L.F.; Bates, F.S. Deformation processes in block copolymer toughened epoxies. Macromolecules 2015, 48, 3672–3684. [Google Scholar] [CrossRef]
- Declet-Perez, C.; Redline, E.M.; Francis, L.F.; Bates, F.S. Role of localized network damage in block copolymer toughened epoxies. ACS Macro Lett. 2012, 1, 338–342. [Google Scholar] [CrossRef]
- Liu, J.; Sue, H.-J.; Thompson, Z.J.; Bates, F.S.; Dettloff, M.; Jacob, G.; Verghese, N.; Pham, H. Nanocavitation in self-assembled amphiphilic block copolymer-modified epoxy. Macromolecules 2008, 41, 7616–7624. [Google Scholar] [CrossRef]
- Liu, J.; Thompson, Z.J.; Sue, H.-J.; Bates, F.S.; Hillmyer, M.A.; Dettloff, M.; Jacob, G.; Verghese, N.; Pham, H. Toughening of epoxies with block copolymer micelles of wormlike morphology. Macromolecules 2010, 43, 7238–7243. [Google Scholar] [CrossRef]
- Wu, S.; Guo, Q.; Peng, S.; Hameed, N.; Kraska, M.; Stühn, B.; Mai, Y.W. Toughening epoxy thermosets with block ionomer complexes: A nanostructure–mechanical property correlation. Macromolecules 2012, 45, 3829–3840. [Google Scholar] [CrossRef]
- Cao, L.; Manners, I.; Winnik, M.A. Influence of the interplay of crystallization and chain stretching on micellar morphologies: solution self-assembly of coil-crystalline poly(isoprene-block-ferrocenylsilane). Macromolecules 2002, 35, 8258–8260. [Google Scholar] [CrossRef]
- Hailes, R.L.; Oliver, A.M.; Gwyther, J.; Whittell, G.R.; Manners, I. Polyferrocenylsilanes: Synthesis, properties, and applications. Chem. Soc. Rev. 2016, 45, 5358–8407. [Google Scholar] [CrossRef]
- Lotz, B.; Kovacs, A.J. Propriétés des copolymères biséquencés polyoxyéthylène-polystyrène. Kolloid-Z. Z. Für Polym. 1966, 209, 97–114. [Google Scholar] [CrossRef]
- Lin, E.K.; Gast, A.P. Semicrystalline diblock copolymer platelets in dilute solution. Macromolecules 1996, 29, 4432–4441. [Google Scholar] [CrossRef]
- Lotz, B.; Kovacs, A.J.; Bassett, G.A.; Keller, A. Properties of copolymers composed of one poly(ethylene oxide) and one polystyrene block. Kolloid-Z. Z. Für Polym. 1966, 209, 115–128. [Google Scholar] [CrossRef]
- MacFarlane, L.; Zhao, C.; Cai, J.; Qiu, H.; Manners, I. Emerging applications for living crystallization-driven self-assembly. Chem. Sci. 2021, 12, 4661–4682. [Google Scholar] [CrossRef]
- Massey, J.; Power, K.N.; Manners, I.; Winnik, M.A. Self-assembly of a novel organometallic−inorganic block copolymer in solution and the solid state: Nonintrusive observation of novel wormlike poly(ferrocenyldimethylsilane)-b- poly(dimethylsiloxane) micelles. J. Am. Chem. Soc. 1998, 120, 9533–9540. [Google Scholar] [CrossRef]
- Massey, J.A.; Temple, K.; Cao, L.; Rharbi, Y.; Raez, J.; Winnik, M.A.; Manners, I. Self-assembly of organometallic block copolymers: The role of crystallinity of the core-forming polyferrocene block in the micellar morphologies formed by poly(ferrocenylsilane-b-dimethylsiloxane) in n-alkane solvents. J. Am. Chem. Soc. 2000, 122, 11577–11584. [Google Scholar] [CrossRef]
- Ramzi, A.; Prager, M.; Richter, D.; Efstratiadis, V.; Hadjichristidis, N.; Young, R.N.; Allgaier, J.B. Influence of polymer architecture on the formation of micelles of miktoarm star copolymers polyethylene/poly(ethylenepropylene) in the selective solvent decane. Macromolecules 1997, 30, 7171–7182. [Google Scholar] [CrossRef]
- Richter, D.; Schneiders, D.; Monkenbusch, M.; Willner, L.; Fetters, L.J.; Huang, J.S.; Lin, M.; Mortensen, K.; Farago, B. Polymer aggregates with crystalline cores: The system polyethylene-poly(ethylenepropylene). Macromolecules 1997, 30, 1053–1068. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, C.; Li, L.; Zheng, S. Polystyrene-block-polyethylene-block- polystyrene triblock copolymers: Synthesis and crystallization-driven self-assembly behavior. Polymer 2017, 128, 1–11. [Google Scholar] [CrossRef]
- MaryMichell, R.; Müller, A.J. Confined crystallization of polymeric materials. Prog. Polym. Sci. 2016, 54, 183–213. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Zheng, S. Formation and confined crystallization of polyethylene nanophases in epoxy thermosets. Macromolecules 2013, 46, 2740–2753. [Google Scholar] [CrossRef]
