
Macromolecular Insights into the Altered Mechanical Deformation Mechanisms of Non-Polyolefin Contaminated Polyolefins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Mechanical Characterization
2.4. Differential Scanning Calorimetry (DSC)
2.5. Scanning Electron Microscopy (SEM)
3. Results
3.1. Crystallinity
3.1.1. Crystallinity of Mono Materials
3.1.2. Crystallinity of the Blend Series
3.2. Tensile Deformation and Properties
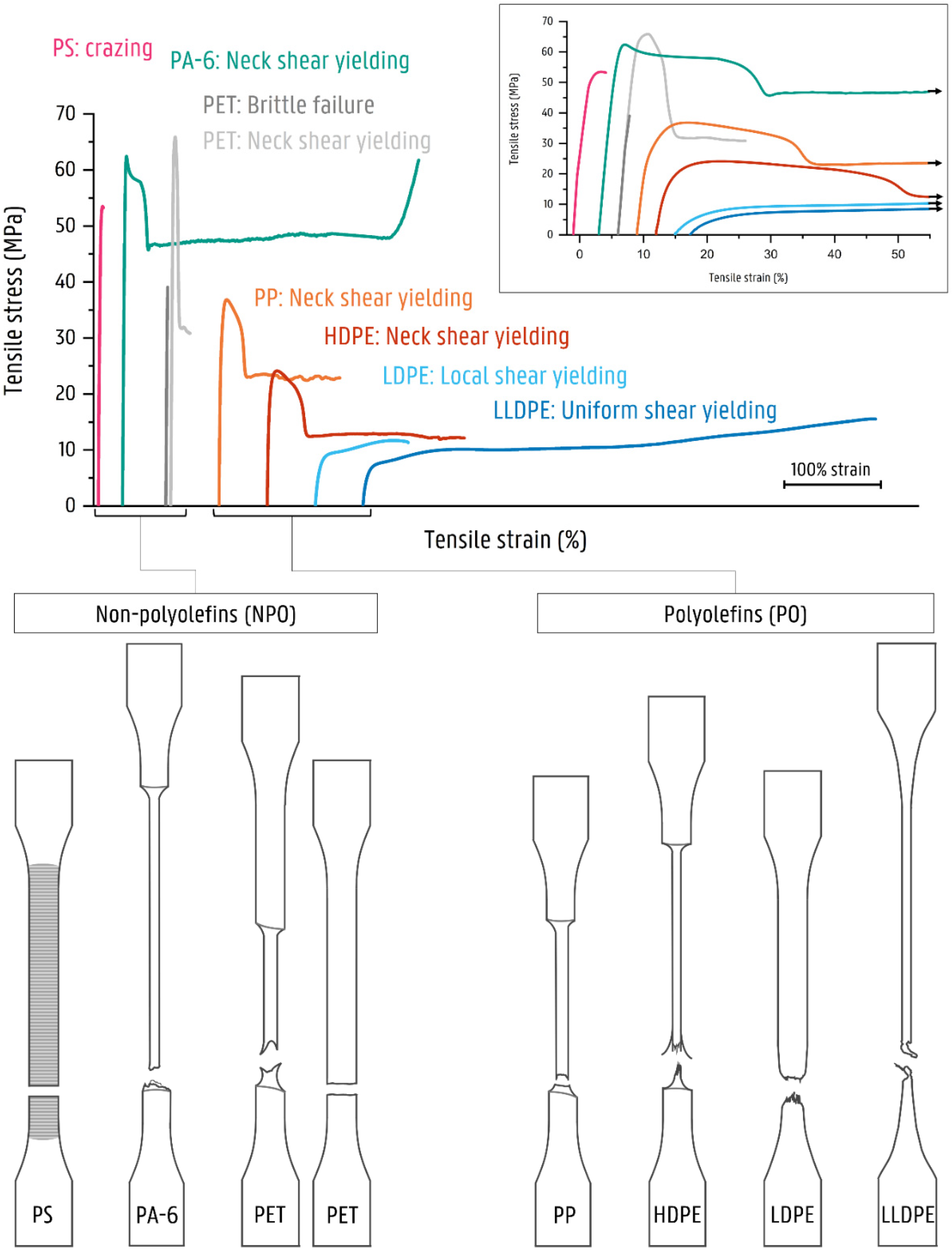
3.2.1. Mechanical Properties of the Mono Materials
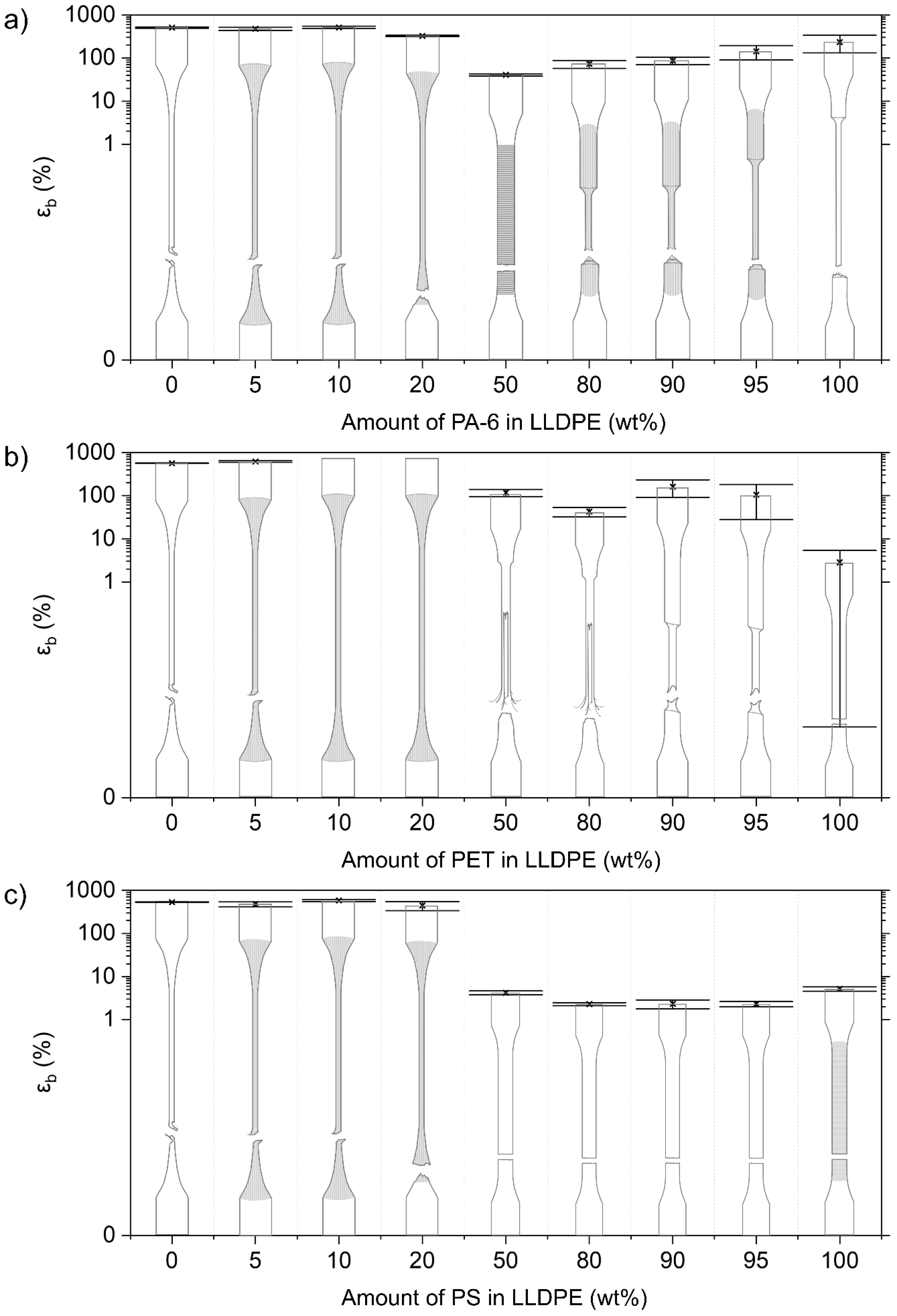
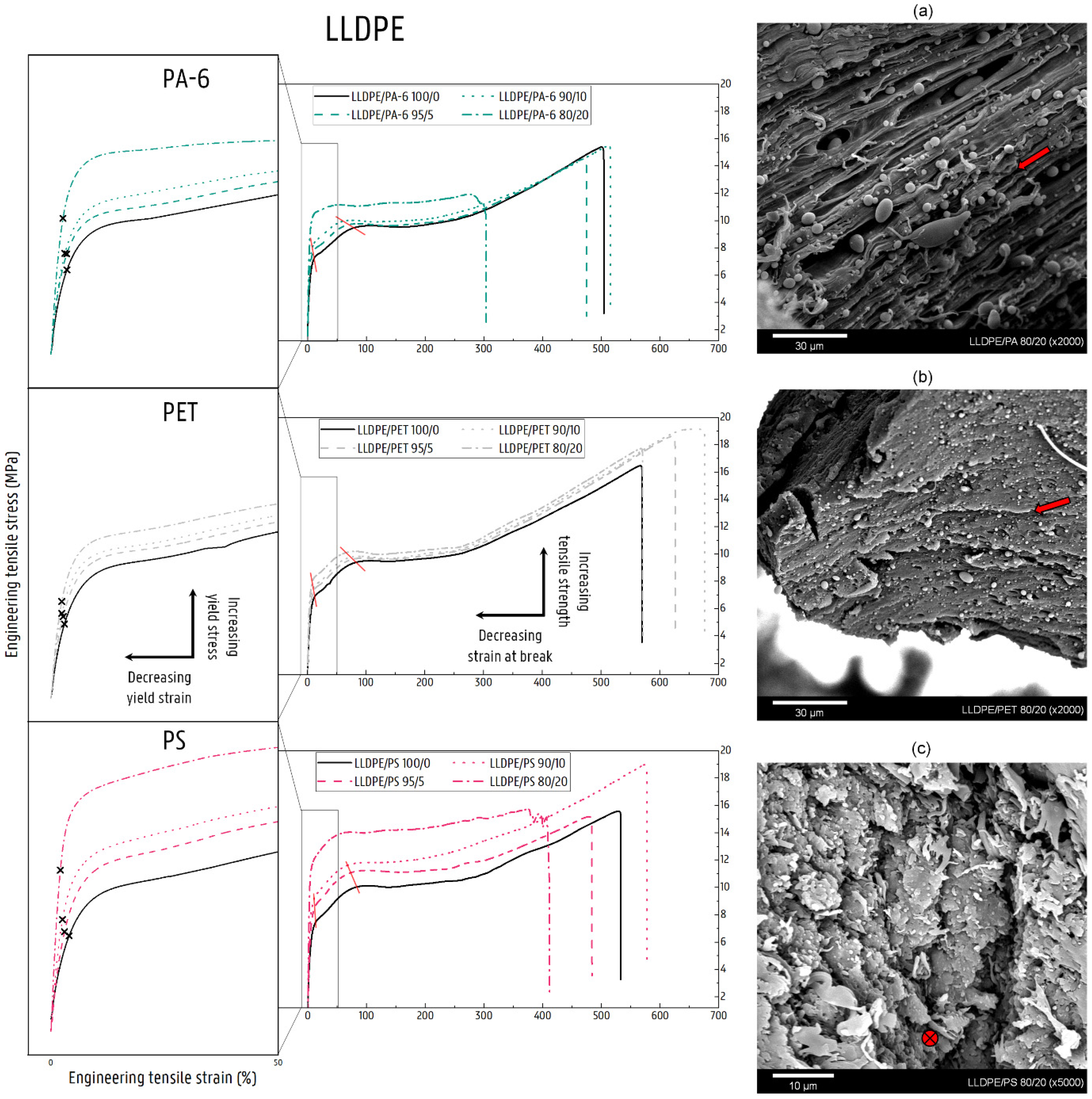
3.2.2. Mechanical Properties of LLDPE Blends
LLDPE/PA-6
LLDPE/PET Blends
LLDPE/PS Blends
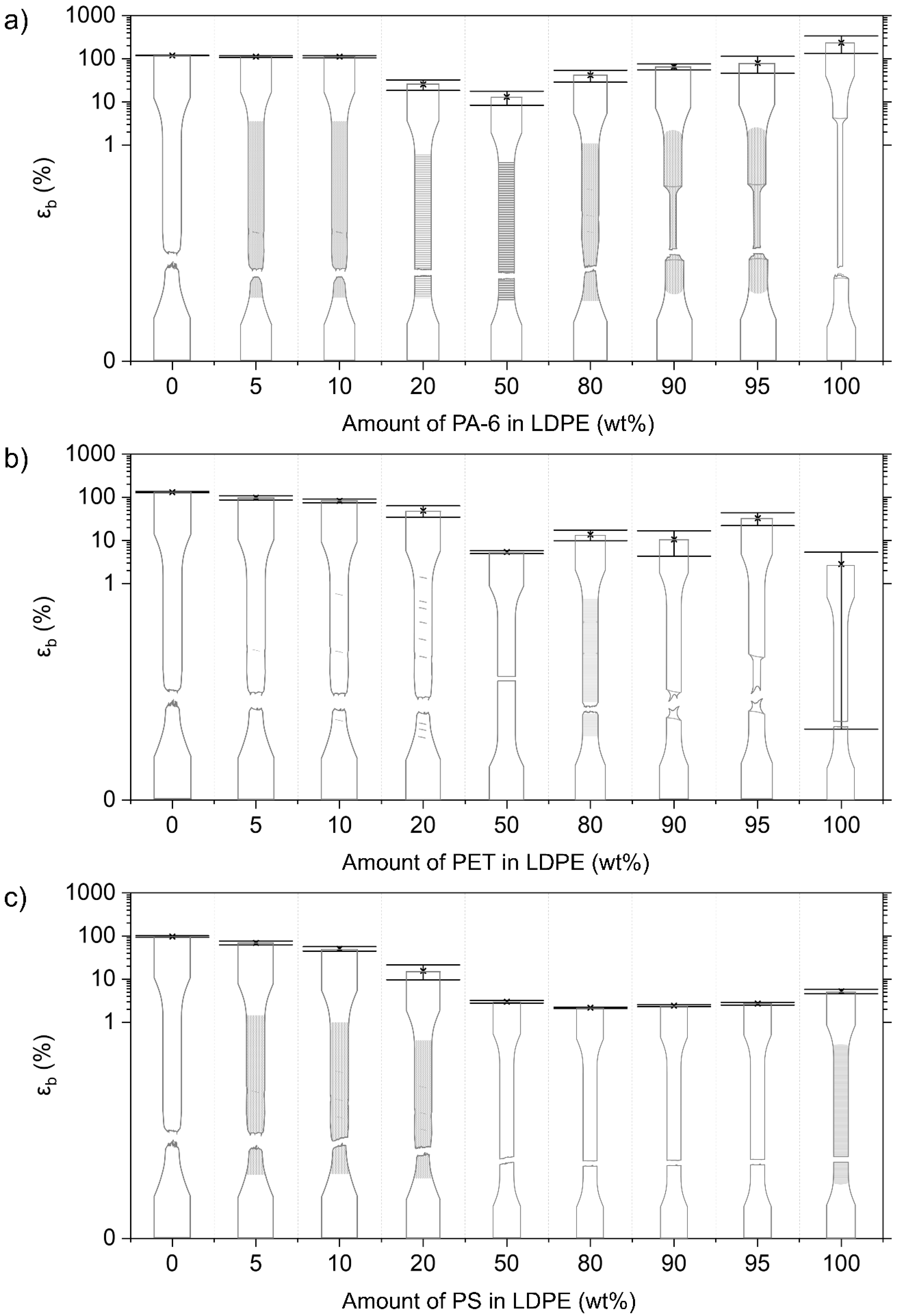
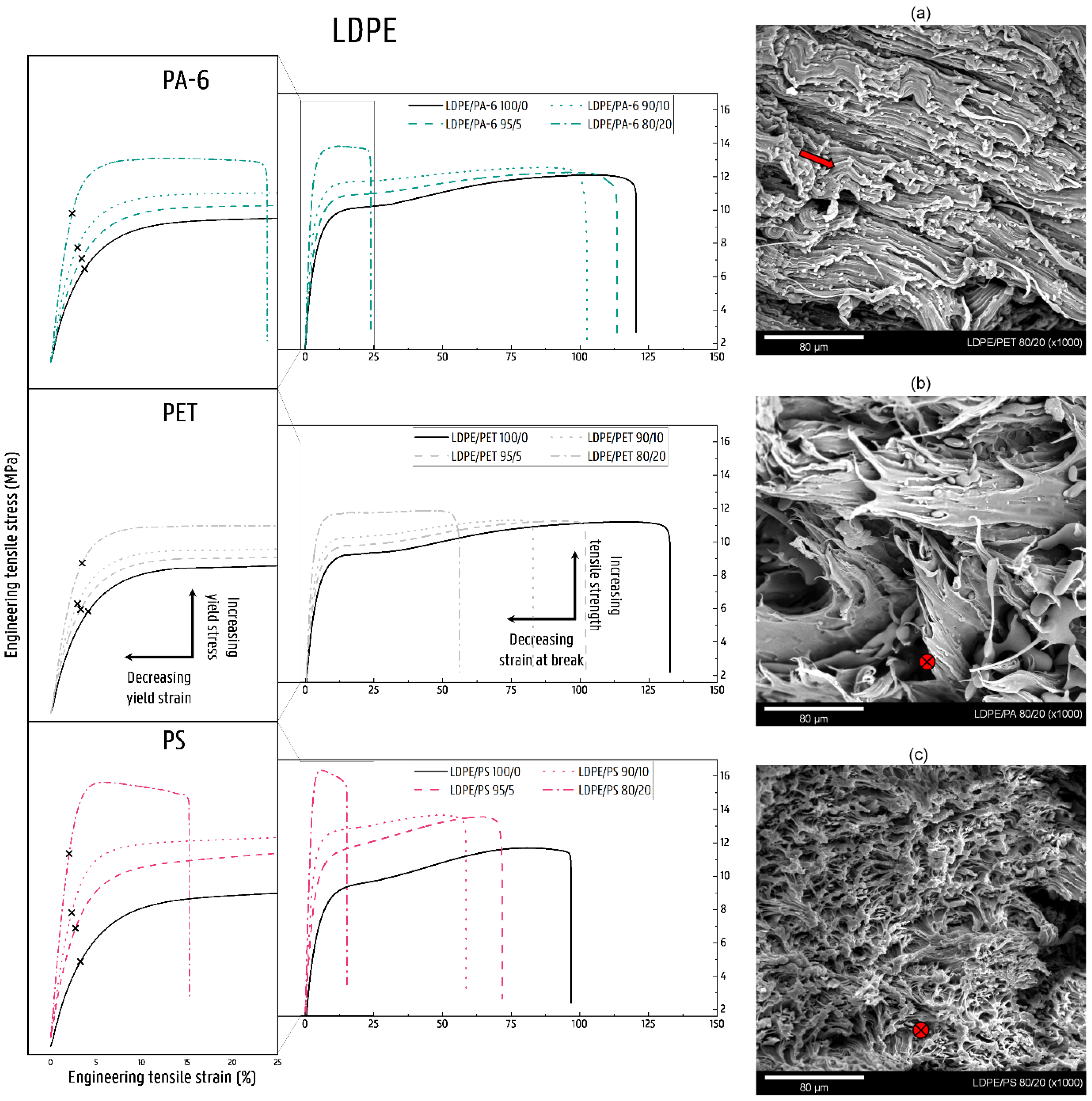
3.2.3. Mechanical Properties of LDPE Blends
LDPE/PA-6 Blends
LDPE/PET Blends
LDPE/PS Blends
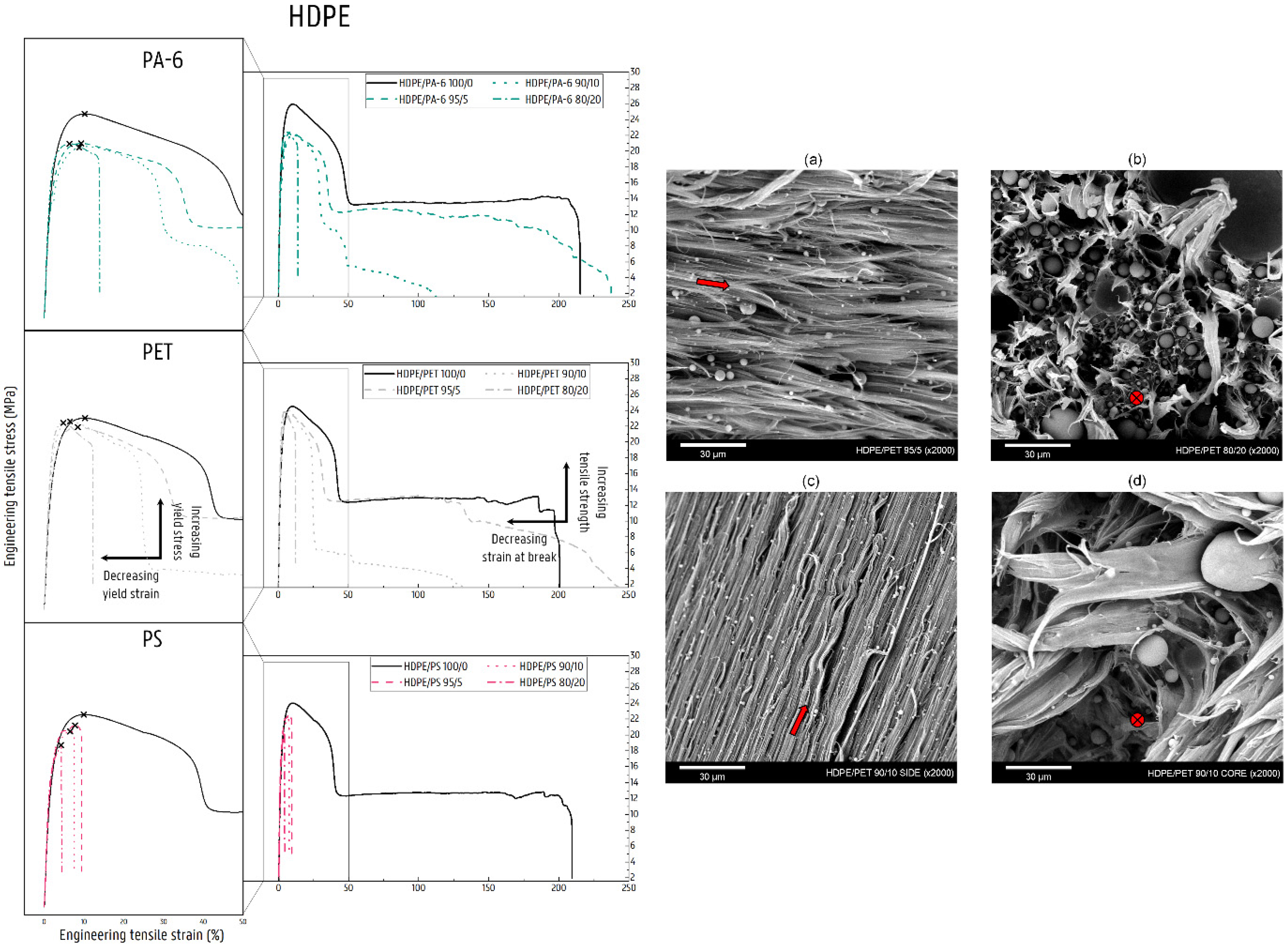
3.2.4. Mechanical Properties of HDPE Blends
HDPE/PA-6 Blends
HDPE/PET Blends
HDPE/PS Blends
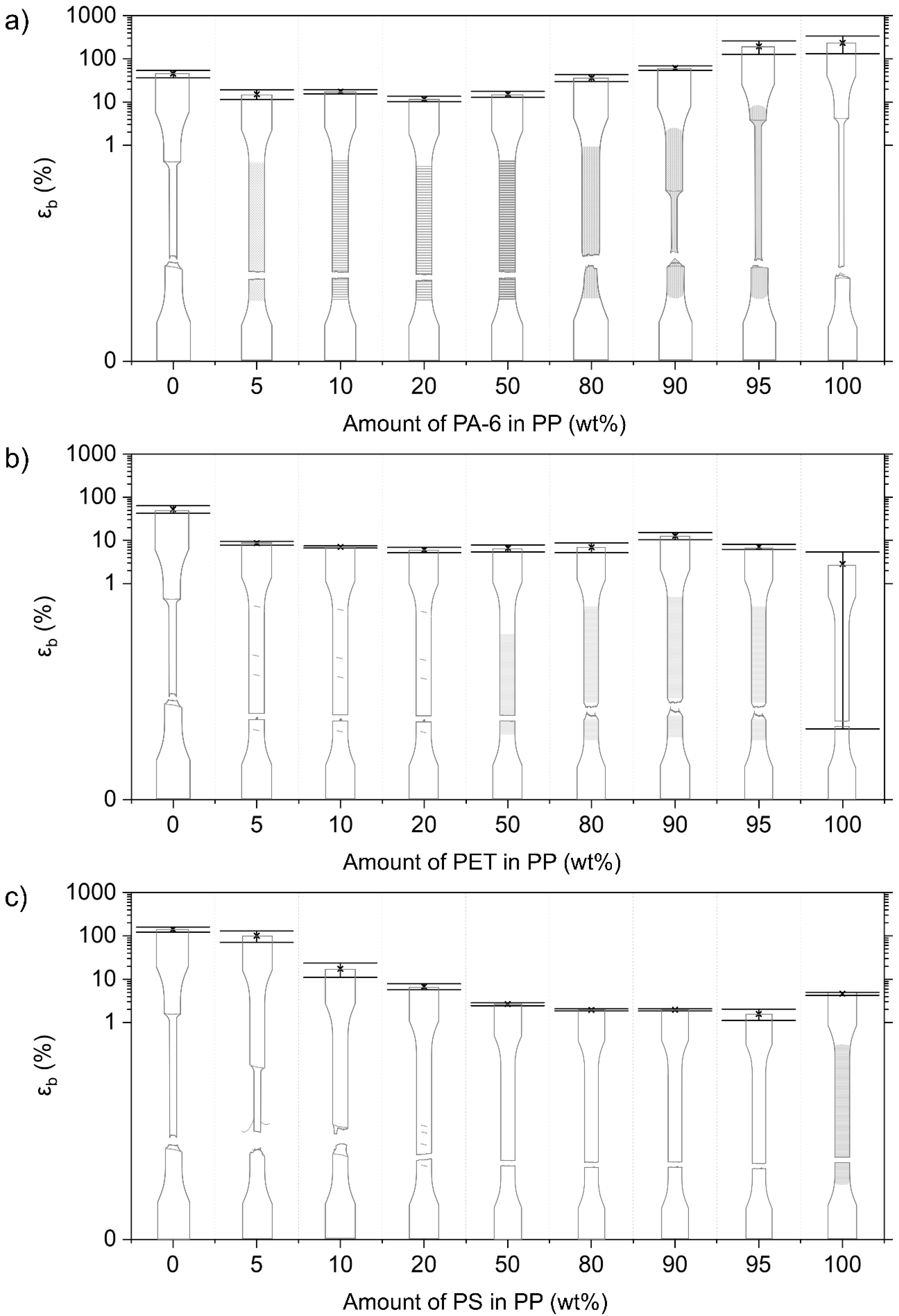
3.2.5. Mechanical Properties of PP Blends
PP/PA-6 Blends
PP/PET Blends
PP/PS Blends
4. Discussion
4.1. Deformation of Contaminated LLDPE Matrix Blends
4.2. Deformation of Contaminated LDPE Matrix Blends
4.3. Deformation of Contaminated HDPE Matrix Blends
4.4. Deformation of Contaminated PP Matrix Blends
