
Blending PLA with Polyesters Based on 2,5-Furan Dicarboxylic Acid: Evaluation of Physicochemical and Nanomechanical Properties

 , ,
, ,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
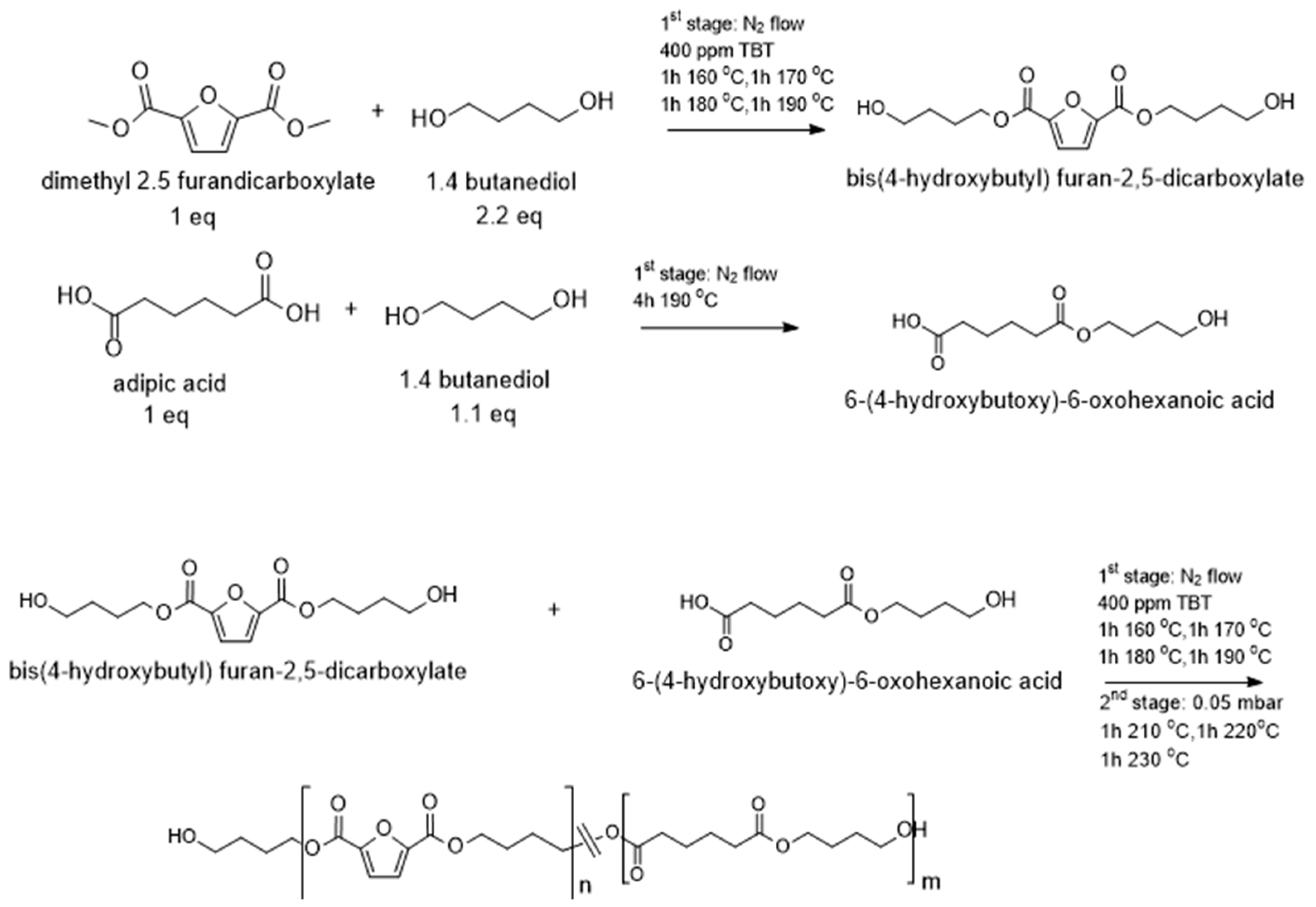
2.2. Synthesis of the Polyesters
2.3. Blend Preparation
2.4. Characterization
3. Results
3.1. Synthesis of the FDCA-Based Polyesters
3.2. Characterization of the FDCA-Based Polyesters
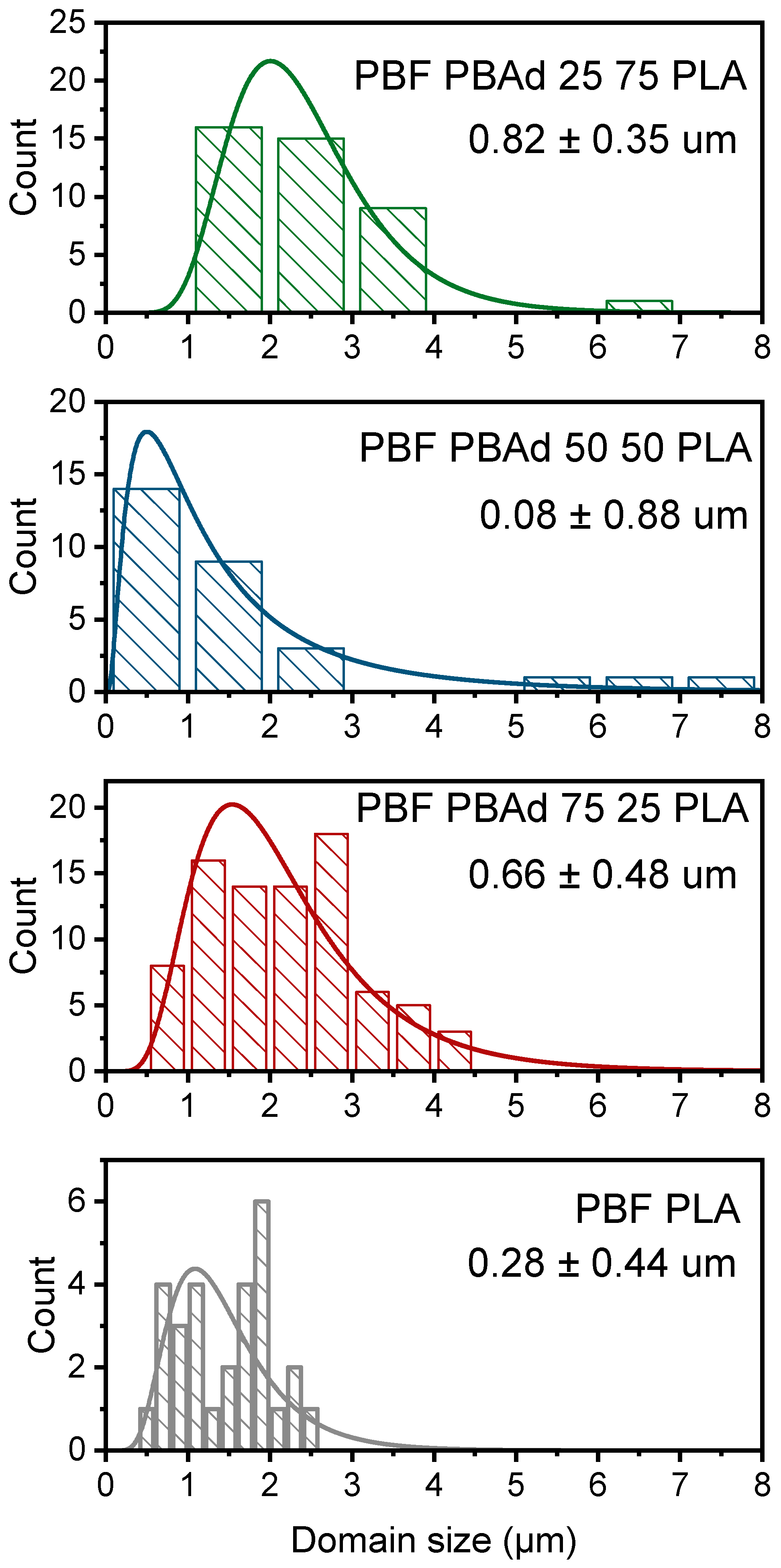
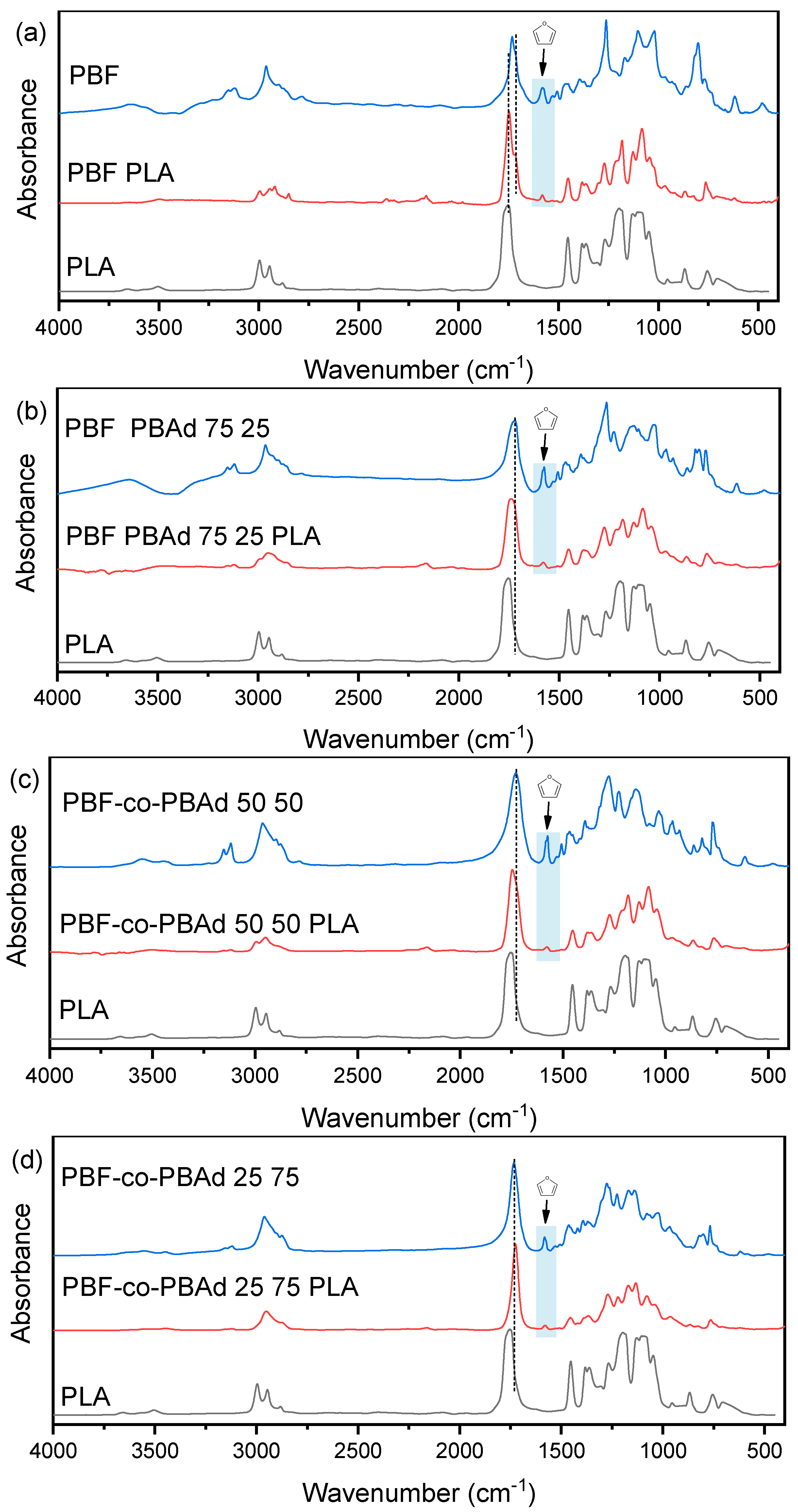
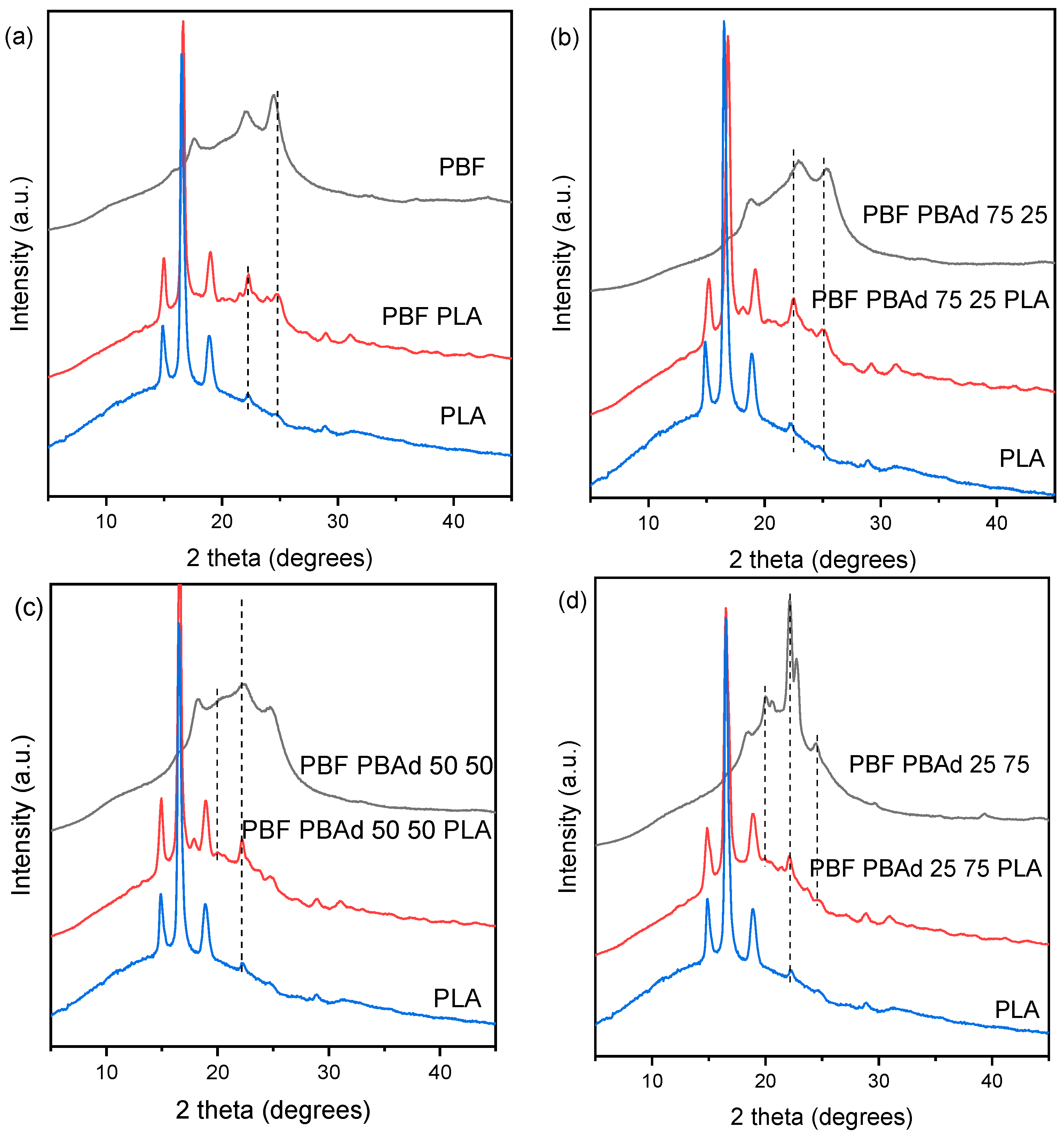
3.3. Microstructural Features and Spectroscopic Analysis of the Blends
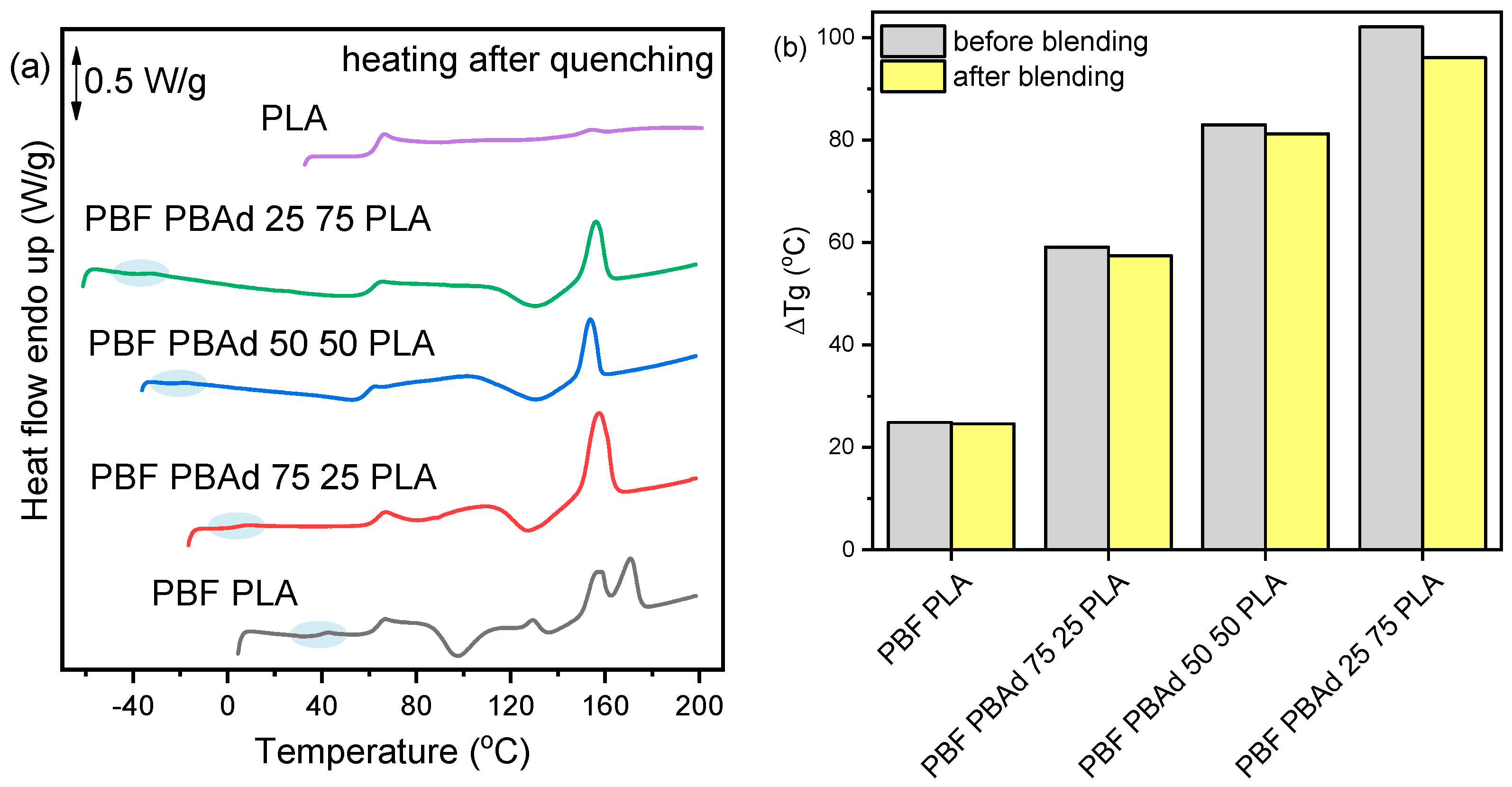
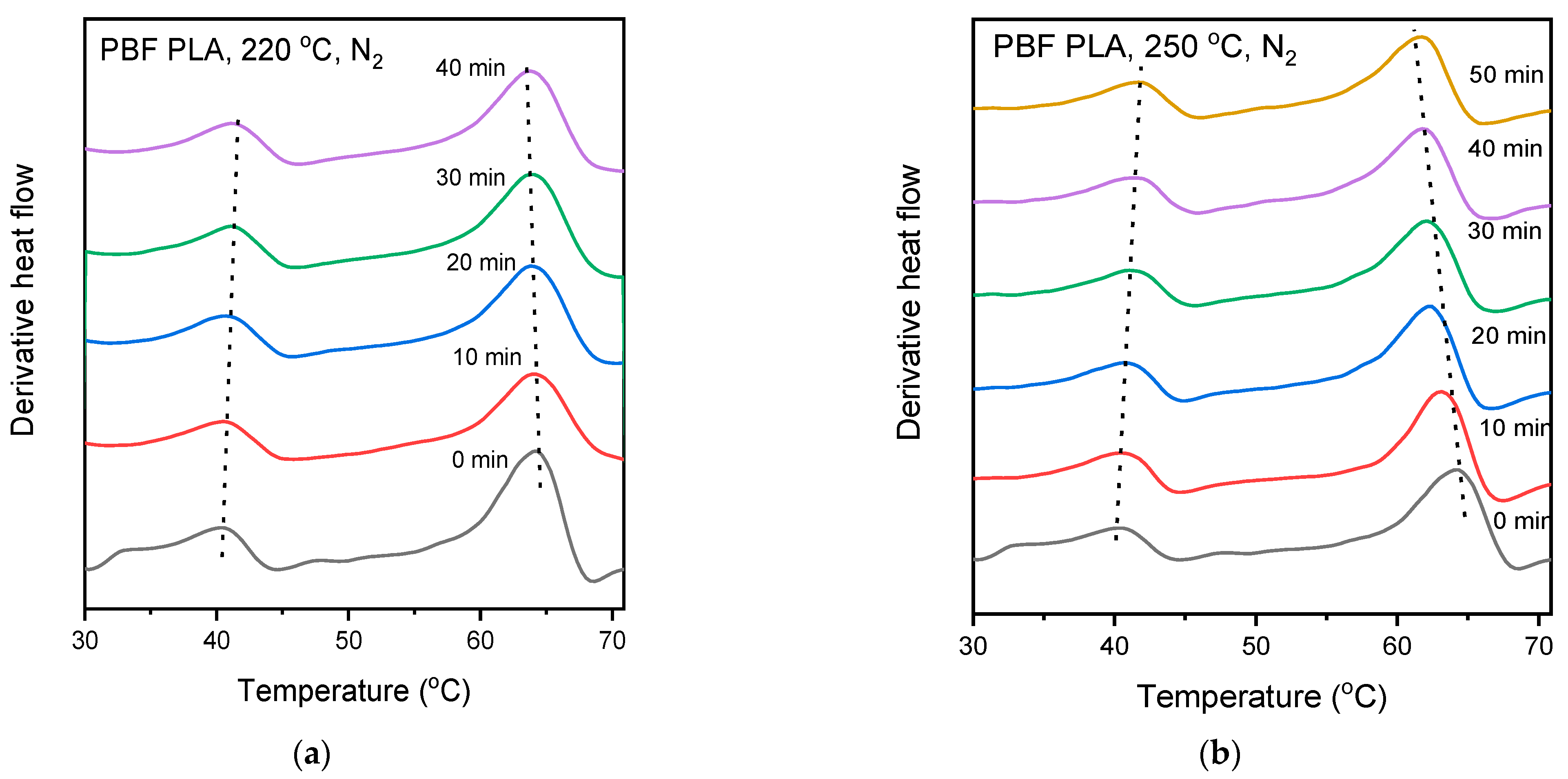
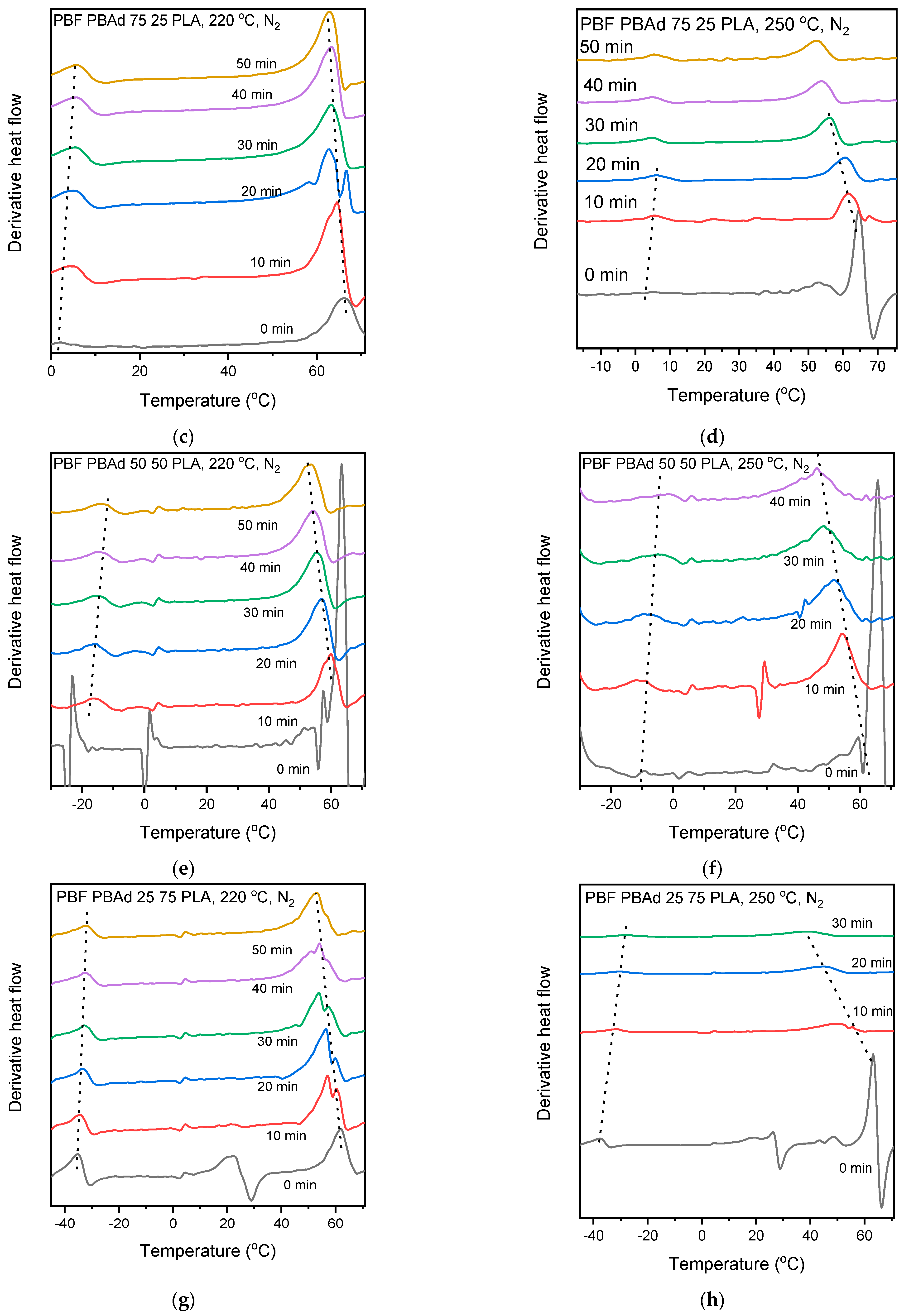
