
Reversible Protein Capture and Release by Redox-Responsive Hydrogel in Microfluidics

 , , ,
, , ,  , and
, and
Abstract
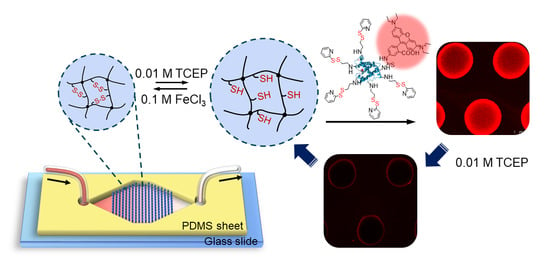
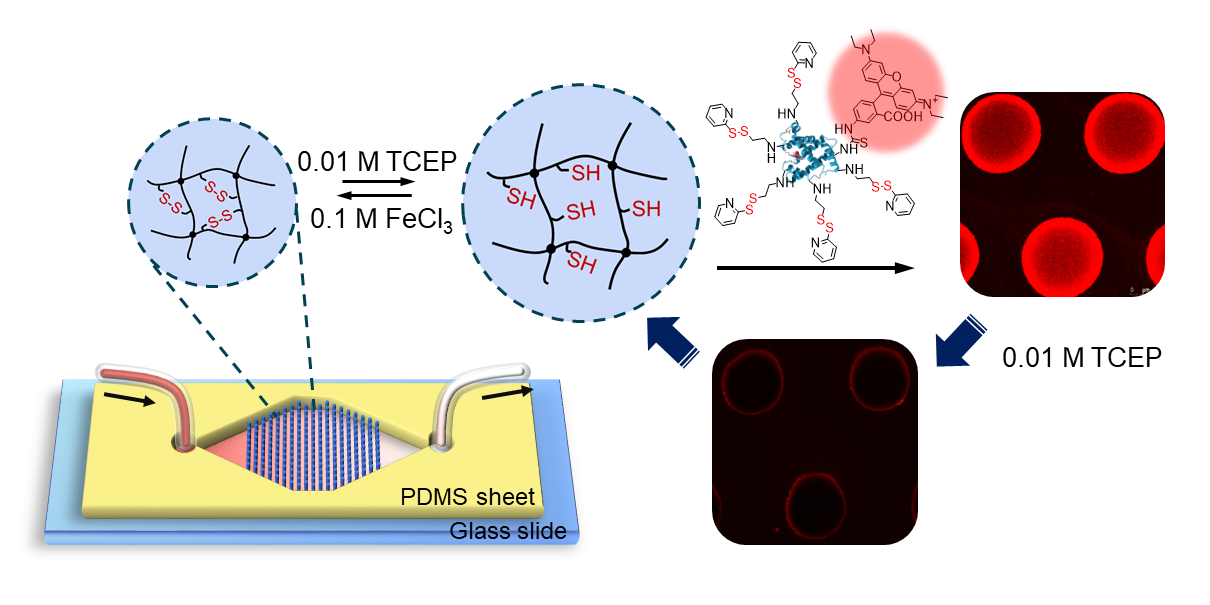
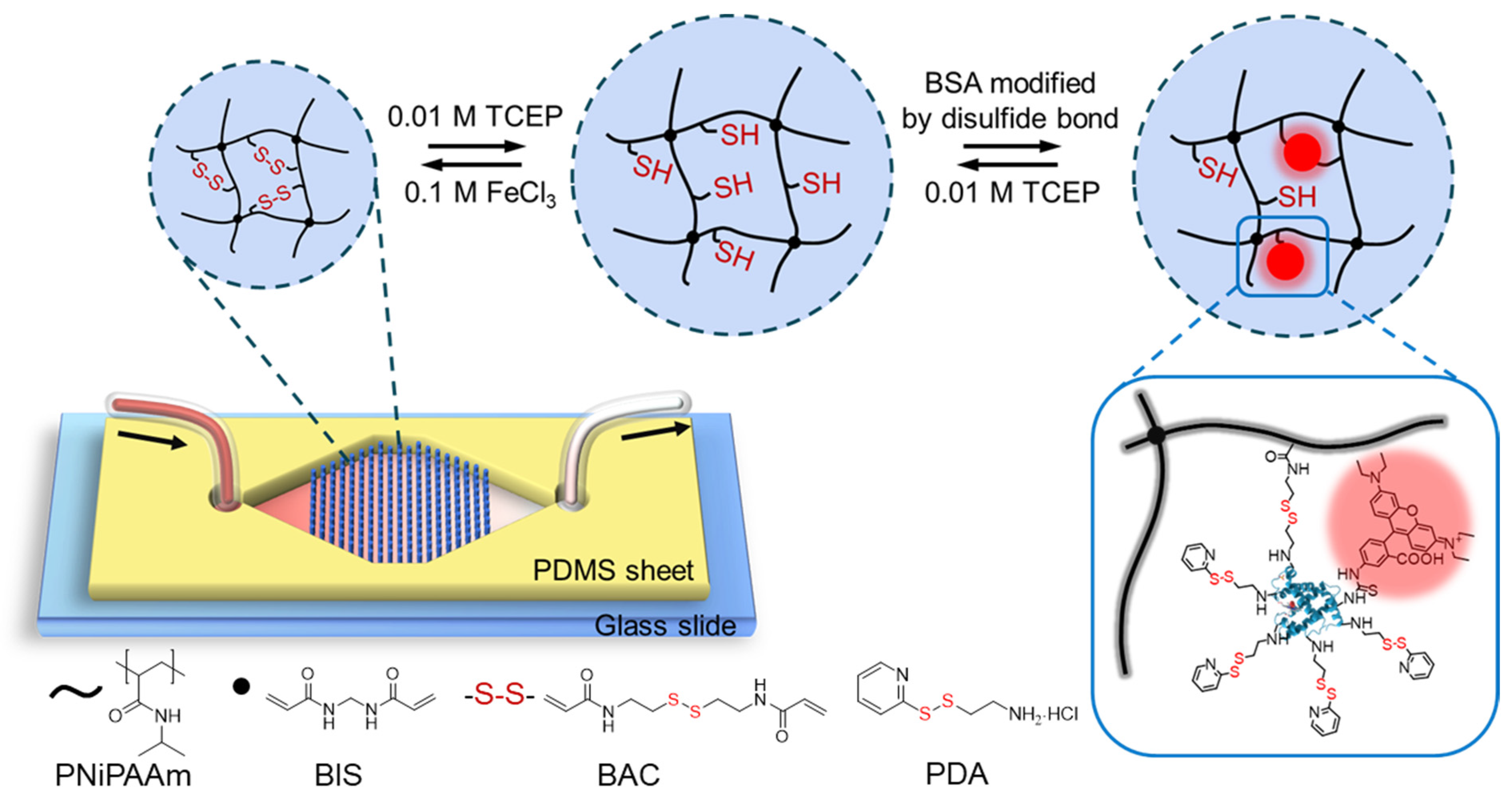
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of 2-(2-pyridyldithio)-ethylamine (PDA)
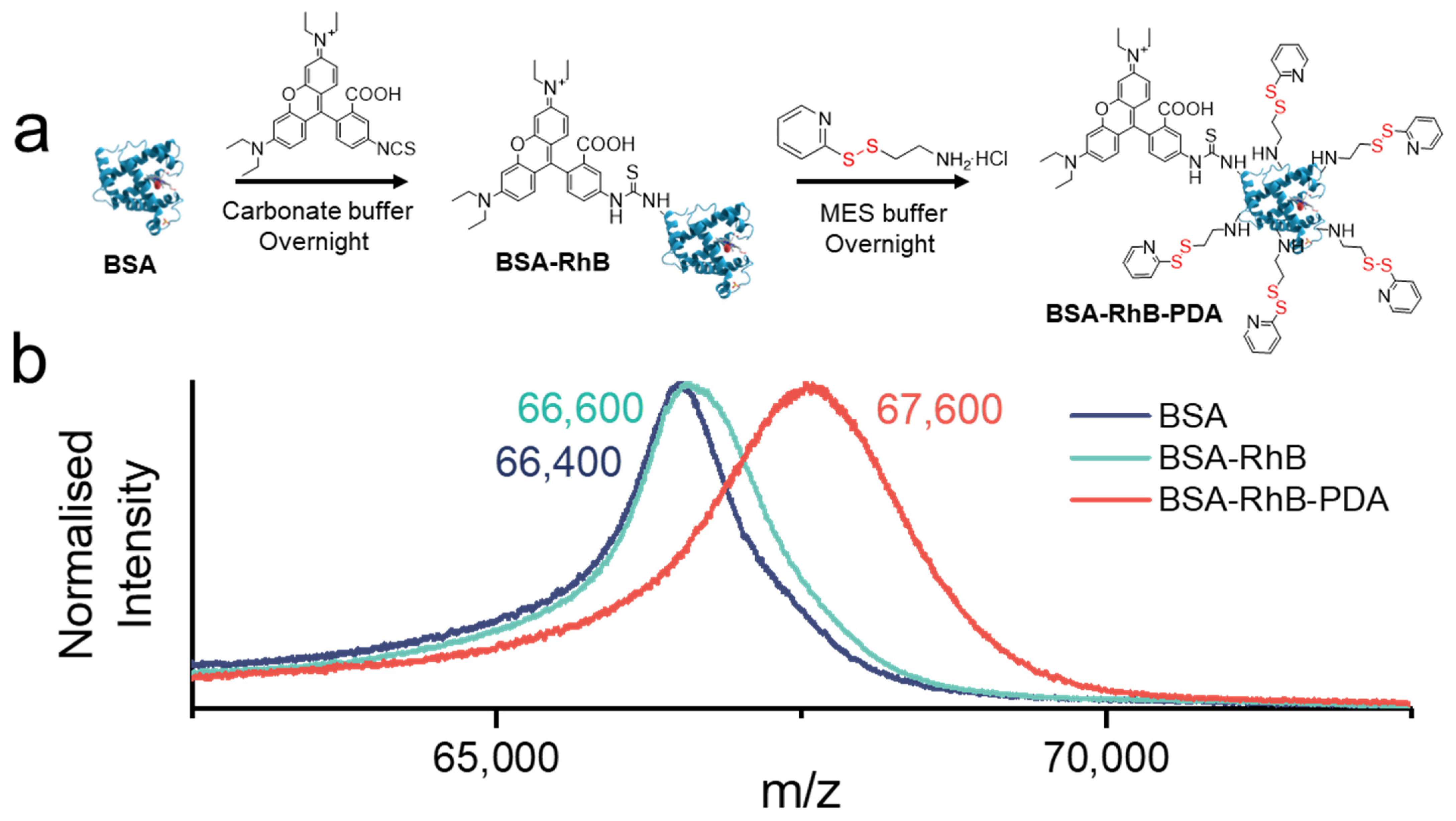
2.3. Synthesis of BSA-RhB
2.4. Synthesis of BSA-RhB-PDA
2.5. Preparation of Hydrogel Arrays
2.6. Microfluidic Testing
2.7. MALDI-TOF
2.8. NMR Measurement
3. Results and Discussion
3.1. Experimental Design and Microfluidic Chip Design
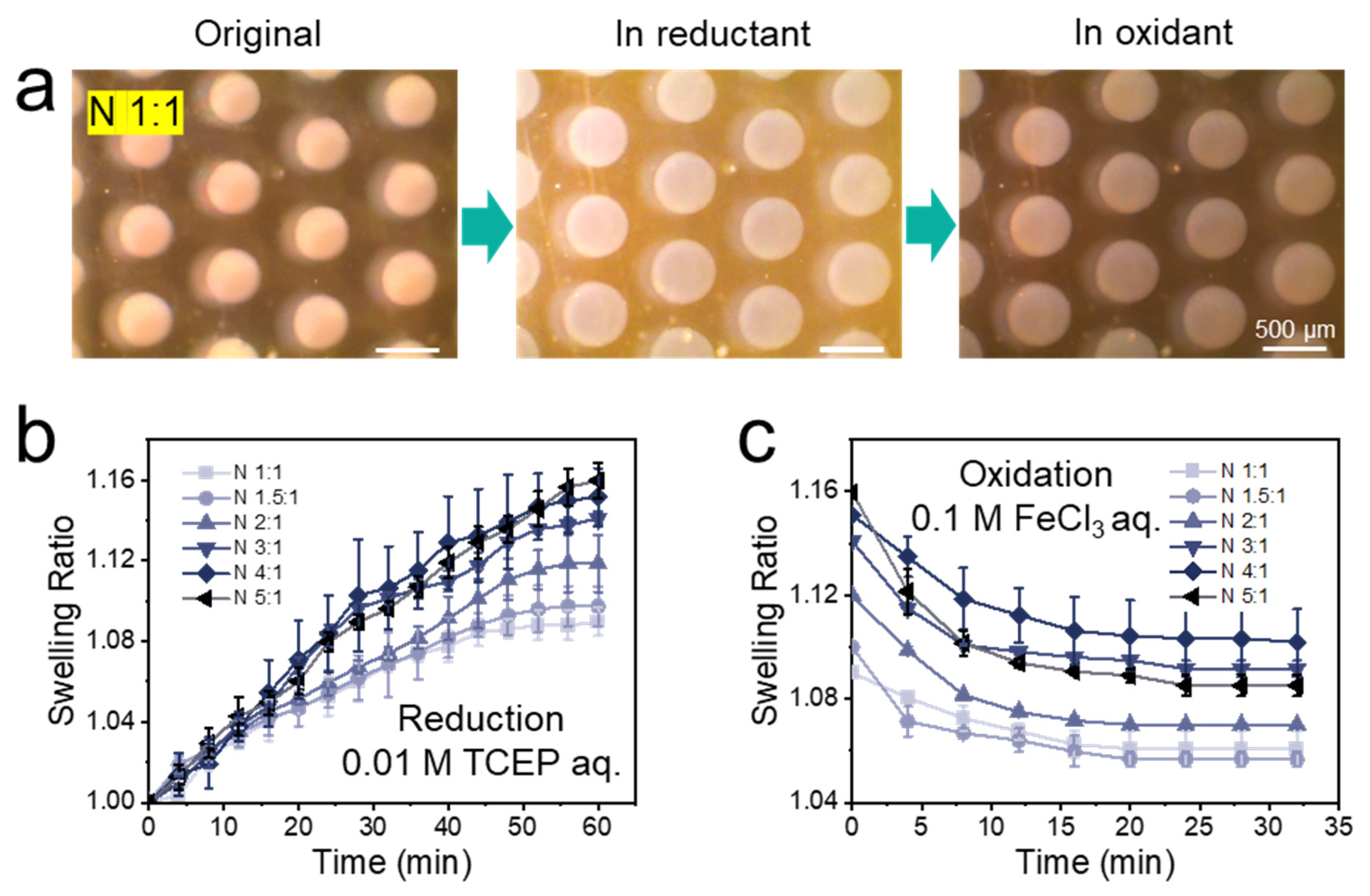
3.2. Optimizing the Cross-Linker Composition of the Hydrogel
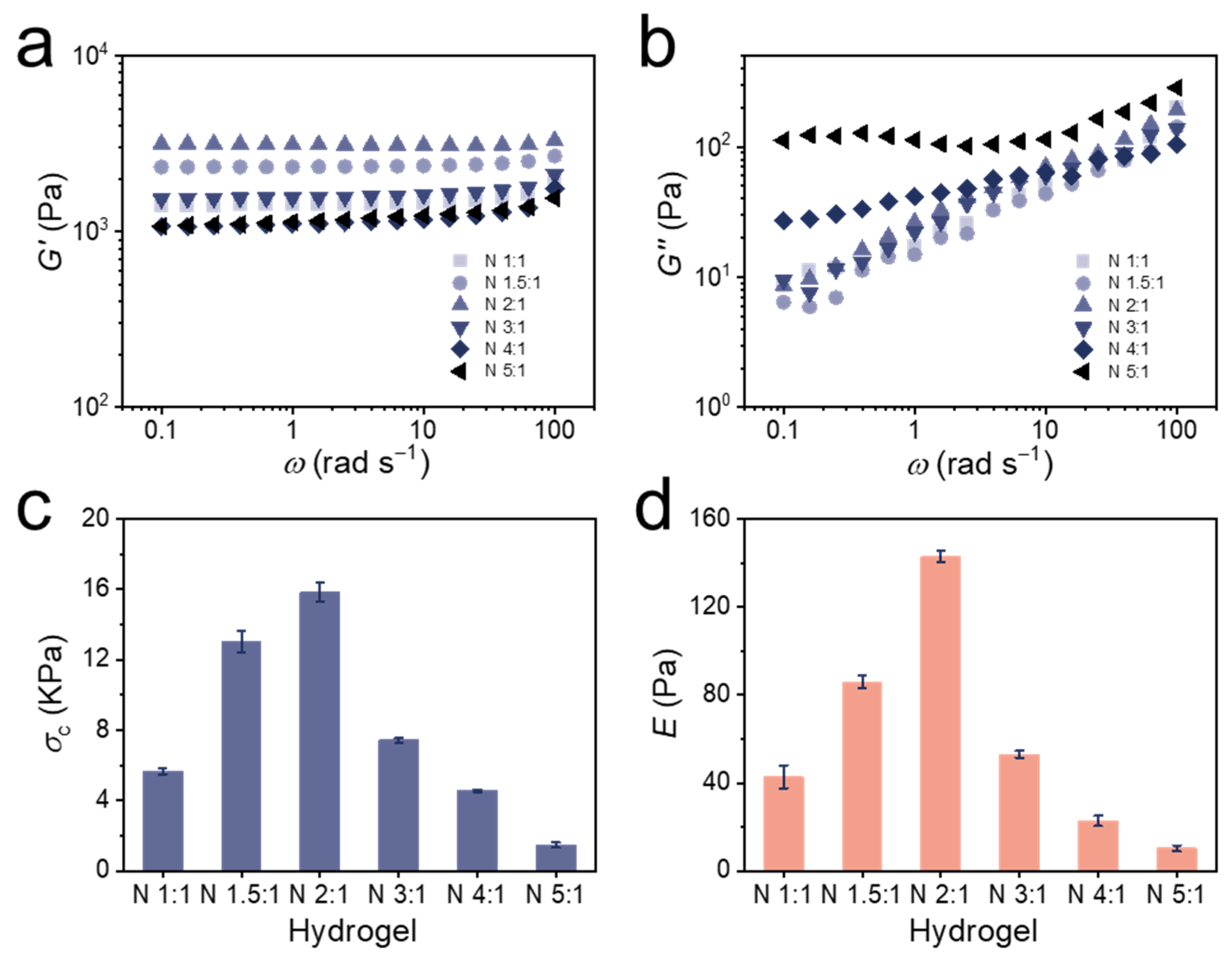
3.3. Mechanical Properties of Bulk Hydrogels
3.4. Modifications of BSA
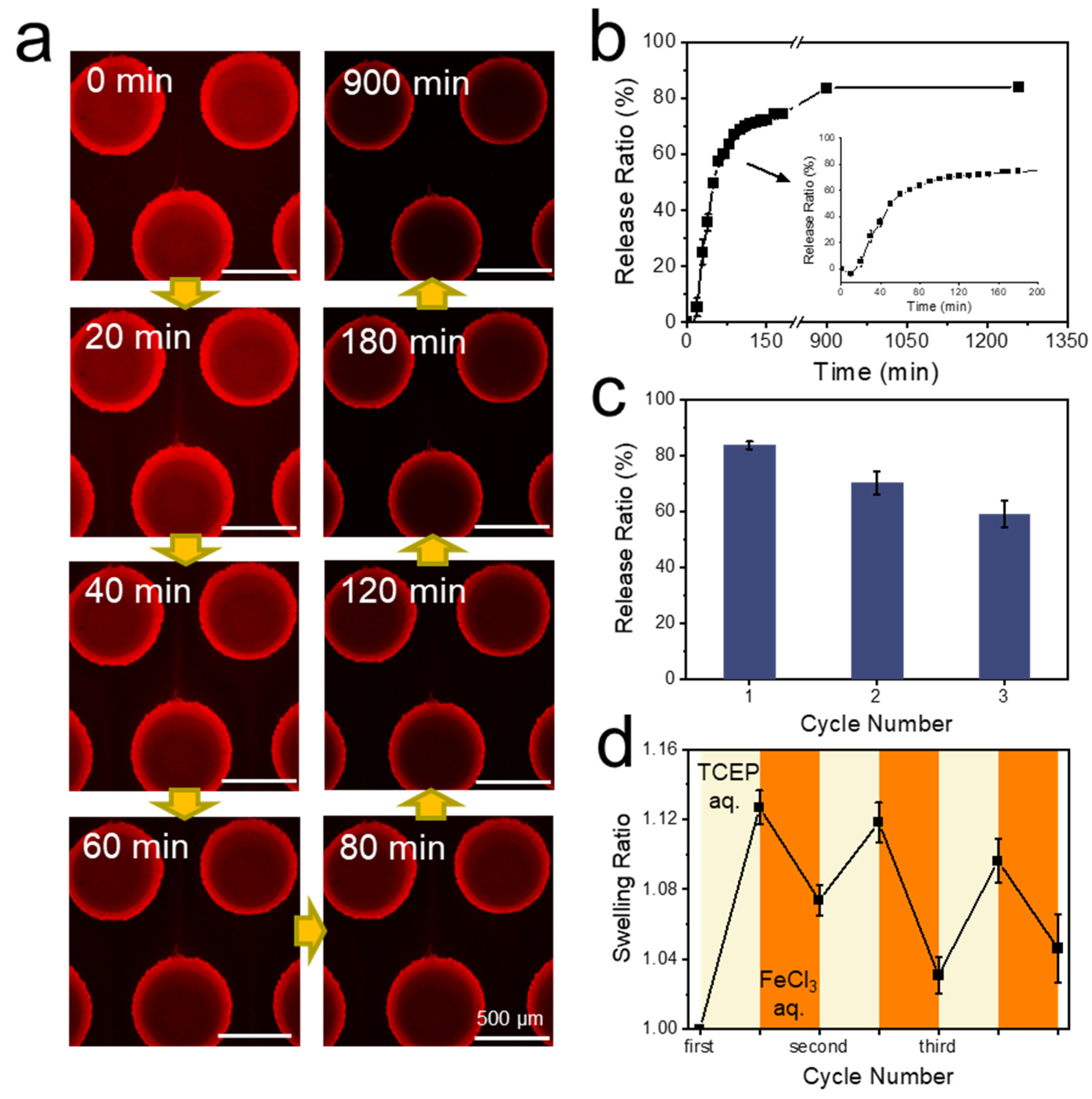
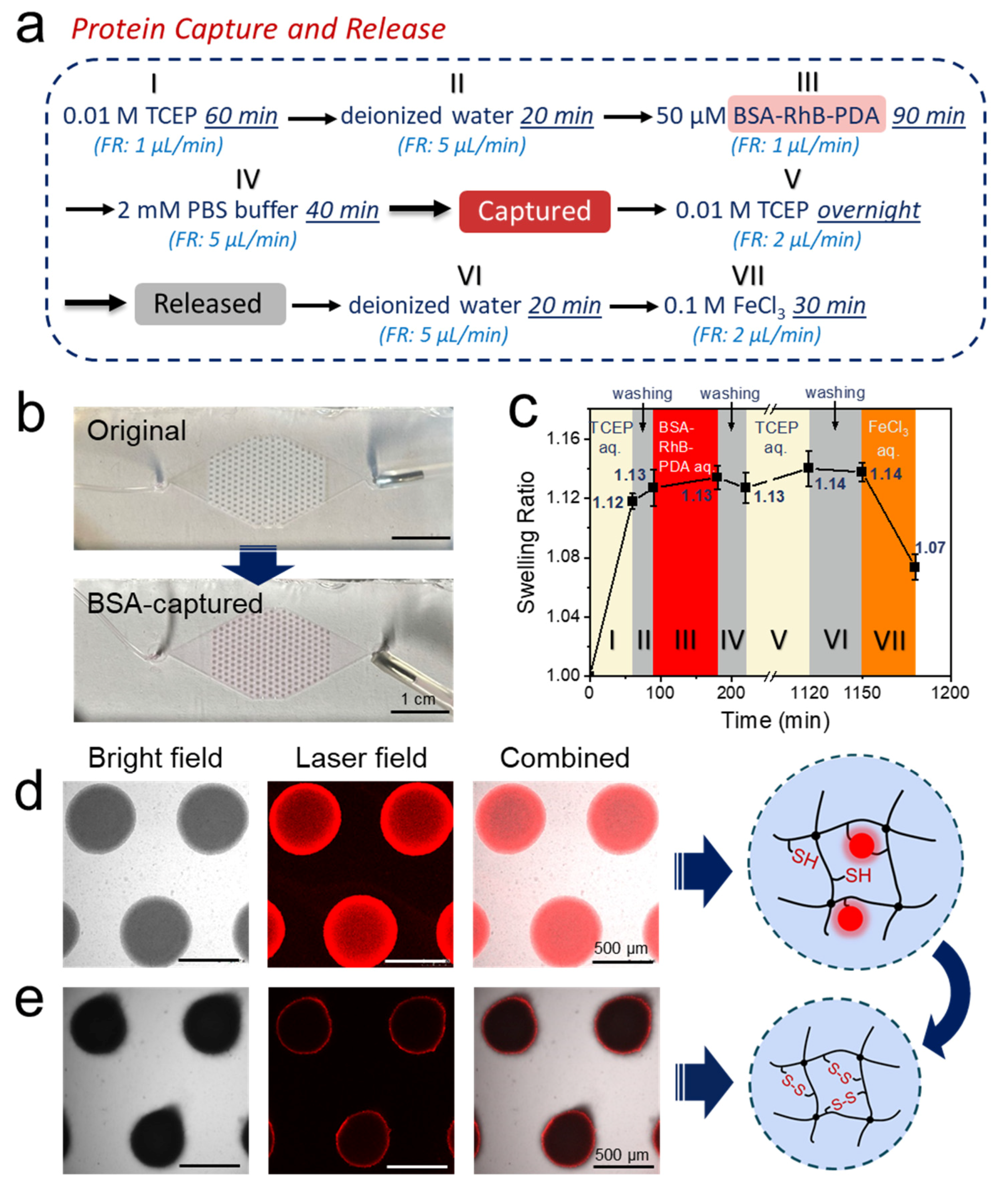
3.5. Protein Capture and Release by Hydrogel Dots in Microfluidics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, L.; Zhang, H.; Shang, L.; Zhao, Y. Microfluidics for drug development: From synthesis to evaluation. Chem. Rev. 2021, 121, 7468–7529. [Google Scholar] [CrossRef]
- Shang, L.; Cheng, Y.; Zhao, Y. Emerging droplet microfluidics. Chem. Rev. 2017, 117, 7964–8040. [Google Scholar] [CrossRef]
- Li, W.; Zhang, L.; Ge, X.; Xu, B.; Zhang, W.; Qu, L.; Choi, C.H.; Xu, J.; Zhang, A.; Lee, H.; et al. Microfluidic fabrication of microparticles for biomedical applications. Chem. Soc. Rev. 2018, 47, 5646–5683. [Google Scholar] [CrossRef]
- Choi, T.M.; Lee, G.H.; Kim, Y.S.; Park, J.G.; Hwang, H.; Kim, S.H. Photonic microcapsules containing single-crystal colloidal arrays with optical anisotropy. Adv. Mater. 2019, 31, e1900693. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Chen, Z.; Sun, L.; Zhao, Y. Anisotropic structural color particles from colloidal phase separation. Sci. Adv. 2020, 6, eaay1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieviet, B.D.; Schon, P.M.; Vancso, G.J. Stimulus-responsive polymers and other functional polymer surfaces as components in glass microfluidic channels. Lab Chip 2014, 14, 4159–4170. [Google Scholar] [CrossRef]