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations and Symbols
| Abbreviations | |
| DSC | differential scanning calorimetry |
| HDPE | high-density polyethylene |
| LD | longitudinal direction |
| LDPE | low-density polyethylene |
| LLDPE | linear low-density polyethylene |
| MFI | melt flow index |
| NPO | non-polyolefin |
| PA-6 | polyamide-6 |
| PET | polyethylene terephthalate |
| PO | polyolefine |
| PP | polypropylene |
| PS | polystyrene |
| SEM | scanning electron microscopy |
| TD | transversal direction |
| Symbols | |
| °C | degrees Celsius |
| E | elastic modulus |
| J/g | joule per gram |
| kN | kilonewton |
| kV | kilovolt |
| mm | millimeter |
| mm/min | millimeter per minute |
| MPa | megapascal |
| N | newton |
| Tprocessing | processing temperature |
| wt% | weight fraction |
| melting enthalpy of the 100% crystalline polymer | |
| ΔHcc | cold crystallization enthalpy |
| ΔHm | melting enthalpy |
| εb | strain at break |
| εy | strain at yield |
| σy | yield strength |
| Χc | degree of crystallinity |
References
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef]
- PlasticsEurope Plastics–the Facts. 2019. Available online: https://plasticseurope.org/wp-content/uploads/2021/10/2019-Plastics-the-facts.pdf (accessed on 29 November 2021).
- Ebnesajjad, S.; Ebnesajjad, C. Surface Treatment of Materials for Adhesive Bonding, 2nd ed.; Elsevier Science & Technology Books: Norwich, CT, USA, 2014; ISBN 9780323264358. [Google Scholar]