3.4. Thermal Properties and Crystallization of the Blends
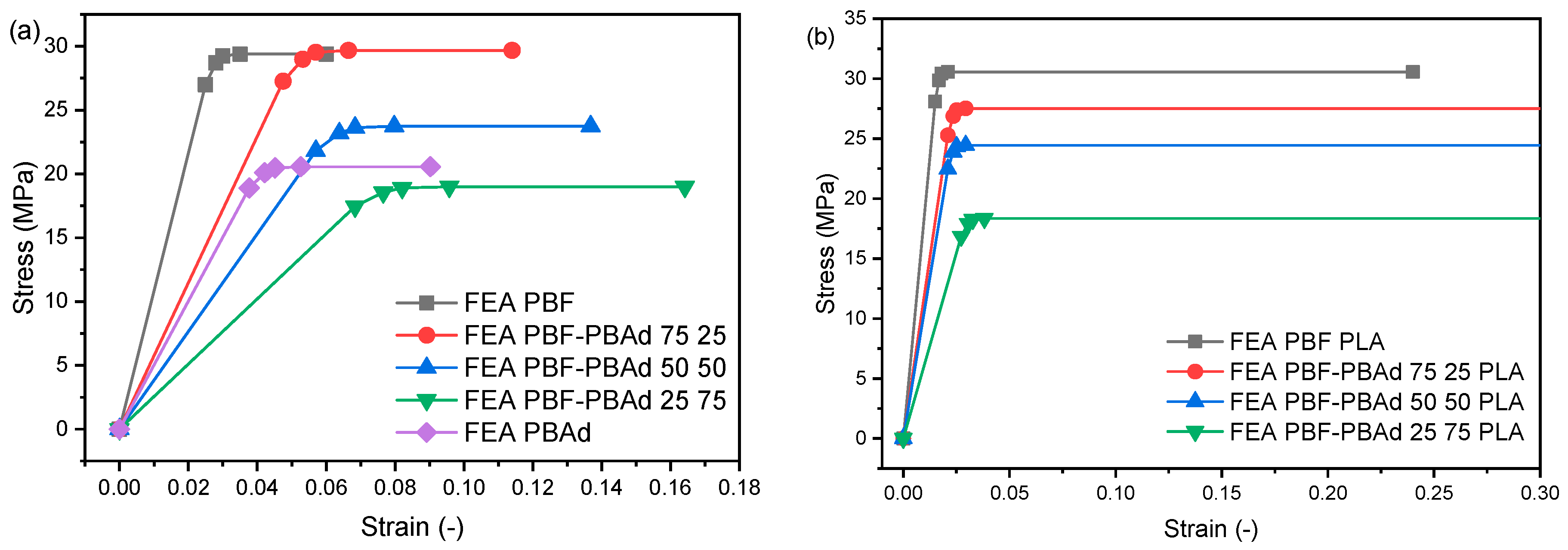
3.5. Nanomechanical Properties
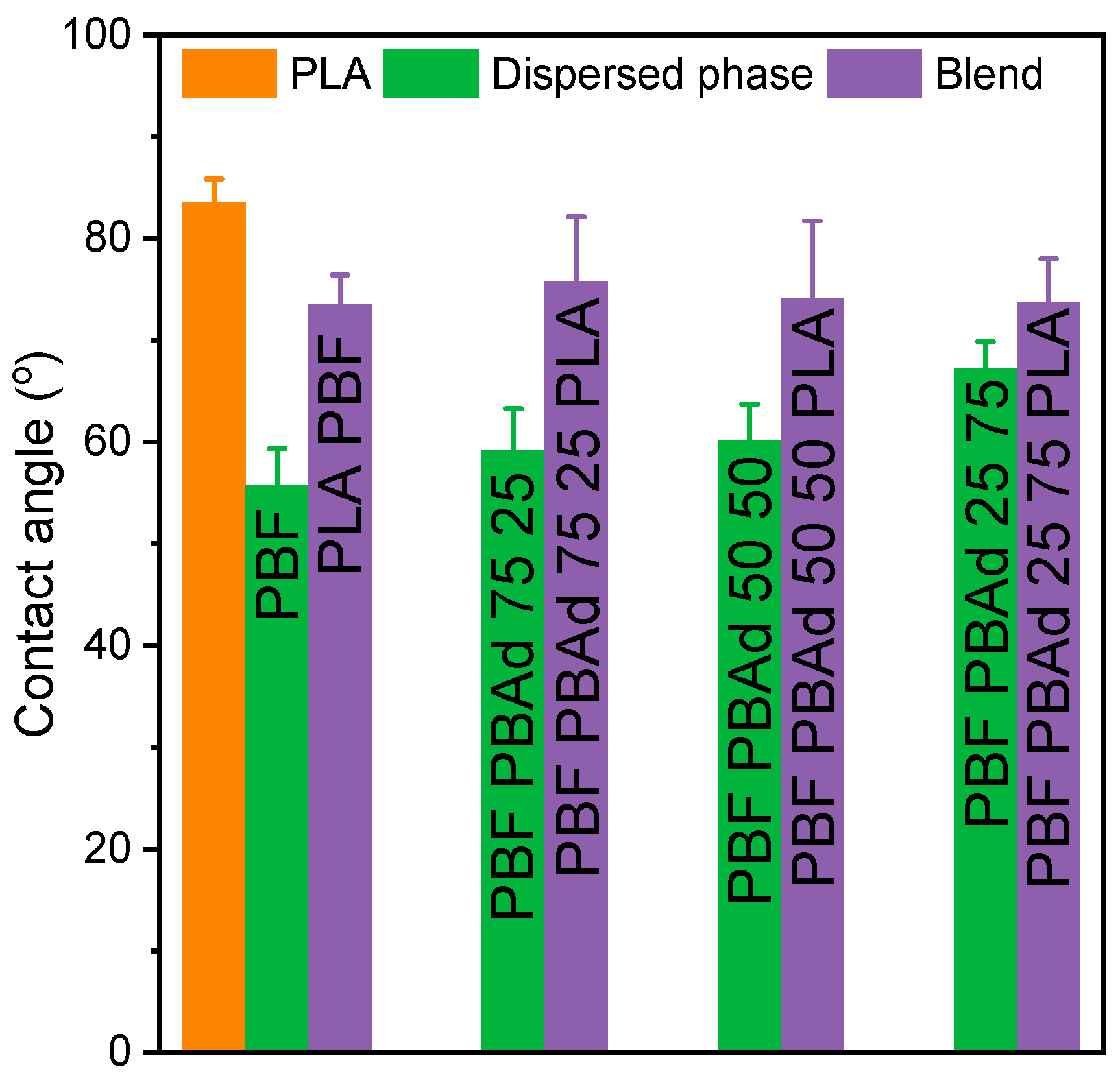
3.6. Wettability and Soil Burial Test
3.7. Reactive Blending
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamad, K.; Kaseem, M.; Ayyoob, M.; Joo, J.; Deri, F. Polylactic acid blends: The future of green, light and tough. Prog. Polym. Sci. 2018, 85, 83–127. [Google Scholar] [CrossRef]
- Papadopoulos, L.; Klonos, P.; Terzopoulou, Z.; Psochia, E.; Sanusi, O.M.; Hocine, N.A.; Benelfellah, A.; Giliopoulos, D.; Triantafyllidis, K.; Kyritsis, A.; et al. Comparative study of crystallization, semicrystalline morphology, and