
L-Cysteine Modified Chitosan Nanoparticles and Carbon-Based Nanostructures for the Intranasal Delivery of Galantamine


 ,
,  , and
, and 
Abstract
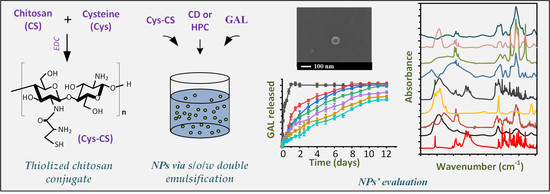
:
1. Introduction
2. Results and Discussion
2.1. Cys-CS Characterization
2.1.1. H-NMR
2.1.2. FTIR Spectroscopy
2.1.3. Degree of Deacetylation (DD)
2.1.4. Physical State Evaluation via pXRD
2.1.5. Solubility Result
2.2. Characterization of GAL-Loaded NPs
2.2.1. Drug Loading, EE and NP Yield
2.2.2. NPs’ Morphology via SEM
2.2.3. PSD and ζ-Potential Results
2.2.4. Evaluation of Drug’s Crystalline State after NP Production via pXRD
2.2.5. Evaluation of Molecular Interactions after NP Production via FTIR Spectroscopy
2.2.6. In Vitro Dissolution Release Results
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Materials and NPs
3.2.1. Synthesis of Cys-CS
3.2.2. Synthesis of HPC
3.2.3. Synthesis of CDs
3.2.4. HPC Drug Loading Process
3.2.5. CDs Drug Loading Process
3.2.6. Preparation of NPs
3.3. Characterization
3.3.1. Characterization of HPC
3.3.2. Characterization of CDs
3.3.3. Characterization of Cys-CS
3.3.4. Characterization of NPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bores, G.M.; Huger, F.P.; Petko, W.; Mutlib, A.E.; Camacho, F.; Rush, D.K.; Selk, D.E.; Wolf, V.; Kosley, R.W., Jr.; Davis, L.; et al. Pharmacological evaluation of novel alzheimer’s disease therapeutics: Acetylcholinesterase inhibitors related to galanthamine. J. Pharmacol. Exp. Ther. 1996, 277, 728–738. [Google Scholar]
- Thomsen, T.; Kewitz, H. Selective inhibition of human acetylcholinesterase by galanthamine in vitro and in vivo. Life Sci. 1990, 46, 1553–1558. [Google Scholar] [CrossRef]
- Olin, J.; Schneider, L. Galantamine for alzheimer’s disease. Cochrane Database Syst. Rev. 2002, Cd001747. [Google Scholar] [CrossRef]
- Koola, M.M. Galantamine-memantine combination in the treatment of alzheimer’s disease and beyond. Psychiatry Res. 2020, 293, 113409. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar] [PubMed]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. JAD 2014, 41, 615–631. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Chapman, C.D.; Frey, W.H.; Craft, S.; Danielyan, L.; Hallschmid, M.; Schiöth, H.B.; Benedict, C. Intranasal treatment of central nervous system dysfunction in humans. Pharm. Res. 2013, 30, 2475–2484. [Google Scholar] [CrossRef]
- Aderibigbe, B.A.; Naki, T. Design and efficacy of nanogels formulations for intranasal administration. Molecules 2018, 23, 1241. [Google Scholar] [CrossRef]
- Cunha, S.; Amaral, M.H.; Lobo, J.M.S.; Silva, A.C. Lipid nanoparticles for nasal/intranasal drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 257–282. [Google Scholar] [CrossRef]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, Y.; Zhao, N.; Hao, B.; Wang, X.; Kong, P. Pharmacokinetic behavior and efficiency of acetylcholinesterase inhibition in rat brain after intranasal administration of galanthamine hydrobromide loaded flexible liposomes. Environ. Toxicol. Pharmacol. 2012, 34, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, A.S.; Farid, R.M.; ElGamal, S.S. Complexation as an approach to entrap cationic drugs into cationic nanoparticles administered intranasally for alzheimer’s disease management: Preparation and detection in rat brain. Drug Dev. Ind. Pharm. 2015, 41, 2055–2068. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Maelicke, A.; Montag, D. Nasal application of the galantamine pro-drug memogain slows down plaque deposition and ameliorates behavior in 5x familial alzheimer’s disease mice. J. Alzheimer’s Dis. 2015, 46, 123–136. [Google Scholar] [CrossRef]