- Umwelt Bundesamt Aufkommen und Verwertung von Verpackungsabfällen in Deutschland im Jahr. 2018. Available online: https://www.umweltbundesamt.de/sites/default/files/medien/479/publikationen/texte_166-2020_aufkommen_und_verwertung_von_verpackungsabfaellen_in_deutschland_im_jahr_2018.pdf (accessed on 29 November 2021).
- Tartakowski, Z. Recycling of packaging multilayer films: New materials for technical products. Resour. Conserv. Recycl. 2010, 55, 167–170. [Google Scholar] [CrossRef]
- CEFLEX A Roadmap for Flexible Packaging in a Circular Economy. Available online: https://ceflex.eu/flexible-packaging-in-europe/ (accessed on 29 November 2021).
- Anukiruthika, T.; Sethupathy, P.; Wilson, A.; Kashampur, K.; Moses, J.A.; Anandharamakrishnan, C. Multilayer packaging: Advances in preparation techniques and emerging food applications. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1156–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maga, D.; Hiebel, M.; Aryan, V. A Comparative Life Cycle Assessment of Meat Trays Made of Various Packaging Materials. Sustainability 2019, 11, 5324. [Google Scholar] [CrossRef] [Green Version]
- Kunchimon, S.Z.; Tausif, M.; Goswami, P.; Cheung, V. From hybrid fibers to microfibers: The characteristics of polyamide 6/polypropylene blend via one-step twin-screw melt extrusion. Polym. Eng. Sci. 2020, 60, 690–699. [Google Scholar] [CrossRef]