- Nge, P.N.; Rogers, C.I.; Woolley, A.T. Advances in microfluidic materials, functions, integration, and applications. Chem. Rev. 2013, 113, 2550–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obst, F.; Beck, A.; Bishayee, C.; Mehner, P.J.; Richter, A.; Voit, B.; Appelhans, D. Hydrogel microvalves as control elements for parallelized enzymatic cascade reactions in microfluidics. Micromachines 2020, 11, 167. [Google Scholar] [CrossRef] [Green Version]
- Obst, F.; Simon, D.; Mehner, P.J.; Neubauer, J.W.; Beck, A.; Stroyuk, O.; Richter, A.; Voit, B.; Appelhans, D. One-step photostructuring of multiple hydrogel arrays for compartmentalized enzyme reactions in microfluidic devices. React. Chem. Eng. 2019, 4, 2141–2155. [Google Scholar] [CrossRef]
- Obst, F.; Mertz, M.; Mehner, P.J.; Beck, A.; Castiglione, K.; Richter, A.; Voit, B.; Appelhans, D. Enzymatic synthesis of sialic acids in microfluidics to overcome cross-inhibitions and substrate supply limitations. ACS Appl. Mater. Interfaces 2021, 13, 49433–49444. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.; McGregor, L.M.; Liu, Y.; Harris, V.; Nan, H.; Mujica, M.; Vasquez, Y.; Bhattacharya, A.; Ma, Y.; Aizenberg, M.; et al. An aptamer-functionalized chemomechanically modulated biomolecule catch-and-release system. Nat. Chem. 2015, 7, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Chen, T.; Kamath, R.; Xiong, X.; Tan, W.; Fan, Z.H. Aptamer-enabled efficient isolation of cancer cells from whole blood using a microfluidic device. Anal. Chem. 2012, 84, 4199–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, A.A.; Okagbare, P.I.; Feng, J.; Hupert, M.L.; Patterson, D.; Gottert, J.; McCarley, R.L.; Nikitopoulos, D.; Murphy, M.C.; Soper, S.A. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J. Am. Chem. Soc. 2008, 130, 8633–8641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Yu, M.; Nakamura, Z.; Ciciliano, J.; Ulman, M.; Kotz, K.; Stott, S.L.; Maheswaran, S.; Haber, D.A.; Toner, M. Biopolymer system for cell recovery from microfluidic cell capture devices. Anal. Chem. 2012, 84, 3682–3688. [Google Scholar] [CrossRef] [Green Version]