- Hanafy, A.S.; Farid, R.M.; Helmy, M.W.; ElGamal, S.S. Pharmacological, toxicological and neuronal localization assessment of galantamine/chitosan complex nanoparticles in rats: Future potential contribution in alzheimer’s disease management. Drug Deliv. 2016, 23, 3111–3122. [Google Scholar] [CrossRef]
- Kandil, L.S.; Farid, R.M.; ElGamal, S.S.; Hanafy, A.S. Intranasal galantamine/chitosan complex nanoparticles elicit neuroprotection potentials in rat brains via antioxidant effect. Drug Dev. Ind. Pharm. 2021, 47, 735–740. [Google Scholar] [CrossRef]
- Desai, K.G. Chitosan nanoparticles prepared by ionotropic gelation: An overview of recent advances. Crit. Rev. Ther. Drug Carr. Syst. 2016, 33, 107–158. [Google Scholar] [CrossRef]
- Quiñones, J.P.; Peniche, H.; Peniche, C. Chitosan based self-assembled nanoparticles in drug delivery. Polymers 2018, 10, 235. [Google Scholar] [CrossRef]
- Rashki, S.; Asgarpour, K.; Tarrahimofrad, H.; Hashemipour, M.; Ebrahimi, M.S.; Fathizadeh, H.; Khorshidi, A.; Khan, H.; Marzhoseyni, Z.; Salavati-Niasari, M.; et al. Chitosan-based nanoparticles against bacterial infections. Carbohydr. Polym. 2021, 251, 117108. [Google Scholar] [CrossRef]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, bioapplications, and toxicity evaluation of chitosan-based nanoparticles. Int. J. Mol. Sci. 2019, 20, 5776. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Feng, J.; Liu, M.; Hu, K. Chitosan and chitosan coating nanoparticles for the treatment of brain disease. Int. J. Pharm. 2019, 560, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Xing, R.; Liu, S.; Qin, Y.; Li, K.; Li, P. Advances in chitosan-based nanoparticles for oncotherapy. Carbohydr. Polym. 2019, 222, 115004. [Google Scholar] [CrossRef] [PubMed]
- Baldrick, P. The safety of chitosan as a pharmaceutical excipient. Regul. Toxicol. Pharmacol. 2010, 56, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Elgadir, M.A.; Uddin, M.S.; Ferdosh, S.; Adam, A.; Chowdhury, A.J.K.; Sarker, M.Z.I. Impact of chitosan composites and chitosan nanoparticle composites on various drug delivery systems: A review. J. Food Drug Anal. 2015, 23, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Khan, J.; Saraf, S.; Saraf, S. Formulation and evaluation of chitosan-based long-acting injectable hydrogel for pegylated melphalan conjugate. J. Pharm. Pharmacol. 2014, 66, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- M. Ways, T.M.; Lau, W.M.; Khutoryanskiy, V.V. Chitosan and its derivatives for application in mucoadhesive drug delivery systems. Polymers 2018, 10, 267. [Google Scholar] [CrossRef]
- Caramella, C.; Ferrari, F.; Bonferoni, M.C.; Rossi, S.; Sandri, G. Chitosan and its derivatives as drug penetration enhancers. J. Drug Deliv. Sci. Technol. 2010, 20, 5–13. [Google Scholar] [CrossRef]
- Sogias, I.A.; Khutoryanskiy, V.V.; Williams, A.C. Exploring the factors affecting the solubility of chitosan in water. Macromol. Chem. Phys. 2010, 211, 426–433. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Schwarz, V.; Steininger, S. Polymers with thiol groups: A new generation of mucoadhesive polymers? Pharm Res 1999, 16, 876–881. [Google Scholar] [CrossRef]