- Van Bruggen, E.P.A.; Koster, R.P.; Picken, S.J.; Ragaert, K. Influence of Processing Parameters and Composition on the Effective Compatibilization of Polypropylene–Poly(ethylene terephthalate) Blends. Int. Polym. Process. 2016, 31, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Utracki, L.A.; Wilkie, C.A. Polymer Blends Handbook; Utracki, L.A., Ed.; Springer: Dordrecht, The Netherlands, 2014; ISBN 9789400760646. [Google Scholar]
- Kratofil Krehula, L.; Hrnjak-Murgić, Z.; Jelenčić, J. Study of masterbatch effect on miscibility and morphology in PET/HDPE blends. J. Adhes. Sci. Technol. 2015, 29, 74–93. [Google Scholar] [CrossRef]
- Robeson, L.M. Polymer Blends: A Comprehensive Review; Hanser: Munich, Germnay, 2007; ISBN 978-3-446-43650-3. [Google Scholar]
- Van Puyvelde, P.; Velankar, S.; Moldenaers, P. Rheology and morphology of compatibilized polymer blends. Curr. Opin. Colloid Interface Sci. 2001, 6, 457–463. [Google Scholar] [CrossRef]
- Grosso, M.; Luca, P. Fourier Transform Rheology: A New Tool to Characterize Material Properties. In Fourier Transforms-New Analytical Approaches and FTIR Strategies; Nikolic, G., Ed.; InTech: Rijeka, Croatia, 2011; ISBN 978-953-307-232-6. [Google Scholar]
