
Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
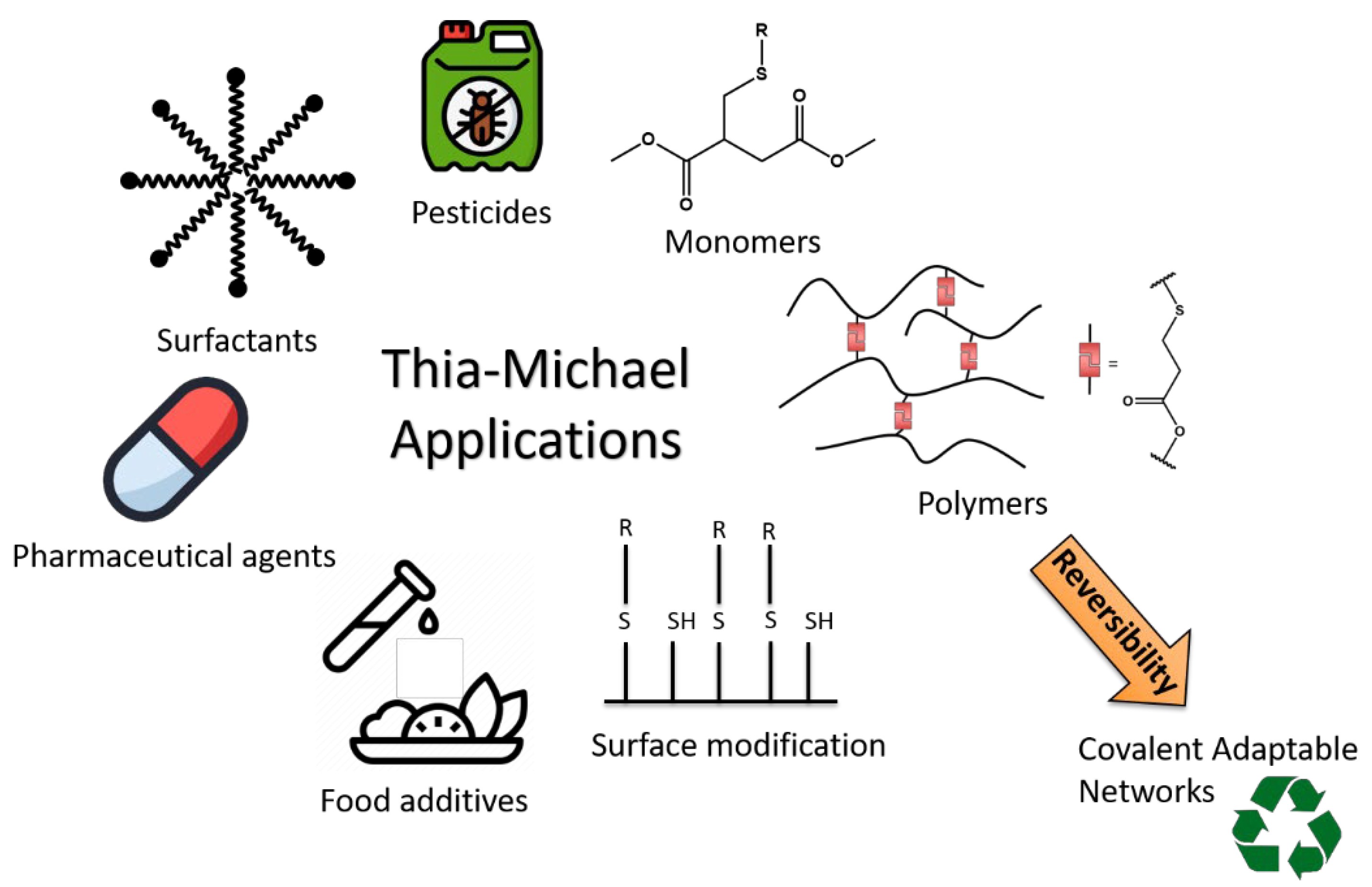
1. Introduction
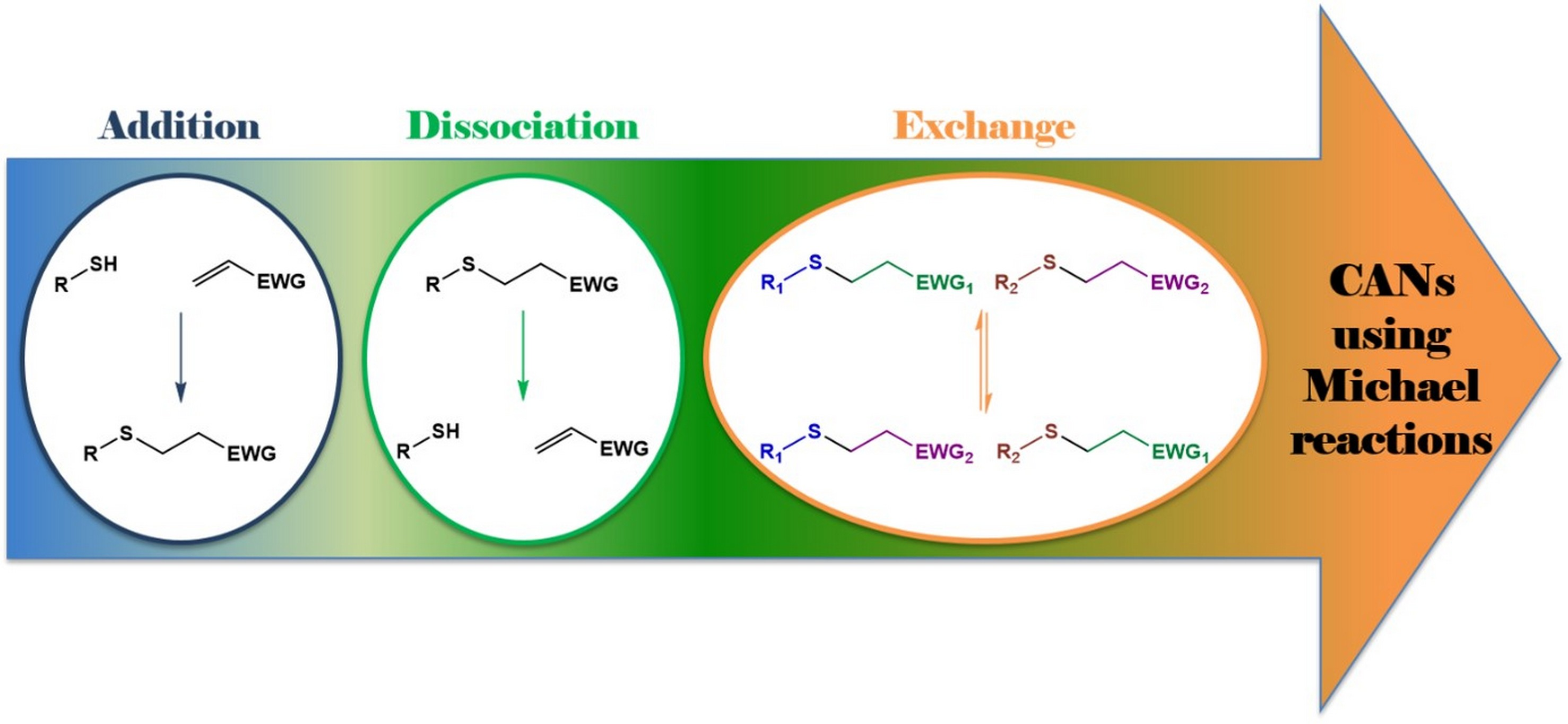
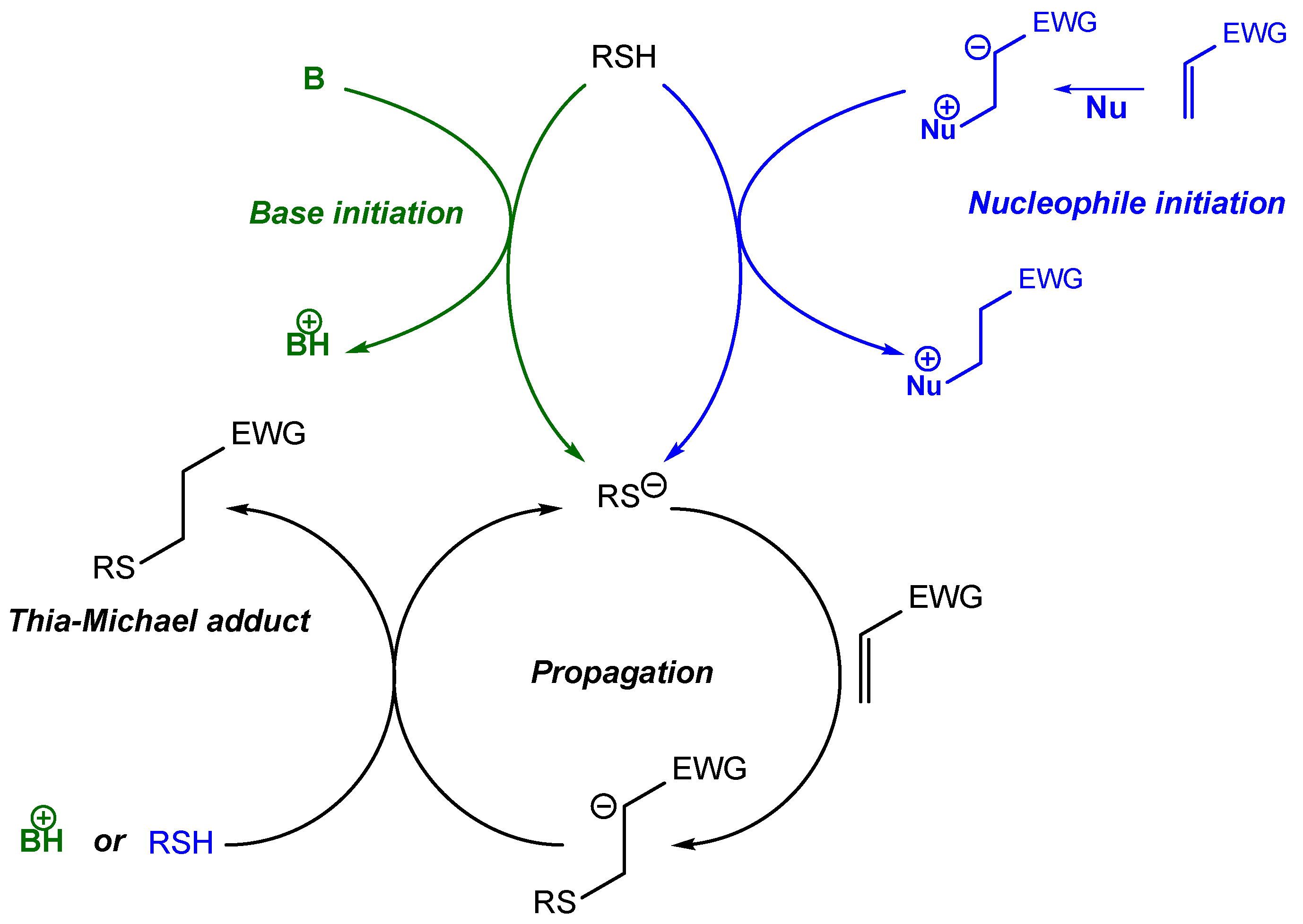
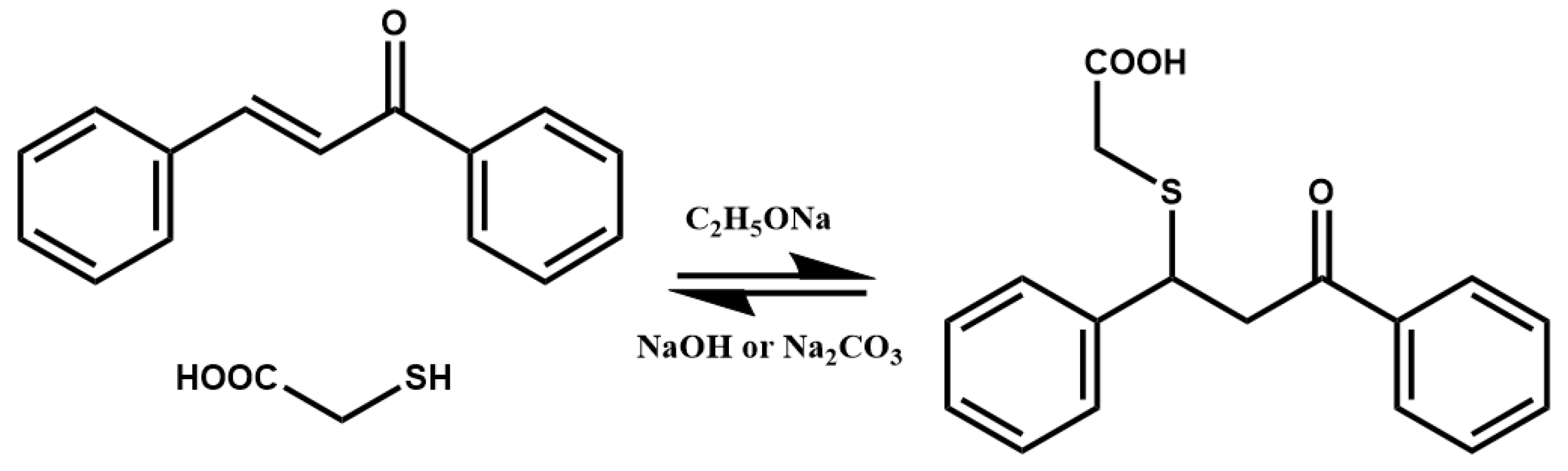
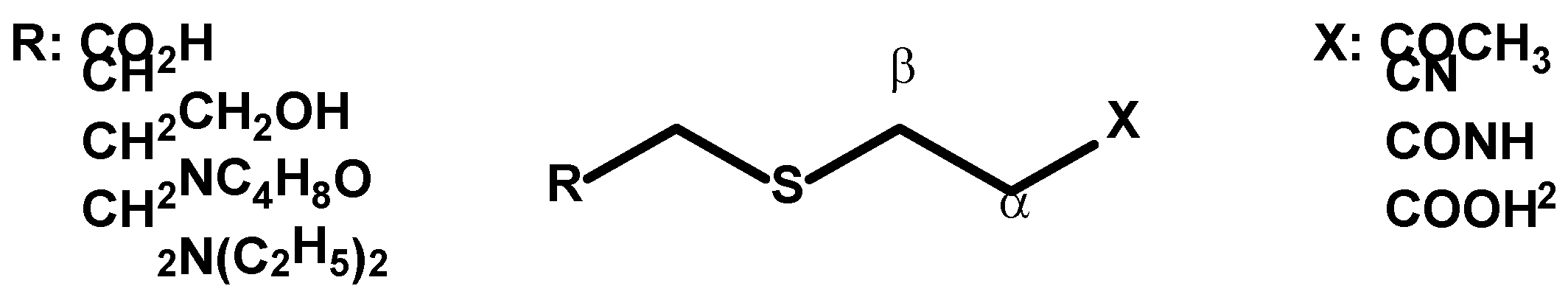
2. Thia-Michael Reaction
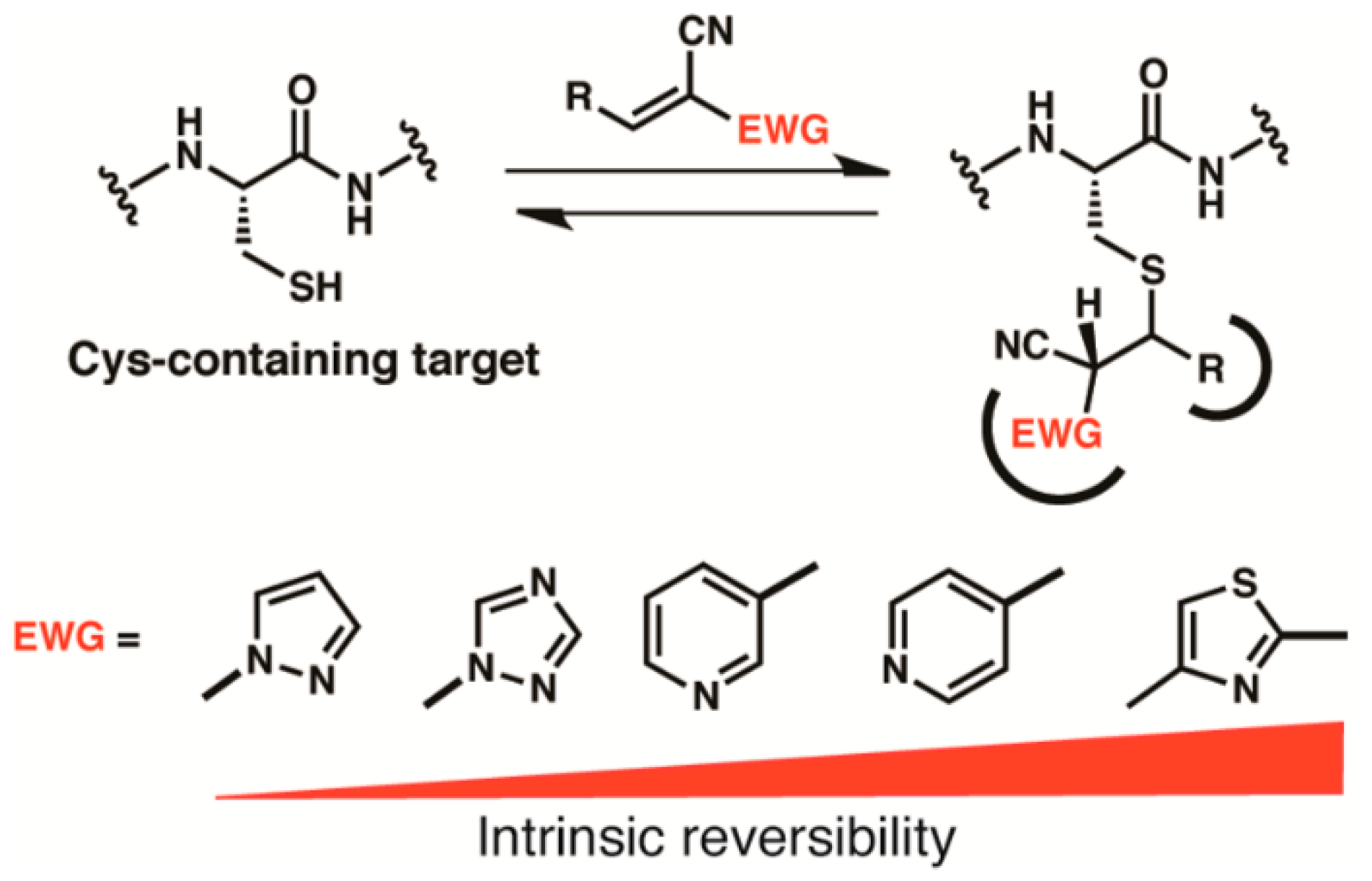
3. Thia-Michael Equilibrium and Exchange Kinetics
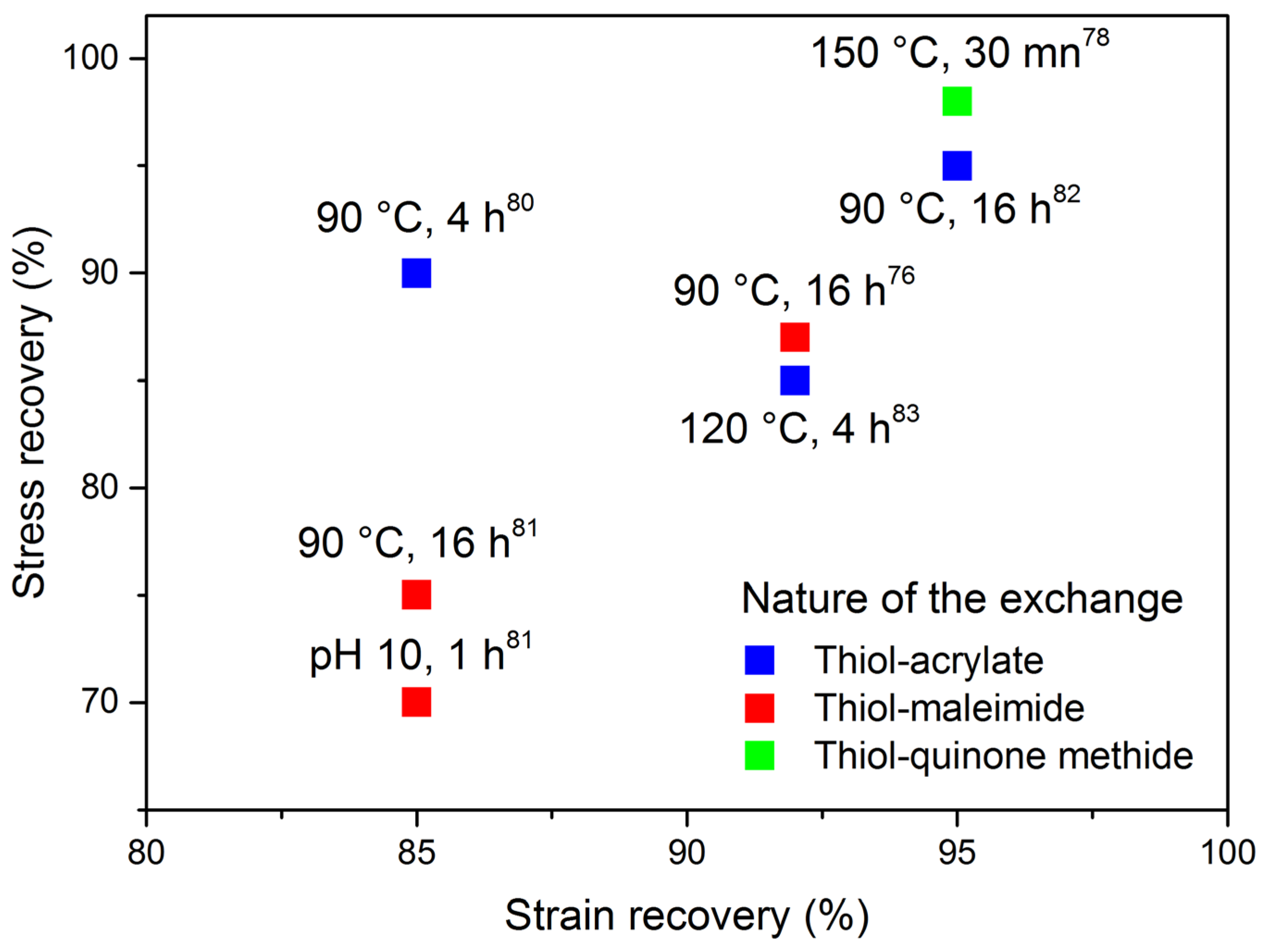
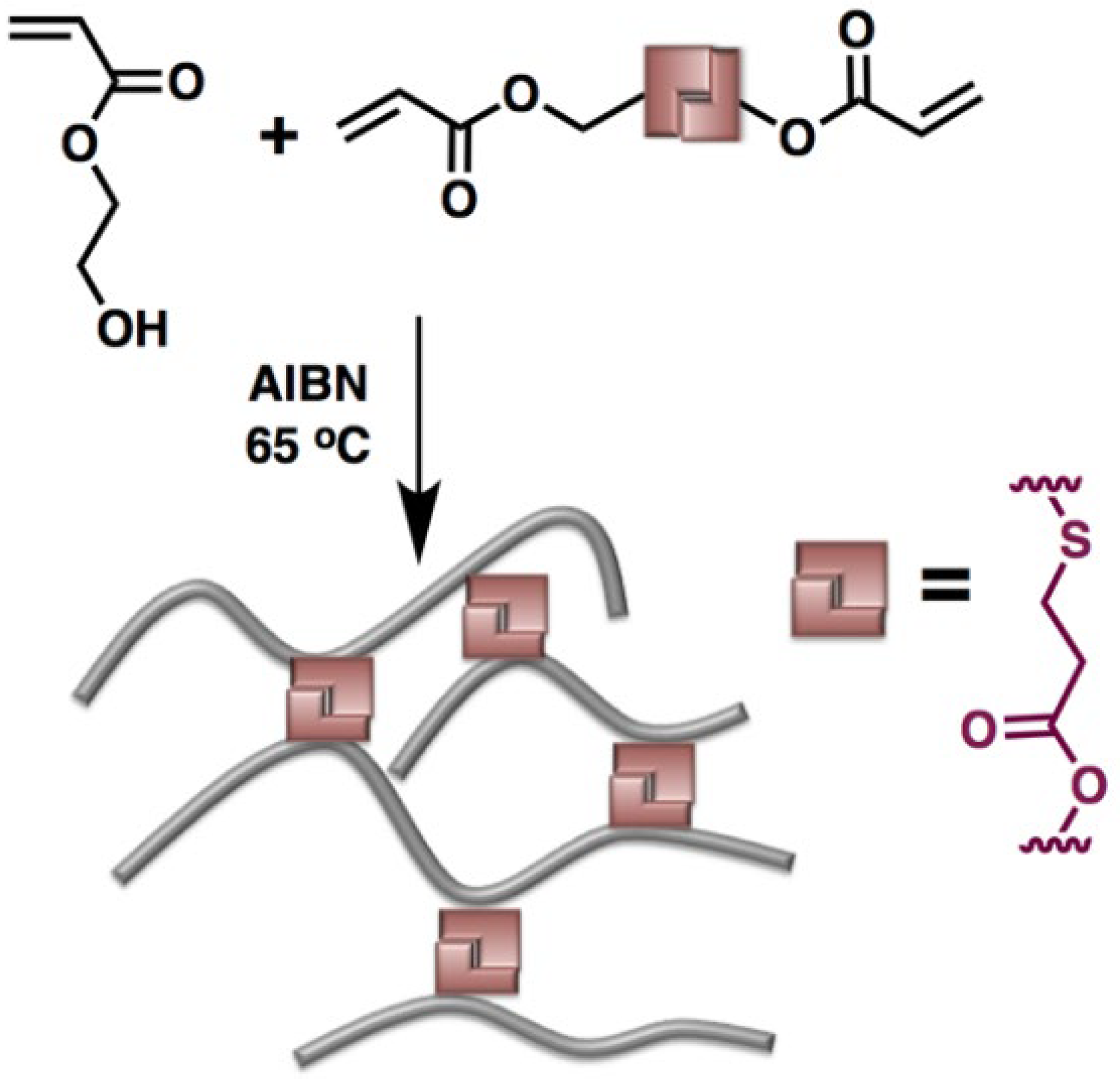
4. Thia-Michael Exchange in CANs
5. Perspectives on the Development of Aza-Michael Exchange Reactions in CANs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fotouhi, L.; Heravi, M.M.; Zadsirjan, V.; Atoi, P.A. Electrochemically Induced Michael Addition Reaction: An Overview. Chem. Rec. 2018, 18, 1633–1657. [Google Scholar] [CrossRef] [PubMed]
- Michael, A. On the Addition of Sodium Acetacetic Ether and Analogous Sodium Compounds to Unsaturated Organic Ethers. Am. Chem. J. 1887, 9, 112. [Google Scholar]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael Addition Reactions in Macromolecular Design for Emerging Technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Patrick, J.F.; Robb, M.J.; Sottos, N.R.; Moore, J.S.; White, S.R. Polymers with Autonomous Life-Cycle Control. Nature 2016, 540, 363–370. [Google Scholar] [CrossRef]
- Zheng, N.; Xu, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing. Chem. Rev. 2021, 121, 1716–1745. [Google Scholar] [CrossRef]
- Tobolsky, A.V.; Prettyman, I.B.; Dillon, J.H. Stress Relaxation of Natural and Synthetic Rubber Stocks. Rubber Chem. Technol. 1944, 17, 551–575. [Google Scholar] [CrossRef]
- Stern, M.D.; Tobolsky, A.V. Stress-Time-Temperature Relations in Polysulfide Rubbers. Rubber Chem. Technol. 1946, 19, 1178–1192. [Google Scholar] [CrossRef]
- Zheng, P.; McCarthy, T.J. A Surprise from 1954: Siloxane Equilibration Is a Simple, Robust, and Obvious Polymer Self-Healing Mechanism. J. Am. Chem. Soc. 2012, 134, 2024–2027. [Google Scholar] [CrossRef]
- Osthoff, R.C.; Bueche, A.M.; Grubb, W.T. Chemical Stress-Relaxation of Polydimethylsiloxane Elastomers 1. J. Am. Chem. Soc. 1954, 76, 4659–4663. [Google Scholar] [CrossRef]
- Lyon, G.B.; Baranek, A.; Bowman, C.N. Scaffolded Thermally Remendable Hybrid Polymer Networks. Adv. Funct. Mater. 2016, 26, 1477–1485. [Google Scholar] [CrossRef]
- Reutenauer, P.; Buhler, E.; Boul, P.J.; Candau, S.J.; Lehn, J.-M. Room Temperature Dynamic Polymers Based on Diels-Alder Chemistry. Chem.-A Eur. J. 2009, 15, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A Thermally Re-Mendable Cross-Linked Polymeric Material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Digby, Z.A.; Flum, J.A.; Foster, E.M.; Sparks, J.L.; Konkolewicz, D. Self-Healing, Malleable and Creep Limiting Materials Using Both Supramolecular and Reversible Covalent Linkages. Polym. Chem. 2015, 6, 7368–7372. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Han, J.; Liu, T.; Hao, C.; Zhang, S.; Guo, B.; Zhang, J. A Catalyst-Free Epoxy Vitrimer System Based on Multifunctional Hyperbranched Polymer. Macromolecules 2018, 51, 6789–6799. [Google Scholar] [CrossRef]