- Hatch, A.; Hansmann, G.; Murthy, S.K. Engineered alginate hydrogels for effective microfluidic capture and release of endothelial progenitor cells from whole blood. Langmuir 2011, 27, 4257–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zhang, X.; Li, Y. Synthesis and application of modulated polymer gels. Science 1995, 269, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Tanaka, T. Phase transition in polymer gels induced by visible light. Nature 1990, 346, 345–347. [Google Scholar] [CrossRef]
- Miyamae, K.; Nakahata, M.; Takashima, Y.; Harada, A. Self-healing, expansion-contraction, and shape-memory properties of a preorganized supramolecular hydrogel through host-guest interactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 8984–8987. [Google Scholar] [CrossRef]
- Lin, C.Y.; Battistoni, C.M.; Liu, J.C. Redox-responsive hydrogels with decoupled initial stiffness and degradation. Biomacromolecules 2021, 22, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Kawaguchi, H.; Endo, T. Characteristics of pH-sensitive hydrogel microsphere of poly(acrylamide-co-methacrylic acid) with sharp pH–volume transition. Colloid Polym. Sci. 2007, 285, 873–879. [Google Scholar] [CrossRef]
- Grafe, D.; Frank, P.; Erdmann, T.; Richter, A.; Appelhans, D.; Voit, B. Tetra-sensitive graft copolymer gels as active material of chemomechanical valves. ACS Appl. Mater. Interfaces 2017, 9, 7565–7576. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Zhang, J.; Liu, T.; Peng, X.; Wang, H. Mechanically strong, tough, and shape deformable poly(acrylamide-co-vinylimidazole) hydrogels based on Cu2+ complexation. ACS Appl. Mater. Interfaces 2020, 12, 44205–44214. [Google Scholar] [CrossRef] [PubMed]