- Chen, L.; Pan, D.; He, H. Morphology Development of Polymer Blend Fibers along Spinning Line. Fibers 2019, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Charfeddine, I.; Majesté, J.C.; Carrot, C.; Lhost, O. A model for the prediction of the morphology of immiscible blends of polymers. Polymer 2020, 193, 122334. [Google Scholar] [CrossRef]
- Lee, J.K.; Han, C.D. Evolution of polymer blend morphology during compounding in a twin-screw extruder. Polymer 2000, 41, 1799–1815. [Google Scholar] [CrossRef]
- Willemse, R.C.; Ramaker, E.J.J.; van Dam, J.; Posthuma de Boer, A. Morphology development in immiscible polymer blends: Initial blend morphology and phase dimensions. Polymer 1999, 40, 6651–6659. [Google Scholar] [CrossRef]
- Van Kets, K. Effectiveness of Maleic Anhydride Grafted Compatibilizers for Multiple Mechanical Recycling of Poly(Propylene)-Poly(Ethylene Terephthalate) Blends. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2020. ISBN 978-94-6355-432-9. [Google Scholar]
- Van Kets, K.; Delva, L.; Ragaert, K. Onset critical strains as an effective parameter for compatibilizer efficiency in a polypropylene-poly(ethylene terephthalate) blend. J. Appl. Polym. Sci. 2021, 138, 50656. [Google Scholar] [CrossRef]
- Dixon, J. Packaging Materials: 9. Multilayer Packaging for Food and Beverages. Available online: https://www.pac.gr/bcm/uploads/9-multilayer-packaging-for-food-and-beverages.pdf (accessed on 29 November 2021).