molecular mobility in nanocomposites based on polylactide and various inclusions at low filler loadings. Polymer 2021, 217, 123457. [Google Scholar] [CrossRef]
- Sousa, A.F.; Patrício, R.; Terzopoulou, Z.; Bikiaris, D.N.; Stern, T.; Wenger, J.; Loos, K.; Lotti, N.; Siracusa, V.; Szymczyk, A.; et al. Recommendations for replacing PET on packaging, fiber, and film materials with biobased counterparts. Green Chem. 2021, 23, 8795–8820. [Google Scholar] [CrossRef]
- Fei, X.; Wang, J.; Zhang, X.; Jia, Z.; Jiang, Y.; Liu, X. Recent Progress on Bio-Based Polyesters Derived from 2,5-Furandicarbonxylic Acid (FDCA). Polymers 2022, 14, 625. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Terzopoulou, Z.; Bikiaris, D.N. Production of bio-based 2,5-furan dicarboxylate polyesters: Recent progress and critical aspects in their synthesis and thermal properties. Eur. Polym. J. 2016, 83, 202–229. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Papadopoulos, L.; Zamboulis, A.; Papageorgiou, D.G.; Papageorgiou, G.Z.; Bikiaris, D.N. Tuning the properties of furandicarboxylic acid-based polyesters with copolymerization: A review. Polymers 2020, 12, 1209. [Google Scholar] [CrossRef]
- Loos, K.; Zhang, R.; Pereira, I.; Agostinho, B.; Hu, H.; Maniar, D.; Sbirrazzuoli, N.; Silvestre, A.J.D.; Guigo, N.; Sousa, A.F. A Perspective on PEF Synthesis, Properties, and End-Life. Front. Chem. 2020, 8, 585. [Google Scholar] [CrossRef]