- Chimisso, V.; Conti, S.; Kong, P.; Fodor, C.; Meier, W.P. Metal cation responsive anionic microgels: Behaviour towards biologically relevant divalent and trivalent ions. Soft Matter 2021, 17, 715–723. [Google Scholar] [CrossRef]
- Tanaka, T.; Nishio, I.; Sun, S.T.; Ueno-Nishio, S. Collapse of gels in an electric field. Science 1982, 218, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y.; Pan, L.; Shi, Y.; Cheng, W.; Shi, Y.; Yu, G. A nanostructured conductive hydrogels-based biosensor platform for human metabolite detection. Nano Lett. 2015, 15, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ji, C.; Lu, B.; Rodin, M.; Paradies, J.; Yin, M.; Kuckling, D. Dually crosslinked supramolecular hydrogel for cancer biomarker sensing. ACS Appl. Mater. Interfaces 2020, 12, 36873–36881. [Google Scholar] [CrossRef] [PubMed]
- Beebe, D.J.; Moore, J.S.; Bauer, J.M.; Yu, Q.; Liu, R.H.; Devadoss, C.; Jo, B.H. Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 2000, 404, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for protein delivery. Chem. Rev. 2012, 112, 2853–2888. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Elkassih, S.A.; Kos, P.; Xiong, H.; Siegwart, D.J. Degradable redox-responsive disulfide-based nanogel drug carriers via dithiol oxidation polymerization. Biomater. Sci. 2019, 7, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Kuckling, D.; Howitz, S.; Gehring, T.; Arndt, K. Electronically controllable microvalves based on smart hydrogels: Magnitudes and potential applications. J. Microelectromech. Syst. 2003, 12, 748–753. [Google Scholar] [CrossRef]
- Richter, A.; Howitz, S.; Kuckling, D.; Arndt, K.F. Influence of volume phase transition phenomena on the behavior of hydrogel-based valves. Sens. Actuators B Chem. 2004, 99, 451–458. [Google Scholar] [CrossRef]