- Van Belle, A.; Demets, R.; Mys, N.; Van Kets, K.; Dewulf, J.; Van Geem, K.; De Meester, S.; Ragaert, K. Microstructural Contributions of Different Polyolefins to the Deformation Mechanisms of Their Binary Blends. Polymers 2020, 12, 1171. [Google Scholar] [CrossRef]
- Men, Y.; Strobl, G. Critical Strains in Poly(ε -caprolactone) and Blends with Poly(vinyl methyl ether) and Poly(styrene- c o -acrylonitrile). Macromolecules 2003, 36, 1889–1898. [Google Scholar] [CrossRef]
- Fu, Q.; Men, Y.; Strobl, G. Understanding of the tensile deformation in HDPE/LDPE blends based on their crystal structure and phase morphology. Polymer 2003, 44, 1927–1933. [Google Scholar] [CrossRef]
- Deblieck, R.A.C.; van Beek, D.J.M.; Remerie, K.; Ward, I.M. Failure mechanisms in polyolefines: The role of crazing, shear yielding and the entanglement network. Polymer 2011, 52, 2979–2990. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, A.; Galeski, A. Cavitation during Tensile Deformation of Polypropylene. Macromolecules 2008, 41, 2839–2851. [Google Scholar] [CrossRef]
- Patlazhan, S.; Remond, Y. Structural mechanics of semicrystalline polymers prior to the yield point: A review. J. Mater. Sci. 2012, 47, 6749–6767. [Google Scholar] [CrossRef]
- Wu, S. Calculation of interfacial tension in polymer systems. J. Polym. Sci. Part C Polym. Symp. 2007, 34, 19–30. [Google Scholar] [CrossRef]
- Fu, S.-Y.; Feng, X.-Q.; Lauke, B.; Mai, Y.-W. Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate–polymer composites. Compos. Part B Eng. 2008, 39, 933–961. [Google Scholar] [CrossRef]
- G’Sell, C.; Bai, S.-L.; Hiver, J.-M. Polypropylene/polyamide 6/polyethylene–octene elastomer blends. Part 2: Volume dilatation during plastic deformation under uniaxial tension. Polymer 2004, 45, 5785–5792. [Google Scholar] [CrossRef]
- Michler, G.H. Atlas of Polymer Structures: Morphology, Deformation and Fracture Structures; Hanser: Munich, Germany, 2016; ISBN 978-1-56990-557-9. [Google Scholar]
- Men, Y.; Rieger, J.; Strobl, G. Role of the Entangled Amorphous Network in Tensile Deformation of Semicrystalline Polymers. Phys. Rev. Lett. 2003, 91, 095502. [Google Scholar] [CrossRef]
- Galeski, A. Strength and toughness of crystalline polymer systems. Prog. Polym. Sci. 2003, 28, 1643–1699. [Google Scholar] [CrossRef]
- Hobeika, S.; Men, Y.; Strobl, G. Temperature and Strain Rate Independence of Critical Strains in Polyethylene and Poly(ethylene- c o -vinyl acetate). Macromolecules 2000, 33, 1827–1833. [Google Scholar] [CrossRef]
- Hiss, R.; Hobeika, S.; Lynn, C.; Strobl, G. Network Stretching, Slip Processes, and Fragmentation of Crystallites during Uniaxial Drawing of Polyethylene and Related Copolymers. A Comparative Study. Macromolecules 1999, 32, 4390–4403. [Google Scholar] [CrossRef]
- Demets, R.; Van Kets, K.; Huysveld, S.; Dewulf, J.; De Meester, S.; Ragaert, K. Addressing the complex challenge of understanding and quantifying substitutability for recycled plastics. Resour. Conserv. Recycl. 2021, 174, 105826. [Google Scholar] [CrossRef]
- Netzsch. Handbook DSC; NETZSCH: Selb, Germany, 2015. [Google Scholar]
- Spalding, M.A.; Chatterjee, A.M. Handbook of Industrial Polyethylene Technology; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2016; ISBN 9781119159797. [Google Scholar]
- Bandla, S.; Allahkarami, M.; Hanan, J.C. Thermal Crystallinity and Mechanical Behavior of Polyethylene Terephthalate. In Challenges in Mechanics of Time-Dependent Materials; Springer: Cham, Switerland, 2015; Volume 2, pp. 141–146. [Google Scholar]
- Gehring, F.; Bouvard, J.-L.; Billon, N. Modeling of time dependent mechanical behavior of polymers: Comparison between amorphous and semicrystalline polyethylene terephthalate. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Rastogi, R.; Vellinga, W.P.; Rastogi, S.; Schick, C.; Meijer, H.E.H. The three-phase structure and mechanical properties of poly(ethylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 2092–2106. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A.; Rozanski, A. Cavitation during deformation of semicrystalline polymers. Prog. Polym. Sci. 2014, 39, 921–958. [Google Scholar] [CrossRef]
- Seguela, R. Critical review of the molecular topology of semicrystalline polymers: The origin and assessment of intercrystalline tie molecules and chain entanglements. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1729–1748. [Google Scholar] [CrossRef]
- Seguela, R.; Rietsch, F. Tensile drawing behaviour of ethylene/α-olefin copolymers: Influence of the co-unit concentration. Polymer 1986, 27, 703–708. [Google Scholar] [CrossRef]
- Seguela, R.; Rietsch, F. Molecular topology in ethylene copolymers studied by means of mechanical testing. J. Mater. Sci. 1988, 23, 415–421. [Google Scholar] [CrossRef]