- Papadopoulos, L.; Magaziotis, A.; Nerantzaki, M.; Terzopoulou, Z.; Papageorgiou, G.Z.; Bikiaris, D.N. Synthesis and characterization of novel poly(ethylene furanoate-co-adipate) random copolyesters with enhanced biodegradability. Polym. Degrad. Stab. 2018, 156, 32–42. [Google Scholar] [CrossRef]
- Pandey, S.; Dumont, M.J.; Orsat, V.; Rodrigue, D. Biobased 2,5-furandicarboxylic acid (FDCA) and its emerging copolyesters’ properties for packaging applications. Eur. Polym. J. 2021, 160, 110778. [Google Scholar] [CrossRef]
- Wu, B.; Xu, Y.; Bu, Z.; Wu, L.; Li, B.G.; Dubois, P. Biobased poly(butylene 2,5-furandicarboxylate) and poly(butylene adipate-co-butylene 2,5-furandicarboxylate)s: From synthesis using highly purified 2,5-furandicarboxylic acid to thermo-mechanical properties. Polymer 2014, 55, 3648–3655. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, X.; Yang, B.; Xu, Y.; Zhang, W.; Zhang, Y.; Ji, J. Synthesis, physical properties and enzymatic degradation of bio-based poly(butylene adipate-co-butylene furandicarboxylate) copolyesters. Polym. Degrad. Stab. 2013, 98, 2177–2183. [Google Scholar] [CrossRef]
- Little, A.; Pellis, A.; Comerford, J.W.; Naranjo-Valles, E.; Hafezi, N.; Mascal, M.; Farmer, T.J. Effects of Methyl Branching on the Properties and Performance of Furandioate-Adipate Copolyesters of Bio-Based Secondary Diols. ACS Sustain. Chem. Eng. 2020, 8, 14471–14483. [Google Scholar] [CrossRef]