- Altuna, F.I.; Hoppe, C.E.; Williams, R.J.J. Shape Memory Epoxy Vitrimers Based on DGEBA Crosslinked with Dicarboxylic Acids and Their Blends with Citric Acid. RSC Adv. 2016, 6, 88647–88655. [Google Scholar] [CrossRef] [Green Version]
- Altuna, F.I.; Casado, U.; Dell’Erba, I.E.; Luna, L.; Hoppe, C.E.; Williams, R.J.J. Epoxy Vitrimers Incorporating Physical Crosslinks Produced by Self-Association of Alkyl Chains-SI. Polym. Chem. 2020, 11, 1337–1347. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Jin, K.; Torkelson, J.M. Reprocessable Polyhydroxyurethane Networks Exhibiting Full Property Recovery and Concurrent Associative and Dissociative Dynamic Chemistry: Via Transcarbamoylation and Reversible Cyclic Carbonate Aminolysis. Polym. Chem. 2017, 8, 6349–6355. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural Effects on the Reprocessability and Stress Relaxation of Crosslinked Polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984. [Google Scholar] [CrossRef] [Green Version]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef]
- Elizalde, F.; Aguirresarobe, R.H.; Gonzalez, A.; Sardon, H. Dynamic Polyurethane Thermosets: Tuning Associative/Dissociative Behavior by Catalyst Selection. Polym. Chem. 2020, 11, 5386–5396. [Google Scholar] [CrossRef]
- Van Lijsebetten, F.; Spiesschaert, Y.; Winne, J.M.; Du Prez, F.E. Reprocessing of Covalent Adaptable Polyamide Networks through Internal Catalysis and Ring-Size Effects. J. Am. Chem. Soc. 2021, 143, 15834–15844. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, H.; Majumdar, S.; van Benthem, R.A.T.M.; Heuts, J.P.A.; Sijbesma, R.P. Dynamic Polyamide Networks via Amide–Imide Exchange. Macromolecules 2021, 54, 9703–9711. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent Organic Networks with Glass-like Fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elling, B.R.; Dichtel, W.R. Reprocessable Cross-Linked Polymer Networks: Are Associative Exchange Mechanisms Desirable? ACS Cent. Sci. 2020, 6, 1488–1496. [Google Scholar] [CrossRef]
- Zhang, L.; Rowan, S.J. Effect of Sterics and Degree of Cross-Linking on the Mechanical Properties of Dynamic Poly(Alkylurea–Urethane) Networks. Macromolecules 2017, 50, 5051–5060. [Google Scholar] [CrossRef]