- Casettari, L.; Vllasaliu, D.; Mantovani, G.; Howdle, S.M.; Stolnik, S.; Illum, L. Effect of pegylation on the toxicity and permeability enhancement of chitosan. Biomacromolecules 2010, 11, 2854–2865. [Google Scholar] [CrossRef]
- Hauptstein, S.; Bonengel, S.; Griessinger, J.; Bernkop-Schnürch, A. Synthesis and characterization of ph tolerant and mucoadhesive (thiol-polyethylene glycol) chitosan graft polymer for drug delivery. J. Pharm. Sci. 2014, 103, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Prabaharan, M.; Nair, S.V.; Tokura, S.; Tamura, H.; Selvamurugan, N. Novel carboxymethyl derivatives of chitin and chitosan materials and their biomedical applications. Prog. Mater. Sci. 2010, 55, 675–709. [Google Scholar] [CrossRef]
- Jintapattanakit, A.; Junyaprasert, V.B.; Kissel, T. The role of mucoadhesion of trimethyl chitosan and pegylated trimethyl chitosan nanocomplexes in insulin uptake. J. Pharm. Sci. 2009, 98, 4818–4830. [Google Scholar] [CrossRef]
- Kafedjiiski, K.; Krauland, A.H.; Hoffer, M.H.; Bernkop-Schnürch, A. Synthesis and in vitro evaluation of a novel thiolated chitosan. Biomaterials 2005, 26, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Kast, C.E.; Frick, W.; Losert, U.; Bernkop-Schnürch, A. Chitosan-thioglycolic acid conjugate: A new scaffold material for tissue engineering? Int. J. Pharm. 2003, 256, 183–189. [Google Scholar] [CrossRef]
- Sajomsang, W.; Rungsardthong Ruktanonchai, U.; Gonil, P.; Nuchuchua, O. Mucoadhesive property and biocompatibility of methylated n-aryl chitosan derivatives. Carbohydr. Polym. 2009, 78, 945–952. [Google Scholar] [CrossRef]
- Upadhyaya, L.; Singh, J.; Agarwal, V.; Tewari, R.P. The implications of recent advances in carboxymethyl chitosan based targeted drug delivery and tissue engineering applications. J. Control. Release Off. J. Control. Release Soc. 2014, 186, 54–87. [Google Scholar] [CrossRef] [PubMed]
- Krauland, A.H.; Leitner, V.M.; Grabovac, V.; Bernkop-Schnürch, A. In vivo evaluation of a nasal insulin delivery system based on thiolated chitosan. J. Pharm. Sci. 2006, 95, 2463–2472. [Google Scholar] [CrossRef]
- Sunena; Singh, S.K.; Mishra, D.N. Nose to brain delivery of galantamine loaded nanoparticles: In-vivo pharmacodynamic and biochemical study in mice. Curr. Drug Deliv. 2019, 16, 51–58. [Google Scholar] [CrossRef]
- Nanaki, S.G.; Spyrou, K.; Bekiari, C.; Veneti, P.; Baroud, T.N.; Karouta, N.; Grivas, I.; Papadopoulos, G.C.; Gournis, D.; Bikiaris, D.N. Hierarchical porous carbon—plla and plga hybrid nanoparticles for intranasal delivery of galantamine for alzheimer’s disease therapy. Pharmaceutics 2020, 12, 227. [Google Scholar] [CrossRef]
- Calabrese, G.; De Luca, G.; Nocito, G.; Rizzo, M.G.; Lombardo, S.P.; Chisari, G.; Forte, S.; Sciuto, E.L.; Conoci, S. Carbon dots: An innovative tool for drug delivery in brain tumors. Int. J. Mol. Sci. 2021, 22, 11783. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, Z.; Zeng, G.; Zhang, Y.; Xue, W.; Liu, Z. Polyethylenimine-modified fluorescent carbon dots as vaccine delivery system for intranasal immunization. ACS Biomater. Sci. Eng. 2018, 4, 142–150. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, A. Carbon quantum dots: Synthesis, properties and applications. J. Mater. Chem. C 2014, 2, 6921–6939. [Google Scholar] [CrossRef]