- Lee, E.; Lee, H.; Yoo, S.I.; Yoon, J. Photothermally triggered fast responding hydrogels incorporating a hydrophobic moiety for light-controlled microvalves. ACS Appl. Mater. Interfaces 2014, 6, 16949–16955. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Klatt, S.; Paschew, G.; Klenke, C. Micropumps operated by swelling and shrinking of temperature-sensitive hydrogels. Lab Chip 2009, 9, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiao, Y.; Lin, L.; Zhu, X.; Du, L.; Shi, X. A microfluidic chip integrated with hyaluronic acid-functionalized electrospun chitosan nanofibers for specific capture and nondestructive release of CD44-overexpressing circulating tumor cells. Bioconjug. Chem. 2018, 29, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Yang, Z.; Lim, C.W.; Lee, Y.H.; Dongbang, S.; Kang, C.; Kim, J.S. Disulfide-cleavage-triggered chemosensors and their biological applications. Chem. Rev. 2013, 113, 5071–5109. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 2010, 35, 278–301. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, R.D.; Dmitrenko, O.; Thorpe, C. Mechanism of thiolate-disulfide interchange reactions in biochemistry. J. Org. Chem. 2008, 73, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Houk, J.; Whitesides, G.M. Structure-reactivity relations for thiol-disulfide interchange. J. Am. Chem. Soc. 2002, 109, 6825–6836. [Google Scholar] [CrossRef]
- Pires, M.M.; Chmielewski, J. Fluorescence imaging of cellular glutathione using a latent rhodamine. Org. Lett. 2008, 10, 837–840. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, M.; Lin, L.; Du, L.; Shen, M.; Shi, X. Integration of aligned polymer nanofibers within a microfluidic chip for efficient capture and rapid release of circulating tumor cells. Mater. Chem. Front. 2018, 2, 891–900. [Google Scholar] [CrossRef]
- Chong, S.F.; Chandrawati, R.; Stadler, B.; Park, J.; Cho, J.; Wang, Y.; Jia, Z.; Bulmus, V.; Davis, T.P.; Zelikin, A.N.; et al. Stabilization of polymer-hydrogel capsules via thiol-disulfide exchange. Small 2009, 5, 2601–2610. [Google Scholar] [CrossRef]