- Hayashi, M.; Chen, L. Functionalization of Triblock Copolymer Elastomers by Cross-Linking the End Blocks via Trans-N -Alkylation-Based Exchangeable Bonds. Polym. Chem. 2020, 11, 1713–1719. [Google Scholar] [CrossRef]
- Jourdain, A.; Asbai, R.; Anaya, O.; Chehimi, M.M.; Drockenmuller, E.; Montarnal, D. Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks. Macromolecules 2020, 53, 1884–1900. [Google Scholar] [CrossRef] [Green Version]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Winne, J.M.; Du Prez, F.E. Vitrimers: Directing Chemical Reactivity to Control Material Properties. Chem. Sci. 2020, 11, 4855–4870. [Google Scholar] [CrossRef] [Green Version]
- Cuminet, F.; Caillol, S.; Dantras, É.; Leclerc, É.; Ladmiral, V. Neighboring Group Participation and Internal Catalysis Effects on Exchangeable Covalent Bonds: Application to the Thriving Field of Vitrimer Chemistry. Macromolecules 2021, 54, 3927–3961. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, B.; Zhang, J. Recent Development of Repairable, Malleable and Recyclable Thermosetting Polymers through Dynamic Transesterification. Polymer 2020, 194, 122392. [Google Scholar] [CrossRef]
- Bakkali-Hassani, C.; Berne, D.; Ladmiral, V.; Caillol, S. Transcarbamoylation in Polyurethanes: Underestimated Exchange Reactions? Macromolecules 2022, 55, 7974–7991. [Google Scholar] [CrossRef]
- Liu, G.; Link, J.T.; Pei, Z.; Reilly, E.B.; Leitza, S.; Nguyen, B.; Marsh, K.C.; Okasinski, G.F.; Von Geldern, T.W.; Ormes, M.; et al. Discovery of Novel P-Arylthio Cinnamides as Antagonists of Leukocyte Function-Associated Antigen-1/Intracellular Adhesion Molecule-1 Interaction. 1. Identification of an Additional Binding Pocket Based on an Anilino Diaryl Sulfide Lead. J. Med. Chem. 2000, 43, 4025–4040. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.J.; Chen, B. Simultaneous Determination of Nonnutritive Sweeteners in Foods by HPLC/ESI-MS. J. Agric. Food Chem. 2009, 57, 3022–3027. [Google Scholar] [CrossRef]
- Matsuno, R.; Takami, K.; Ishihara, K. Simple Synthesis of a Library of Zwitterionic Surfactants via Michael-Type Addition of Methacrylate and Alkane Thiol Compounds. Langmuir 2010, 26, 13028–13032. [Google Scholar] [CrossRef]
- Alswieleh, A.M.; Cheng, N.; Canton, I.; Ustbas, B.; Xue, X.; Ladmiral, V.; Xia, S.; Ducker, R.E.; El Zubir, O.; Cartron, M.L.; et al. Zwitterionic Poly(Amino Acid Methacrylate) Brushes. J. Am. Chem. Soc. 2014, 136, 9404–9413. [Google Scholar] [CrossRef]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon–Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5521–5577. [Google Scholar] [CrossRef]
- Giacobazzi, G.; Gioia, C.; Colonna, M.; Celli, A. Thia-Michael Reaction for a Thermostable Itaconic-Based Monomer and the Synthesis of Functionalized Biopolyesters. ACS Sustain. Chem. Eng. 2019, 7, 5553–5559. [Google Scholar] [CrossRef]