- Li, R.; Liu, Q.; Wu, H.; Wang, K.; Li, L.; Zhou, C.; Ao, N. Preparation and characterization of in-situ formable liposome/chitosan composite hydrogels. Mater. Lett. 2018, 220, 289–292. [Google Scholar] [CrossRef]
- Meena, L.K.; Raval, P.; Kedaria, D.; Vasita, R. Study of locust bean gum reinforced cyst-chitosan and oxidized dextran based semi-ipn cryogel dressing for hemostatic application. Bioact. Mater. 2018, 3, 370–384. [Google Scholar] [CrossRef]
- Arif, M.; Dong, Q.J.; Raja, M.A.; Zeenat, S.; Chi, Z.; Liu, C.G. Development of novel ph-sensitive thiolated chitosan/pmla nanoparticles for amoxicillin delivery to treat helicobacter pylori. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 83, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, S.; Mortazavian, E.; Mohammadi, Z.; Samadi, F.Y.; Samadikhah, H.; Taheritarigh, S.; Tehrani, N.R.; Rafiee-Tehrani, M. Thiolated methylated dimethylaminobenzyl chitosan: A novel chitosan derivative as a potential delivery vehicle. Int. J. Biol. Macromol. 2017, 95, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, X.; Zhang, R.; Wang, L.; Feng, Q. A novel dual microsphere based on water-soluble thiolated chitosan/mesoporous calcium carbonate for controlled dual drug delivery. Mater. Lett. 2021, 285, 129142. [Google Scholar] [CrossRef]
- Nanaki, S.G.; Christodoulou, E.; Bikiaris, N.D.; Kapourani, A.; Kontogiannopoulos, K.N.; Vergkizi-Nikolakaki, S.; Barmpalexis, P. Leflunomide sustained skin delivery based on sulfobetaine-modified chitosan nanoparticles embedded in biodegradable polyesters films. Polymers 2021, 13, 960. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Medeiros Borsagli, F.G.L.; Carvalho, I.C.; Mansur, H.S. Amino acid-grafted and n-acylated chitosan thiomers: Construction of 3d bio-scaffolds for potential cartilage repair applications. Int. J. Biol. Macromol. 2018, 114, 270–282. [Google Scholar] [CrossRef]
- Baroud, T.N.; Giannelis, E.P. Role of mesopore structure of hierarchical porous carbons on the electrosorption performance of capacitive deionization electrodes. ACS Sustain. Chem. Eng. 2019, 7, 7580–7596. [Google Scholar] [CrossRef]
- Mauro, N.; Utzeri, M.A.; Drago, S.E.; Buscarino, G.; Cavallaro, G.; Giammona, G. Carbon nanodots as functional excipient to develop highly stable and smart plga nanoparticles useful in cancer theranostics. Pharmaceutics 2020, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Nanaki, S.G.; Andrianidou, S.; Barmpalexis, P.; Christodoulou, E.; Bikiaris, D.N. Leflunomide loaded chitosan nanoparticles for the preparation of aliphatic polyester based skin patches. Polymers 2021, 13, 1539. [Google Scholar] [CrossRef] [PubMed]
- Kouloumpis, A.; Thomou, E.; Chalmpes, N.; Dimos, K.; Spyrou, K.; Bourlinos, A.B.; Koutselas, I.; Gournis, D.; Rudolf, P. Graphene/carbon dot hybrid thin films prepared by a modified langmuir–schaefer method. ACS Omega 2017, 2, 2090–2099. [Google Scholar] [CrossRef]
- Qu, S.; Wang, X.; Lu, Q.; Liu, X.; Wang, L. A biocompatible fluorescent ink based on water-soluble luminescent carbon nanodots. Angew. Chem. Int. Ed. 2012, 51, 12215–12218. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Xie, M.; Hu, B.; Zhou, L.; Saeeduddin, M.; Zeng, X. Enhanced solubility and antioxidant activity of chlorogenic acid-chitosan conjugates due to the conjugation of chitosan with chlorogenic acid. Carbohydr. Polym. 2017, 170, 206–216. [Google Scholar] [CrossRef]