- Liang, J.Z. Reinforcement and quantitative description of inorganic particulate-filled polymer composites. Compos. Part B Eng. 2013, 51, 224–232. [Google Scholar] [CrossRef]
- Dorigato, A. Recycling of polymer blends. Adv. Ind. Eng. Polym. Res. 2021, 4, 53–69. [Google Scholar] [CrossRef]
- Wu, S. Formation of dispersed phase in incompatible polymer blends: Interfacial and rheological effects. Polym. Eng. Sci. 1987, 27, 335–343. [Google Scholar] [CrossRef]
- Van Puyvelde, P.; Vananroye, A.; Cardinaels, R.; Moldenaers, P. Review on morphology development of immiscible blends in confined shear flow. Polymer 2008, 49, 5363–5372. [Google Scholar] [CrossRef] [Green Version]
- Utracki, L.A.; Shi, Z.H. Development of polymer blend morphology during compounding in a twin-screw extruder. Part I: Droplet dispersion and coalescence?a review. Polym. Eng. Sci. 1992, 32, 1824–1833. [Google Scholar] [CrossRef]
- Perilla, J.E.; Jana, S.C. Coalescence of immiscible polymer blends in chaotic mixers. AIChE J. 2005, 51, 2675–2685. [Google Scholar] [CrossRef]
- Lerdwijitjarud, W.; Sirivat, A.; Larson, R.G. Influence of elasticity on dispersed-phase droplet size in immiscible polymer blends in simple shearing flow. Polym. Eng. Sci. 2002, 42, 798–809. [Google Scholar] [CrossRef]
- González-Nuñez, R.; Arellano, M.; Moscoso, F.J.; González-Romero, V.M.; Favis, B.D. Determination of a limiting dispersed phase concentration for coalescence in PA6/HDPE blends under extensional flow. Polymer 2001, 42, 5485–5489. [Google Scholar] [CrossRef]
- Fortelný, I.; Jůza, J. Description of the Droplet Size Evolution in Flowing Immiscible Polymer Blends. Polymers 2019, 11, 761. [Google Scholar] [CrossRef] [Green Version]
- Fortelný, I.; Kovář, J. Theory of coalescence in immiscible polymer blends. Polym. Compos. 1988, 9, 119–124. [Google Scholar] [CrossRef]
- Willemse, R.C.; Speijer, A.; Langeraar, A.E.; Posthuma de Boer, A. Tensile moduli of co-continuous polymer blends. Polymer 1999, 40, 6645–6650. [Google Scholar] [CrossRef]
- Veenstra, H.; Verkooijen, P.C.; van Lent, B.J.; van Dam, J.; de Boer, A.P.; Nijhof, A.P.H. On the mechanical properties of co-continuous polymer blends: Experimental and modelling. Polymer 2000, 41, 1817–1826. [Google Scholar] [CrossRef]
- Bartczak, Z.; Galeski, A. Mechanical Properties of Polymer Blends. In Polymer Blends Handbook; Springer: Dordrecht, The Netherlands, 2014; pp. 1203–1297. ISBN 978-94-007-6064-6. [Google Scholar]
- Brooks, N.W.; Duckett, R.A.; Ward, I.M. Investigation into double yield points in polyethylene. Polymer 1992, 33, 1872–1880. [Google Scholar] [CrossRef]
- Brooks, N.W.J.; Unwin, A.P.; Duckett, R.A.; Ward, I.M. Double yield points in polyethylene: Structural changes under tensile deformation. J. Macromol. Sci. Part B 1995, 34, 29–54. [Google Scholar] [CrossRef]
- Schrauwen, B.A.G.G.; Janssen, R.P.M.M.; Govaert, L.E.; Meijer, H.E.H.H. Intrinsic Deformation Behavior of Semicrystalline Polymers. Macromolecules 2004, 37, 6069–6078. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.F.; Donald, A.M.; Ryan, A.J. Time resolved simultaneous small- and wide-angle X-ray scattering during polyethylene deformation—II. Cold drawing of linear polyethylene. Polymer 1998, 39, 39–52. [Google Scholar] [CrossRef]
- Shroff, R.N.; Mavridis, H. Assessment of NMR and Rheology for the Characterization of LCB in Essentially Linear Polyethylenes. Macromolecules 2001, 34, 7362–7367. [Google Scholar] [CrossRef]
- Brown, N.; Ward, I.M. The influence of morphology and molecular weight on ductile-brittle transitions in linear polyethylene. J. Mater. Sci. 1983, 18, 1405–1420. [Google Scholar] [CrossRef]
- Lu, X.; Qian, R.; Brown, N. The effect of crystallinity on fracture and yielding of polyethylenes. Polymer 1995, 36, 4239–4244. [Google Scholar] [CrossRef]
- Tarin, P.M.; Thomas, E.L. The role of inter- and intra-links in the transformation of folded chain lamellae into microfibrils. Polym. Eng. Sci. 1979, 19, 1017–1022. [Google Scholar] [CrossRef]
- Shroff, R.; Prasad, A.; Lee, C. Effect of molecular structure on rheological and crystallization properties of polyethylenes. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 2317–2333. [Google Scholar] [CrossRef]
- Guichon, O.; Séguéla, R.; David, L.; Vigier, G. Influence of the molecular architecture of low-density polyethylene on the texture and mechanical properties of blown films. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 327–340. [Google Scholar] [CrossRef]
- Pawlak, A. Cavitation during tensile deformation of isothermally crystallized polypropylene and high-density polyethylene. Colloid Polym. Sci. 2013, 291, 773–787. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, A. Cavitation during tensile deformation of high-density polyethylene. Polymer 2007, 48, 1397–1409. [Google Scholar] [CrossRef]