- Chatani, S.; Podgórski, M.; Wang, C.; Bowman, C.N. Facile and Efficient Synthesis of Dendrimers and One-Pot Preparation of Dendritic-Linear Polymer Conjugates via a Single Chemistry: Utilization of Kinetically Selective Thiol-Michael Addition Reactions. Macromolecules 2014, 47, 4894–4900. [Google Scholar] [CrossRef]
- Khire, V.S.; Lee, T.Y.; Bowman, C.N. Surface Modification Using Thiol−Acrylate Conjugate Addition Reactions. Macromolecules 2007, 40, 5669–5677. [Google Scholar] [CrossRef]
- Chan, J.W.; Wei, H.; Zhou, H.; Hoyle, C.E. The Effects of Primary Amine Catalyzed Thio-Acrylate Michael Reaction on the Kinetics, Mechanical and Physical Properties of Thio-Acrylate Networks. Eur. Polym. J. 2009, 45, 2717–2725. [Google Scholar] [CrossRef]
- Chan, J.W.; Hoyle, C.E.; Lowe, A.B. Sequential Phosphine-Catalyzed, Nucleophilic Thiol−Ene/Radical-Mediated Thiol−Yne Reactions and the Facile Orthogonal Synthesis of Polyfunctional Materials. J. Am. Chem. Soc. 2009, 131, 5751–5753. [Google Scholar] [CrossRef] [PubMed]
- Kasetaite, S.; De la Flor, S.; Serra, A.; Ostrauskaite, J. Effect of Selected Thiols on Cross-Linking of Acrylated Epoxidized Soybean Oil and Properties of Resulting Polymers. Polymers 2018, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Digby, Z.A.; Flum, J.A.; Chakma, P.; Saul, J.M.; Sparks, J.L.; Konkolewicz, D. Dynamic Thiol–Michael Chemistry for Thermoresponsive Rehealable and Malleable Networks. Macromolecules 2016, 49, 6871–6878. [Google Scholar] [CrossRef]
- Chakma, P.; Rodrigues Possarle, L.H.; Digby, Z.A.; Zhang, B.; Sparks, J.L.; Konkolewicz, D. Dual Stimuli Responsive Self-Healing and Malleable Materials Based on Dynamic Thiol-Michael Chemistry. Polym. Chem. 2017, 8, 6534–6543. [Google Scholar] [CrossRef]
- Li, G.Z.; Randev, R.K.; Soeriyadi, A.H.; Rees, G.; Boyer, C.; Tong, Z.; Davis, T.P.; Becer, C.R.; Haddleton, D.M. Investigation into Thiol-(Meth)Acrylate Michael Addition Reactions Using Amine and Phosphine Catalysts. Polym. Chem. 2010, 1, 1196–1204. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Chatani, S.; Nair, D.P.; Bowman, C.N. Relative Reactivity and Selectivity of Vinyl Sulfones and Acrylates towards the Thiol–Michael Addition Reaction and Polymerization. Polym. Chem. 2013, 4, 1048–1055. [Google Scholar] [CrossRef]
- Chan, J.W.; Hoyle, C.E.; Lowe, A.B.; Bowman, M. Nucleophile-Initiated Thiol-Michael Reactions: Effect of Organocatalyst, Thiol, and Ene. Macromolecules 2010, 43, 6381–6388. [Google Scholar] [CrossRef]
- Kilambi, H.; Stansbury, J.W.; Bowman, C.N. Enhanced Reactivity of Monovinyl Acrylates Characterized by Secondary Functionalities toward Photopolymerization and Michael Addition: Contribution of Intramolecular Effects. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3452–3458. [Google Scholar] [CrossRef]