- Moura, C.M.d.; Moura, J.M.d.; Soares, N.M.; Pinto, L.A.d.A. Evaluation of molar weight and deacetylation degree of chitosan during chitin deacetylation reaction: Used to produce biofilm. Chem. Eng. Processing Process Intensif. 2011, 50, 351–355. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Drug Loading (%) | EE (%) | NPs’ Yield (%) |
|---|---|---|---|
| Nano-CS-GAL | 14.49 ± 1.06 | 41.83 ± 1.42 | 20.59 ± 0.61 |
| Nano-CS-HPC-GAL | 11.03 ± 0.95 | 38.95 ± 3.49 | 19.35 ± 1.05 |
| Nano-CS-CD-GAL | 10.91 ± 0.86 | 36.54 ± 2.94 | 21.24 ± 0.35 |
| Nano-Cys-CS-GAL | 19.37 ± 1.54 | 42.36 ± 2.68 | 18.22 ± 0.64 |
| Nano-Cys-CS-HPC-GAL | 17.32 ± 0.86 | 40.69 ± 2.49 | 20.41 ± 0.38 |
| Nano-Cys-CS-CD-GAL | 16.83 ± 1.48 | 39.28 ± 1.95 | 19.06 ± 1.52 |
| Sample | Particle Size (nm) | PDI | ζ-Potential (mV) |
|---|---|---|---|
| Nano-CS | 328.58 ± 2.29 | 0.92 | 54.6 ± 2.2 |
| Nano-Cys-CS | 508.91 ± 3.10 | 0.81 | 58.3 ± 1.9 |
| Nano-CS-GAL | 386.86 ± 1.37 | 0.96 | 41.9 ± 0.3 |
| Nano-Cys-CS-GAL | 527.24 ± 2.68 | 0.88 | 44.0 ± 1.1 |
| Nano-CS-HPC-GAL | 983.02 ± 4.61 | 0.94 | 51.6 ± 2.9 |
| Nano-Cys-CS-HPC-GAL | 1030.95 ± 5.98 | 0.91 | * |
| Nano-CS-CD-GAL | 792.31 ± 4.08 | 0.87 | 43.9 ± 0.9 |
| Nano-Cys-CS-CD-GAL | 828.84 ± 5.81 | 0.91 | 44.5 ± 1.3 |
| Release Model | NPs’ Formulations | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CS-GAL | Cys-CS-GAL | CS-HPC-GAL | Cys-CS-HPC-GAL | CS-CD-GAL | Cys-CS-CD-GAL | |||||||
| R2 | k | R2 | k | R2 | k | R2 | k | R2 | k | R2 | k | |
| Zero order | 0.87 | 9.03 d⁻1 | 0.90 | 8.63 d⁻1 | 0.80 | 9.09 d⁻1 | 0.93 | 7.38 d⁻1 | 0.97 | 6.67 d⁻1 | 0.99 | 6.56 d⁻1 |
| First order | 0.99 | 0.37 d⁻1 | 0.99 | 0.29 d⁻1 | 0.99 | 0.53 d⁻1 | 0.96 | 0.21 d⁻1 | 0.96 | 0.15 d⁻1 | 0.98 | 0.12 d⁻1 |
| Higuchi | 0.97 | 32.83 d⁻1/2 | 0.98 | 30.33 d⁻1/2 | 0.94 | 35.03 d⁻1/2 | 0.98 | 26.93 d⁻1/2 | 0.97 | 23.40 d⁻1/2 | 0.95 | 20.60 d⁻1/2 |
| Hixson-Crowell | 0.97 | 0.10 d⁻1 | 0.97 | 0.07 d⁻1 | 0.98 | 0.12 d⁻1 | 0.93 | 0.05 d⁻1 | 0.95 | 0.04 d⁻1 | 0.98 | 0.03 d⁻1 |
| Korsmeyer-Peppas | 0.99 | 34.59 d⁻n (n = 0.47) | 0.99 | 30.21 d⁻n (n = 0.50) | 0.95 | 40.60 d⁻n (n = 0.42) | 0.99 | 28.15 d⁻n (n = 0.47) | 0.99 | 20.87 d⁻n (n = 0.56) | 0.99 | 13.14 d⁻n (n = 0.73) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanaki, S.G.; Spyrou, K.; Veneti, P.; Karouta, N.; Gournis, D.; Baroud, T.N.; Barmpalexis, P.; Bikiaris, D.N. L-Cysteine Modified Chitosan Nanoparticles and Carbon-Based Nanostructures for the Intranasal Delivery of Galantamine. Polymers 2022, 14, 4004. https://doi.org/10.3390/polym14194004
Nanaki SG, Spyrou K, Veneti P, Karouta N, Gournis D, Baroud TN, Barmpalexis P, Bikiaris DN. L-Cysteine Modified Chitosan Nanoparticles and Carbon-Based Nanostructures for the Intranasal Delivery of Galantamine. Polymers. 2022; 14(19):4004. https://doi.org/10.3390/polym14194004
Chicago/Turabian StyleNanaki, Stavroula G., Konstantinos Spyrou, Pelagia Veneti, Niki Karouta, Dimitrios Gournis, Turki N. Baroud, Panagiotis Barmpalexis, and Dimitrios N. Bikiaris. 2022. "L-Cysteine Modified Chitosan Nanoparticles and Carbon-Based Nanostructures for the Intranasal Delivery of Galantamine" Polymers 14, no. 19: 4004. https://doi.org/10.3390/polym14194004