- An, S.Y.; Noh, S.M.; Oh, J.K. Multiblock copolymer-based dual dynamic disulfide and supramolecular crosslinked self-healing networks. Macromol. Rapid Commun. 2017, 38, 1600777. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, G. A stimuli-responsive hydrogel with reversible three-state transition controlled by redox stimulation. Macromol. Chem. Phys. 2017, 218, 1700002. [Google Scholar] [CrossRef]
- Deng, G.; Li, F.; Yu, H.; Liu, F.; Liu, C.; Sun, W.; Jiang, H.; Chen, Y. Dynamic hydrogels with an environmental adaptive self-healing ability and dual responsive sol-gel transitions. ACS Macro Lett. 2012, 1, 275–279. [Google Scholar] [CrossRef]
- Lee, W.F.; Lu, Y.Y. Influence of novel crosslinker on the properties of the degradable thermosensitive hydrogels. Macromol. Symp. 2015, 358, 41–51. [Google Scholar] [CrossRef]
- Gaulding, J.C.; Smith, M.H.; Hyatt, J.S.; Fernandez-Nieves, A.; Lyon, L.A. Reversible inter- and intra-microgel cross-linking using disulfides. Macromolecules 2012, 45, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkikas, M.; Avery, R.K.; Mills, C.E.; Nagarajan, R.; Wilusz, E.; Olsen, B.D. Hydrogels that actuate selectively in response to organophosphates. Adv. Funct. Mater. 2016, 27, 1602784. [Google Scholar] [CrossRef]
- Che, Y.; Zschoche, S.; Obst, F.; Appelhans, D.; Voit, B. Double-crosslinked reversible redox-responsive hydrogels based on disulfide–thiol interchange. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 2590–2601. [Google Scholar] [CrossRef]
- Eddington, D.T.; Beebe, D.J. Flow control with hydrogels. Adv. Drug Deliv. Rev. 2004, 56, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Jiang, H. Autonomous microfluidics with stimuli-responsive hydrogels. Soft Matter 2007, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.W.; Ahn, C.H. A review of microvalves. J. Micromech. Microeng. 2006, 16, R13–R39. [Google Scholar] [CrossRef]
- Mirhosseini Moghaddam, M.; Baghbanzadeh, M.; Sadeghpour, A.; Glatter, O.; Kappe, C.O. Continuous-flow synthesis of CdSe quantum dots: A size-tunable and scalable approach. Chem. Eur. J. 2013, 19, 11629–11636. [Google Scholar] [CrossRef]