- Nguyen, L.-T.T.; Gokmen, M.T.; Prez, F.E. Du Kinetic Comparison of 13 Homogeneous Thiol–X Reactions. Polym. Chem. 2013, 4, 5527–5536. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-Ene “Click” Reactions and Recent Applications in Polymer and Materials Synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Rizzi, S.C.; Hubbell, J.A. Recombinant Protein-Co-PEG Networks as Cell-Adhesive and Proteolytically Degradable Hydrogel Matrixes. Part I: Development and Physicochemical Characteristics. Biomacromolecules 2005, 6, 1226–1238. [Google Scholar] [CrossRef]
- Wu, D.; Liu, Y.; He, C.; Chung, T.; Goh, S. Effects of Chemistries of Trifunctional Amines on Mechanisms of Michael Addition Polymerizations with Diacrylates. Macromolecules 2004, 37, 6763–6770. [Google Scholar] [CrossRef]
- Connor, R.; McClellan, W.R. The Michael Condensation. V. The Influence of the Experimental Conditions and the Structure of the Acceptor upon the Condensation. J. Org. Chem. 1939, 3, 570–577. [Google Scholar] [CrossRef]
- Long, K.F.; Wang, H.; Dimos, T.T.; Bowman, C.N. Effects of Thiol Substitution on the Kinetics and Efficiency of Thiol-Michael Reactions and Polymerizations. Macromolecules 2021, 54, 3093–3100. [Google Scholar] [CrossRef]
- Huang, S.; Kim, K.; Musgrave, G.M.; Sharp, M.; Sinha, J.; Stansbury, J.W.; Musgrave, C.B.; Bowman, C.N. Determining Michael Acceptor Reactivity from Kinetic, Mechanistic, and Computational Analysis for the Base-Catalyzed Thiol-Michael Reaction. Polym. Chem. 2021, 12, 3619–3628. [Google Scholar] [CrossRef] [PubMed]
- Long, K.F.; Bongiardina, N.J.; Mayordomo, P.; Olin, M.J.; Ortega, A.D.; Bowman, C.N. Effects of 1°, 2°, and 3° Thiols on Thiol–Ene Reactions: Polymerization Kinetics and Mechanical Behavior. Macromolecules 2020, 53, 5805–5815. [Google Scholar] [CrossRef]
- Gennari, A.; Wedgwood, J.; Lallana, E.; Francini, N.; Tirelli, N. Thiol-Based Michael-Type Addition. A Systematic Evaluation of Its Controlling Factors. Tetrahedron 2020, 76, 131637. [Google Scholar] [CrossRef]
- Kade, M.J.; Burke, D.J.; Hawker, C.J. The Power of Thiol-Ene Chemistry. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 743–750. [Google Scholar] [CrossRef]
- Koo, S.P.S.; Stamenović, M.M.; Prasath, R.A.; Inglis, A.J.; Du Prez, F.E.; Barner-Kowollik, C.; Van Camp, W.; Junker, T. Limitations of Radical Thiol-Ene Reactions for Polymer–Polymer Conjugation. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Derboven, P.; D’hooge, D.R.; Stamenovic, M.M.; Espeel, P.; Marin, G.B.; Du Prez, F.E.; Reyniers, M.-F. Kinetic Modeling of Radical Thiol–Ene Chemistry for Macromolecular Design: Importance of Side Reactions and Diffusional Limitations. Macromolecules 2013, 46, 1732–1742. [Google Scholar] [CrossRef]