- Nitta, K.H.; Kuriyagawa, M. Application of catastrophe theory to neck initiation of metallocene-catalyzed high-density polyethylene. Polym. J. 2012, 44, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Leonov, A.I. A theory of necking in semi-crystalline polymers. Int. J. Solids Struct. 2002, 39, 5913–5926. [Google Scholar] [CrossRef] [Green Version]
- Shan, G.F.; Yang, W.; Yang, M.B.; Xie, B.H.; Fu, Q.; Mai, Y.W. Investigation on tensile deformation behavior of semi-crystalline polymers. J. Macromol. Sci. Part B Phys. 2009, 48, 799–811. [Google Scholar] [CrossRef]
- Bärwinkel, S.; Seidel, A.; Hobeika, S.; Hufen, R.; Mörl, M.; Altstädt, V. Morphology formation in PC/ABS blends during thermal processing and the effect of the viscosity ratio of blend partners. Materials 2016, 9, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellahi, S.; Favis, B.D.; Fisa, B. Morphological stability in injection-moulded high-density polyethylene/polyamide-6 blends. Polymer 1996, 37, 2615–2626. [Google Scholar] [CrossRef]
- Zhong, G.J.; Li, Z.M. Injection molding-induced morphology of thermoplastic polymer blends. Polym. Eng. Sci. 2005, 45, 1655–1665. [Google Scholar] [CrossRef]
- Pawlak, A.; Galeski, A. Crystallization of Polypropylene. In Polypropylene Handbook; Springer International Publishing: Cham, Switzerland, 2019; pp. 185–242. ISBN 978-3-030-12902-6. [Google Scholar]
- Bucknall, C.B.; Paul, D.R. Notched impact behaviour of polymer blends: Part 2: Dependence of critical particle size on rubber particle volume fraction. Polymer 2013, 54, 320–329. [Google Scholar] [CrossRef]
- Dompas, D.; Groeninckx, G.; Isogawa, M.; Hasegawa, T.; Kadokura, M. Toughening behaviour of rubber-modified thermoplastic polymers involving very small rubber particles: 2. Rubber cavitation behaviour in poly(vinyl chloride)/methyl methacrylate-butadiene-styrene graft copolymer blends. Polymer 1994, 35, 4750–4759. [Google Scholar] [CrossRef]
- Inberg, J.P.F. Fracture of Polycarbonate/ABS Blends. Ph.D. Thesis, Universiteit Twente, Enschede, The Netherland, 2001. ISBN 90 365 1553x. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Grade | Producer | MFI (g/10 min) * | Typical Use | |
|---|---|---|---|---|---|
| PO | LLDPE | ExceedTM 1012HA | ExxonMobil | 1.0 | Film |
| LDPE | LD150AC | ExxonMobil | 0.8 | Film | |
| HDPE | 25055E | Dow Chemical Company | 25.0 | Housewares, food containers, toys | |
| PP | PP6272NE1 | ExxonMobil | 2.8 | Cups, containers | |
| NPO | PET | Lighter C93 | Equipolymers | - | Beverage and food containers |
| PA-6 | Akulon F230C | DSM | - | Film, injection-molded articles | |
| PS | Styrolution PS 158K | INEOS | 3.0 | Household goods, containers, expanded sheet and film |
| Blend Combination | Temperature Profile (°C) |
|---|---|
| PET + PO | 250–260–270–280 |
| PA-6 + PO | 245–255–265–275 |
| PS + PP | 200–210–220–230 |
| PS + LLDPE/LDPE/HDPE | 170–180–190–200 |
| Material | Tprocessing (°C) | Χc (%) | E (MPa) | σy (MPa) | εy (%) | εb (%) | |
|---|---|---|---|---|---|---|---|
| NPO | PET | 280 | 27.1 | 2337 ± 218 | 49.4 ± 15.7 | 3.07 ± 1.59 | 2.9 ± 2.5 * |
| PA-6 | 275 | 28.5 | 2260 ± 60 | 62.1 ± 0.4 | 4.09 ± 0.03 | 234 ± 102 | |
| PS | 200 | 0 | 3084 ± 42 | 52.5 ± 0.6 | 4.33 ± 0.17 | 5.2 ± 0.6 | |
| 230 | 0 | 3202 ± 37 | 49.7 ± 0.3 | 3.62 ± 0.07 | 4.6 ± 0.4 | ||
| PO | LLDPE | 200 | 28.9 | 97 ± 3 | 5.3 ± 0.1 | 4.00 ± 0.19 | 533 ± 9 |
| 275 | 28.0 | 109 ± 8 | 5.2 ± 0.2 | 3.51 ± 0.35 | 507 ± 18 | ||
| 280 | 29.0 | 120 ± 4 | 4.4 ± 0.1 | 2.96 ± 0.19 | 569 ± 7 | ||
| LDPE | 200 | 34.2 | 165 ± 9 | 5.5 ± 0.3 | 3.20 ± 0.28 | 97 ± 4 | |
| 275 | 33.5 | 154 ± 5 | 7.1 ± 0.1 | 3.73 ± 0.12 | 120 ± 2 | ||
| 280 | 31.9 | 173 ± 37 | 6.5 ± 0.6 | 4.01 ± 0.88 | 132 ± 5 | ||
| HDPE | 200 | 64.4 | 984 ± 14 | 24.2 ± 0.3 | 10.29 ± 0.71 | 220 ± 13 | |
| 275 | 64.1 | 893 ± 61 | 25.0 ± 0.5 | 10.30 ± 0.21 | 206 ± 32 | ||
| 280 | 65.6 | 868 ± 19 | 24.4 ± 0.2 | 10.29 ± 0.08 | 208 ± 33 | ||
| PP | 230 | 46.5 | 1706 ± 29 | 36.8 ± 0.3 | 7.87 ± 0.07 | 142 ± 20 | |
| 275 | 45.2 | 1672 ± 30 | 36.9 ± 0.2 | 7.90 ± 0.10 | 45 ± 9 | ||
| 280 | 47.8 | 1745 ± 32 | 36.0 ± 0.1 | 7.59 ± 0.07 | 53 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demets, R.; Grodent, M.; Van Kets, K.; De Meester, S.; Ragaert, K. Macromolecular Insights into the Altered Mechanical Deformation Mechanisms of Non-Polyolefin Contaminated Polyolefins. Polymers 2022, 14, 239. https://doi.org/10.3390/polym14020239
Demets R, Grodent M, Van Kets K, De Meester S, Ragaert K. Macromolecular Insights into the Altered Mechanical Deformation Mechanisms of Non-Polyolefin Contaminated Polyolefins. Polymers. 2022; 14(2):239. https://doi.org/10.3390/polym14020239
Chicago/Turabian StyleDemets, Ruben, Marie Grodent, Karen Van Kets, Steven De Meester, and Kim Ragaert. 2022. "Macromolecular Insights into the Altered Mechanical Deformation Mechanisms of Non-Polyolefin Contaminated Polyolefins" Polymers 14, no. 2: 239. https://doi.org/10.3390/polym14020239