- Simon, D.; Obst, F.; Haefner, S.; Heroldt, T.; Peiter, M.; Simon, F.; Richter, A.; Voit, B.; Appelhans, D. Hydrogel/enzyme dots as adaptable tool for non-compartmentalized multi-enzymatic reactions in microfluidic devices. Reat. Chem. Eng. 2019, 4, 67–77. [Google Scholar] [CrossRef]
- Battistella, C.; Klok, H.A. Reversion of P-gp-mediated drug resistance in ovarian carcinoma cells with PHPMA-zosuquidar conjugates. Biomacromolecules 2017, 18, 1855–1865. [Google Scholar] [CrossRef]
- Hanton, S.D.; Owens, K.G. Polymer MALDI sample preparation. In Mass Spectrometry in Polymer Chemistry; Wily: Weinheim, Germany, 2012; pp. 119–147. [Google Scholar]
- Jiao, C.; Chen, Y.; Liu, T.; Peng, X.; Zhao, Y.; Zhang, J.; Wu, Y.; Wang, H. Rigid and strong thermoresponsive shape memory hydrogels transformed from poly(vinylpyrrolidone-co-acryloxy acetophenone) organogels. ACS Appl. Mater. Interfaces 2018, 10, 32707–32716. [Google Scholar] [CrossRef]
- Van Den Bulcke, A.I.; Bogdanov, B.; De Rooze, N.; Schacht, E.H.; Cornelissen, M.; Berghmans, H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules 2000, 1, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: New York, NY, USA, 2003; Volume 23. [Google Scholar]
- Hao, J.; Weiss, R.A. Viscoelastic and mechanical behavior of hydrophobically modified hydrogels. Macromolecules 2011, 44, 9390–9398. [Google Scholar] [CrossRef]
- Chen, T.; Chen, Y.; Rehman, H.U.; Chen, Z.; Yang, Z.; Wang, M.; Li, H.; Liu, H. Ultratough, self-healing, and tissue-adhesive hydrogel for wound dressing. ACS Appl. Mater. Interfaces 2018, 10, 33523–33531. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhao, X.; Ye, L. Facile method to fabricate superstrong and tough poly(vinyl alcohol) hydrogels with high energy dissipation. Ind. Eng. Chem. Res. 2020, 59, 10705–10715. [Google Scholar] [CrossRef]