- Wadhwa, P.; Kharbanda, A.; Sharma, A. Thia-Michael Addition: An Emerging Strategy in Organic Synthesis. Asian J. Org. Chem. 2018, 7, 634–661. [Google Scholar] [CrossRef]
- Nicolet, B.H. The Mechanism of Sulfur Lability in Cysteine and Its Derivatives. I. Some Thio Ethers Readily Split by Alkali. J. Am. Chem. Soc. 1931, 53, 3066–3072. [Google Scholar] [CrossRef]
- Allen, C.F.H.; Humphlett, W.J. The Thermal Reversibility of the Michael Reaction: V. the Effect of the Structure of Certain Thiol Adducts on Cleavage. Can. J. Chem. 1966, 44, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.F.H.; Fournier, J.O.; Humphlett, W.J. The Thermal Reversibility of the Michael Reaction: IV. Thiol Adducts. Can. J. Chem. 1964, 42, 2616–2620. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, R.B.; Lough, C.E.; Currie, D.J.; Holmes, H.L. Equilibrium Reactions of n -Butanethiol with Some Conjugated Heteroenoid Compounds. Can. J. Chem. 1968, 46, 775–781. [Google Scholar] [CrossRef]
- Fishbein, J.C.; Jencks, W.P. Elimination Reactions of β-Cyano Thioethers: Evidence for a Carbanion Intermediate and a Change in Rate-Limiting Step. J. Am. Chem. Soc. 1988, 110, 5075–5086. [Google Scholar] [CrossRef]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible Targeting of Noncatalytic Cysteines with Chemically Tuned Electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Miller, R.M.; Tian, B.; Mullins, R.D.; Jacobson, M.P.; Taunton, J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 2014, 136, 12624–12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenske, E.H.; Petter, R.C.; Houk, K.N. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem. 2016, 81, 11726–11733. [Google Scholar] [CrossRef]
- Herbert, K.M.; Getty, P.T.; Dolinski, N.D.; Hertzog, J.E.; de Jong, D.; Lettow, J.H.; Romulus, J.; Onorato, J.W.; Foster, E.M.; Rowan, S.J. Dynamic Reaction-Induced Phase Separation in Tunable, Adaptive Covalent Networks. Chem. Sci. 2020, 11, 5028–5036. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Lyß, G.; Pahl, H.L.; Merfort, I. Helenanolide Type Sesquiterpene Lactones. Part 5: The Role of Glutathione Addition under Physiological Conditions. Bioorg. Med. Chem. 1999, 7, 2849–2855. [Google Scholar] [CrossRef]
- Shi, B.; Greaney, M.F. Reversible Michael Addition of Thiols as a New Tool for Dynamic Combinatorial Chemistry. Chem. Commun. 2005, 7, 886. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, X.; Xie, Y.; Greenberg, M.M.; Xi, Z.; Zhou, C. Thiol Specific and Tracelessly Removable Bioconjugation via Michael Addition to 5-Methylene Pyrrolones. J. Am. Chem. Soc. 2017, 139, 6146–6151. [Google Scholar] [CrossRef] [Green Version]