- Lipic, P.M.; Bates, F.S.; Hillmyer, M.A. Nanostructured thermosets from self-assembled amphiphilic block copolymer/epoxy resin mixtures. J. Am. Chem. Soc. 1998, 120, 8963–8970. [Google Scholar] [CrossRef]
- Guo, Q.; Thomann, R.; Gronski, W.; Thurn-Albrecht, T. Phase behavior, crystallization, and hierarchical nanostructures in self-organized thermoset blends of epoxy resin and amphiphilic poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) triblock copolymers. Macromolecules 2002, 35, 3133–3144. [Google Scholar] [CrossRef]
- Guo, Q.P.; Dean, J.M.; Grubbs, R.B.; Bates, F.S. Block copolymer modified novolac epoxy resin. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1994–2003. [Google Scholar] [CrossRef]
- Liu, D.Y.; Krogstad, D.V. Self-assembly and phase transformation of block copolymer nanostructures in ionic liquid-cured epoxy. Macromolecules 2021, 54, 988–994. [Google Scholar] [CrossRef]
- Blanco, M.; López, M.; Kortaberria, G.; Mondragon, I. Nanostructured thermosets from self-assembled amphiphilic block copolymer/epoxy resin mixtures: Effect of copolymer content on nanostructures. Polym. Inter. 2010, 59, 523–528. [Google Scholar] [CrossRef]
- Hermel-Davidock, T.J.; Tang, H.S.; Murray, D.J.; Hahn, S.F. Control of the block copolymer morphology in templated epoxy thermosets. J. Polym. Sci. Part B Polym. Phys. 2010, 45, 3338–3348. [Google Scholar] [CrossRef]
- Garate, H.; Mondragon, I.; D’Accorso, N.B.; Goyanes, S. Exploring microphase separation behavior of epoxidized poly(styrene-b-isoprene-b-styrene) block copolymer inside thin epoxy coatings. Macromolecules 2013, 46, 2182–2187. [Google Scholar] [CrossRef]
- He, X.; Liu, Y.; Zhang, R.; Wu, Q.; Chen, T.; Sun, P.; Wang, X.; Xue, G. Unique interphase and cross-linked network controlled by different miscible blocks in nanostructured epoxy/block copolymer blends characterized by solid-state NMR. J. Phys. Chem. C 2014, 118, 13285–13299. [Google Scholar] [CrossRef]
- Li, M.; Zheng, G.; Yang, C.; Zou, H.; Mei, L. High toughness induced by wormlike-nanostructure in epoxy thermoset containing amphiphilic PDMS-PCL block copolymers. Ind. Eng. Chem. Res. 2018, 57, 13036–13047. [Google Scholar] [CrossRef]
- Chong, H.; Taylor, A.C. The microstructure and fracture performance of styrene-butadiene-methylmethacrylate block copolymer-modified epoxy polymers. J. Mater. Sci. 2013, 48, 6762–6777. [Google Scholar] [CrossRef]
- Shen, K.; Zhou, Q.; Xu, Q.; Jiang, D.; Ni, L. Reaction-induced microphase separation in DDS-cured TGDDM thermosets containing PCL-b-PES-b-PCL triblock copolymer. RSC Adv. 2015, 5, 61070–61080. [Google Scholar] [CrossRef]
- Zou, Z.; Liu, X.; Wu, Y.; Tang, B.; Chen, M.; Zhao, X. Hyperbranched polyurethane as a highly efficient toughener in epoxy thermosets with reaction-induced microphase separation. RSC Adv. 2016, 6, 18060–18070. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, F.; Wang, K.; Chen, L. Nanostructured thermoset epoxy resin templated by an amphiphilic poly(ethylene oxide)-block-poly(dimethylsiloxane) diblock copolymer. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 3042–3052. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, J.; Chen, L.; Wang, K. Nanostructures and nanoporosity in thermoset epoxy blends with an amphiphilic polyisoprene-block-poly(4-vinyl pyridine) reactive diblock copolymer. Polymer 2008, 49, 1737–1742. [Google Scholar] [CrossRef]
- Yu, R.; Zheng, S. Morphological transition from spherical to lamellar nanophases in epoxy thermosets containing poly(ethylene oxide)-block-poly(ε-caprolactone)- block-polystyrene triblock copolymer by hardeners. Macromolecules 2011, 44, 8546–8557. [Google Scholar] [CrossRef]
- Li, J.; Xiang, Y.; Zheng, S. Hyperbranched block copolymer from AB2 macromonomer: Synthesis and its reaction-induced microphase separation in epoxy thermosets. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 368–380. [Google Scholar] [CrossRef]