- Zugates, G.T.; Anderson, D.G.; Little, S.R.; Lawhorn, I.E.; Langer, R. Synthesis of poly(β-amino ester)s with thiol-reactive side chains for DNA delivery. J. Am. Chem. Soc. 2006, 128, 12726–12734. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, M.; Green, D.C.; Williams, D.S.; Patil, A.J.; Mann, S. Interfacial assembly of protein-polymer nano-conjugates into stimulus-responsive biomimetic protocells. Nat. Commun. 2013, 4, 2239. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
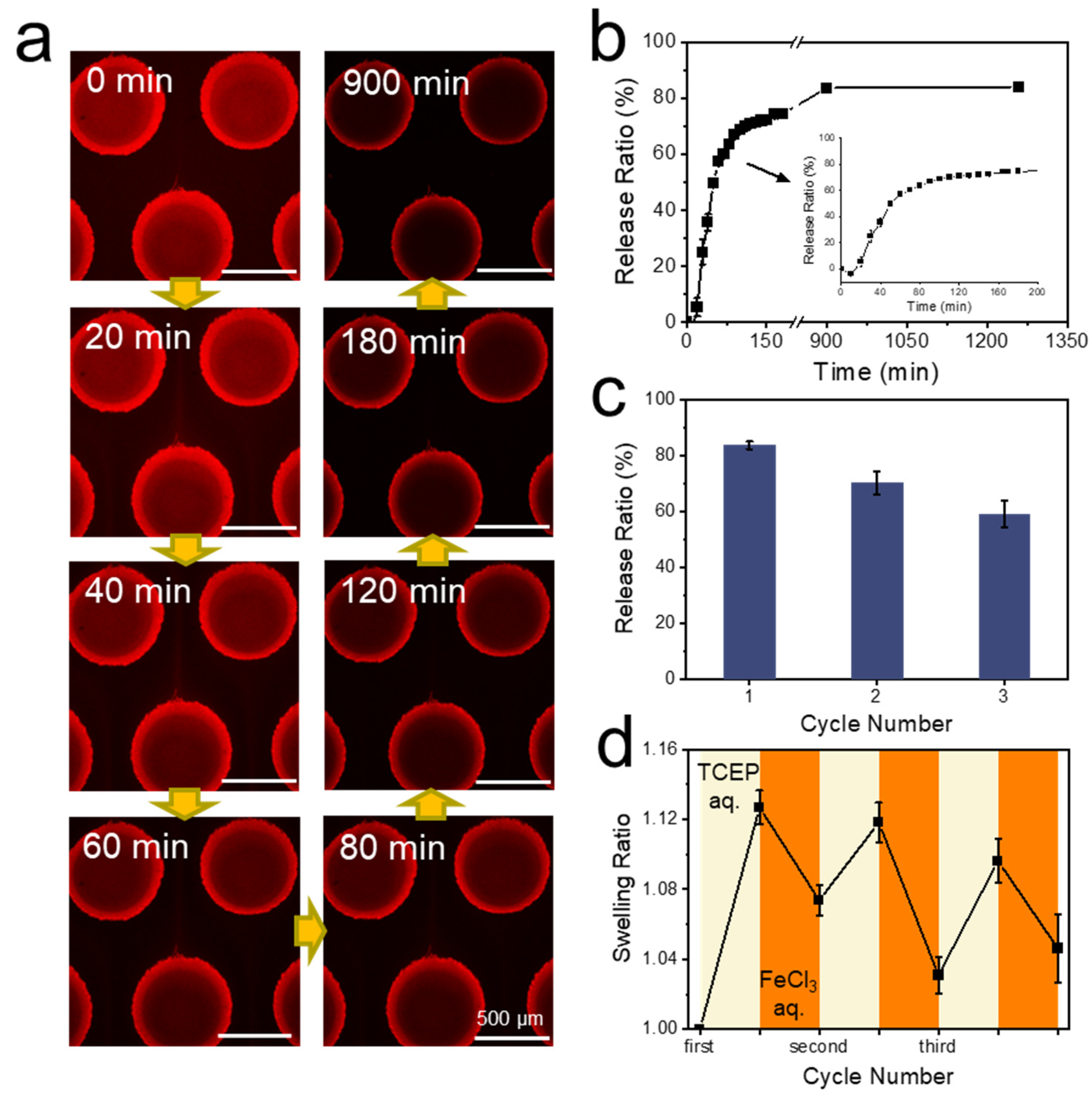{kind=link}
| Hydrogel | Swelling in 0.01 M TCEP aq. a | Residual Swelling in 0.1 M FeCl3 aq. b | Percentage Reduction c | Compression Strength at 50% Strain (kPa) d | Elastic Modulus (Pa) d |
|---|---|---|---|---|---|
| N 1:1 | 1.09 ± 0.007 | 1.06 ± 0.007 | 33% | 5.66 ± 0.17 | 43 ± 5 |
| N 1.5:1 | 1.10 ± 0.010 | 1.06 ± 0.003 | 40% | 13.03 ± 0.60 | 86 ± 3 |
| N 2:1 | 1.12 ± 0.014 | 1.07 ± 0.001 | 42% | 15.85 ± 0.55 | 143 ± 3 |
| N 3:1 | 1.14 ± 0.004 | 1.09 ± 0.003 | 36% | 7.42 ± 0.13 | 53 ± 2 |
| N 4:1 | 1.15 ± 0.014 | 1.10 ± 0.013 | 33% | 4.55 ± 0.08 | 23 ± 2 |
| N 5:1 | 1.16 ± 0.009 | 1.09 ± 0.004 | 44% | 1.47 ± 0.14 | 10 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, C.; Obst, F.; Geisler, M.; Che, Y.; Richter, A.; Appelhans, D.; Gaitzsch, J.; Voit, B. Reversible Protein Capture and Release by Redox-Responsive Hydrogel in Microfluidics. Polymers 2022, 14, 267. https://doi.org/10.3390/polym14020267
Jiao C, Obst F, Geisler M, Che Y, Richter A, Appelhans D, Gaitzsch J, Voit B. Reversible Protein Capture and Release by Redox-Responsive Hydrogel in Microfluidics. Polymers. 2022; 14(2):267. https://doi.org/10.3390/polym14020267
Chicago/Turabian StyleJiao, Chen, Franziska Obst, Martin Geisler, Yunjiao Che, Andreas Richter, Dietmar Appelhans, Jens Gaitzsch, and Brigitte Voit. 2022. "Reversible Protein Capture and Release by Redox-Responsive Hydrogel in Microfluidics" Polymers 14, no. 2: 267. https://doi.org/10.3390/polym14020267
APA StyleJiao, C., Obst, F., Geisler, M., Che, Y., Richter, A., Appelhans, D., Gaitzsch, J., & Voit, B. (2022). Reversible Protein Capture and Release by Redox-Responsive Hydrogel in Microfluidics. Polymers, 14(2), 267. https://doi.org/10.3390/polym14020267