- Chakma, P.; Digby, Z.A.; Via, J.; Shulman, M.P.; Sparks, J.L.; Konkolewicz, D. Tuning Thermoresponsive Network Materials through Macromolecular Architecture and Dynamic Thiol-Michael Chemistry. Polym. Chem. 2018, 9, 4744–4756. [Google Scholar] [CrossRef]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Design of Thiol- and Light-Sensitive Degradable Hydrogels Using Michael-Type Addition Reactions. Polym. Chem. 2015, 6, 5565–5574. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.D.; Kiick, K.L. Reversible Maleimide–Thiol Adducts Yield Glutathione-Sensitive Poly(Ethylene Glycol)–Heparin Hydrogels. Polym. Chem. 2012, 4, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Liu, X.; Zhang, H.; Zhao, N.; Zhang, K. Thermoresponsive Self-Healable and Recyclable Polymer Networks Based on a Dynamic Quinone Methide-Thiol Chemistry. Polym. Chem. 2020, 11, 6157–6162. [Google Scholar] [CrossRef]
- Fitzsimons, T.M.; Oentoro, F.; Shanbhag, T.V.; Anslyn, E.V.; Rosales, A.M. Preferential Control of Forward Reaction Kinetics in Hydrogels Crosslinked with Reversible Conjugate Additions. Macromolecules 2020, 53, 3738–3746. [Google Scholar] [CrossRef]
- Chakma, P.; Wanasinghe, S.V.; Morley, C.N.; Francesconi, S.C.; Saito, K.; Sparks, J.L.; Konkolewicz, D. Heat- and Light-Responsive Materials Through Pairing Dynamic Thiol–Michael and Coumarin Chemistry. Macromol. Rapid Commun. 2021, 42, 2100070. [Google Scholar] [CrossRef] [PubMed]
- Daymon, S.P.; Miller, K.M. Probing the Dynamic and Rehealing Behavior of Crosslinked Polyester Networks Containing Thermoreversible Thiol-Michael Bonds. Polymer 2018, 145, 286–293. [Google Scholar] [CrossRef]
- Kuhl, N.; Geitner, R.; Bose, R.K.; Bode, S.; Dietzek, B.; Schmitt, M.; Popp, J.; Garcia, S.J.; van der Zwaag, S.; Schubert, U.S.; et al. Self-Healing Polymer Networks Based on Reversible Michael Addition Reactions. Macromol. Chem. Phys. 2016, 217, 2541–2550. [Google Scholar] [CrossRef]
- Abdallh, M.; Yoshikawa, C.; Hearn, M.T.W.; Simon, G.P.; Saito, K. Photoreversible Smart Polymers Based on 2π + 2π Cycloaddition Reactions: Nanofilms to Self-Healing Films. Macromolecules 2019, 52, 2446–2455. [Google Scholar] [CrossRef]
- Herbert, K.M.; Dolinski, N.D.; Boynton, N.R.; Murphy, J.G.; Lindberg, C.A.; Sibener, S.J.; Rowan, S.J. Controlling the Morphology of Dynamic Thia-Michael Networks to Target Pressure-Sensitive and Hot Melt Adhesives. ACS Appl. Mater. Interfaces 2021, 13, 27471–27480. [Google Scholar] [CrossRef]
- Schenk, V.; Labastie, K.; Destarac, M.; Olivier, P.; Guerre, M. Vitrimer Composites: Current State and Future Challenges. Mater. Adv. 2022. [Google Scholar] [CrossRef]