- Guo, Q.; Thomann, R.; Gronski, W.; Staneva, R.; Ivanova, R.; Stühn, B. Nanostructures, semicrytalline morphology, and nanoscale confinement effect on the crystallization kinetics in self-organized block copolymer/thermoset blends. Macromolecules 2003, 36, 3635–3645. [Google Scholar] [CrossRef]
- Zucchi, I.A.; Schroeder, W.F. Nanoribbons with semicrystalline core dispersed in a visible-light photopolymerized epoxy network. Polymer 2015, 56, 300–308. [Google Scholar] [CrossRef]
- Puig, J.; Zucchi, I.A.; Ceolin, M.; Schroeder, W.F.; Williams, R.J.J. Evolution of morphologies of a PE-b-PEO block copolymer in an epoxy solvent induced by polymerization followed by crystallization-driven self-assembly of PE blocks during cooling. RSC Adv. 2016, 6, 34903–34912. [Google Scholar] [CrossRef]
- Zhang, J.; Matta, M.E.; Hillmyer, M.A. Synthesis of sequence-specific vinyl copolymers by regioselective ROMP of multiply substituted cyclooctenes. ACS Macro Lett. 2012, 1, 1383–1387. [Google Scholar] [CrossRef]
- Onbulak, S.; Wang, Y.; Brutman, J.P.; Hillmyer, M.A. Synthesis and utility of ethylene (meth)acrylate copolymers prepared by a tandem ring-opening polymerization hydrogenation strategy. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3117–3126. [Google Scholar] [CrossRef]
- Feng, Y.; Jie, S.; Li, B.-G. Synthesis of ethylene/vinyl ester copolymers with pendent linear branches via ring-opening metathesis polymerization of fatty acid-derived cyclooctenes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2211–2220. [Google Scholar] [CrossRef]
- Wang, X.; Guerin, G.; Wang, H.; Wang, Y.; Manners, I.; Winnik, M.A. Cylindrical block copolymer micelles and co-micelles of controlled length and architecture. Science 2007, 317, 644–647. [Google Scholar] [CrossRef] [Green Version]
- Gilroy, J.B.; Gadt, T.; Whittell, G.R.; Chabanne, L.; Mitchels, J.M.; Richardson, R.M.; Winnik, M.A.; Manners, I. Monodisperse cylindrical micelles by crystallization-driven living self-assembly. Nat. Chem. 2010, 2, 566–570. [Google Scholar] [CrossRef]
- Wang, X.S.; Arsenault, A.; Ozin, G.A.; Winnik, M.A.; Manners, I. Shell cross-linked cylinders of polyisoprene-b-ferrocenyldimethylsilane: Formation of magnetic ceramic replicas and microfluidic channel alignment and patterning. J. Am. Chem. Soc. 2003, 125, 12686–12687. [Google Scholar] [CrossRef]
- Kavesh, S.; Schultz, J.M. Lamellar and interlamellar structure in melt-crystallized polyethylene. I. Degree of crystallinity, atomic positions, particle size, and lattice disorder of the first and second kinds. J. Polym. Sci. Part A 1970, 8, 243–276. [Google Scholar] [CrossRef]
- Yi, H.; Wang, X.; Wei, T.; Lin, H.; Zheng, B. Nanoscale-confined crystallization in epoxy resin and polyethylene-block-poly(ethylene oxide) diblock copolymer blends. Colloid. Polym. Sci. 2012, 290, 1347–1352. [Google Scholar]
- Koleske, J.V.; Lundberg, R.D. Lactone polymers. I. Glass transition temperature of poly-ε-caprolactone by means on compatible polymer mixtures. J. Polym. Sci. Part B Polym. Phys. 1969, 7, 795–807. [Google Scholar] [CrossRef]
- Liu, J.; Sue, H.-J.; Thompson, Z.J.; Bates, F.S.; Dettloff, M.; Jacob, G.; Verghese, N.; Pham, H. Strain rate effect on toughening of nano-sized PEP–PEO block copolymer modified epoxy. Acta Mater. 2009, 57, 2691–2701. [Google Scholar] [CrossRef]
- Thio, Y.S.; Wu, J.; Bates, F.S. Epoxy toughening using low molecular weight poly(hexylene oxide)-poly(ethylene oxide) diblock copolymers. Macromolecules 2006, 39, 7187–7189. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, H.; Wang, H.; Li, L.; Zheng, S. Generation of One-Dimensional Fibrous Polyethylene Nanocrystals in Epoxy Thermosets. Polymers 2022, 14, 3921. https://doi.org/10.3390/polym14183921
Mei H, Wang H, Li L, Zheng S. Generation of One-Dimensional Fibrous Polyethylene Nanocrystals in Epoxy Thermosets. Polymers. 2022; 14(18):3921. https://doi.org/10.3390/polym14183921
Chicago/Turabian StyleMei, Honggang, Huaming Wang, Lei Li, and Sixun Zheng. 2022. "Generation of One-Dimensional Fibrous Polyethylene Nanocrystals in Epoxy Thermosets" Polymers 14, no. 18: 3921. https://doi.org/10.3390/polym14183921