- Taplan, C.; Guerre, M.; Du Prez, F.E. Covalent Adaptable Networks Using β-Amino Esters as Thermally Reversible Building Blocks. J. Am. Chem. Soc. 2021, 143, 9140–9150. [Google Scholar] [CrossRef]
- Berne, D.; Coste, G.; Morales, R.; Boursier, M.; Pinaud, J.; Ladmiral, V.; Caillol, S. Taking Advantage of β-Hydroxy Amine Enhanced Reactivity and Functionality for the Synthesis of Dual Covalent Adaptable Networks. Polym. Chem. 2022, 8, 5255–5446. [Google Scholar] [CrossRef]
- Holloway, J.O.; Taplan, C.; Du Prez, F.E. Combining Vinylogous Urethane and β-Amino Ester Chemistry for Dynamic Material Design. Polym. Chem. 2022, 13, 2008–2018. [Google Scholar] [CrossRef]
- Berne, D.; Quienne, B.; Caillol, S.; Leclerc, E.; Ladmiral, V. Biobased Catalyst-Free Covalent Adaptable Networks Based on CF3-Activated Synergistic Aza-Michael Exchange and Transesterification. J. Mater. Chem. A 2022. [Google Scholar] [CrossRef]
- González, G.; Fernández-Francos, X.; Serra, À.; Sangermano, M.; Ramis, X. Environmentally-Friendly Processing of Thermosets by Two-Stage Sequential Aza-Michael Addition and Free-Radical Polymerization of Amine–Acrylate Mixtures. Polym. Chem. 2015, 6, 6987–6997. [Google Scholar] [CrossRef] [Green Version]
- Paramarta, A.; Webster, D.C. The Exploration of Michael-Addition Reaction Chemistry to Create High Performance, Ambient Cure Thermoset Coatings Based on Soybean Oil. Prog. Org. Coat. 2017, 108, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Ecochard, Y.; Auvergne, R.; Boutevin, B.; Caillol, S. Linseed Oil-Based Thermosets by Aza-Michael Polymerization. Eur. J. Lipid Sci. Technol. 2020, 122, 1900145. [Google Scholar] [CrossRef]
- Peyrton, J.; Avérous, L. Aza-Michael Reaction as a Greener, Safer, and More Sustainable Approach to Biobased Polyurethane Thermosets. ACS Sustain. Chem. Eng. 2021, 9, 4872–4884. [Google Scholar] [CrossRef]
- Cornille, A.; Ecochard, Y.; Blain, M.; Boutevin, B.; Caillol, S. Synthesis of Hybrid Polyhydroxyurethanes by Michael Addition. Eur. Polym. J. 2017, 96, 370–382. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berne, D.; Ladmiral, V.; Leclerc, E.; Caillol, S. Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks. Polymers 2022, 14, 4457. https://doi.org/10.3390/polym14204457
Berne D, Ladmiral V, Leclerc E, Caillol S. Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks. Polymers. 2022; 14(20):4457. https://doi.org/10.3390/polym14204457
Chicago/Turabian StyleBerne, Dimitri, Vincent Ladmiral, Eric Leclerc, and Sylvain Caillol. 2022. "Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks" Polymers 14, no. 20: 4457. https://doi.org/10.3390/polym14204457
APA StyleBerne, D., Ladmiral, V., Leclerc, E., & Caillol, S. (2022). Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks. Polymers, 14(20), 4457. https://doi.org/10.3390/polym14204457