- Peng, S.; Wu, B.S.; Wu, L.; Li, B.G.; Dubois, P. Hydrolytic degradation of biobased poly(butylene succinate-co-furandicarboxylate) and poly(butylene adipate-co-furandicarboxylate) copolyesters under mild conditions. J. Appl. Polym. Sci. 2017, 134, 44674. [Google Scholar] [CrossRef]
- Kim, H.; Kim, T.; Choi, S.; Jeon, H.; Oh, D.X.; Park, J.; Eom, Y.; Hwang, S.Y.; Koo, J.M. Remarkable elasticity and enzymatic degradation of bio-based poly(butylene adipate-: Co-furanoate): Replacing terephthalate. Green Chem. 2020, 22, 7778–7787. [Google Scholar] [CrossRef]
- Fredi, G.; Rigotti, D.; Bikiaris, D.N.; Dorigato, A. Tuning thermo-mechanical properties of poly(lactic acid) films through blending with bioderived poly(alkylene furanoate)s with different alkyl chain length for sustainable packaging. Polymer 2021, 218, 123527. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Kantoutsis, G.; Bikiaris, D.N.; Achilias, D.S.; Kapnisti, M.; Papageorgiou, G.Z. Biobased engineering thermoplastics: Poly(butylene 2,5-furandicarboxylate) blends. Polymers 2019, 11, 937. [Google Scholar] [CrossRef] [Green Version]
- Fredi, G.; Karimi Jafari, M.; Dorigato, A.; Bikiaris, D.N.; Pegoretti, A. Improving the Thermomechanical Properties of Poly(lactic acid) via Reduced Graphene Oxide and Bioderived Poly(decamethylene 2,5-furandicarboxylate). Materials 2022, 15, 1316. [Google Scholar] [CrossRef]
- Perin, D.; Rigotti, D.; Fredi, G.; Papageorgiou, G.Z.; Bikiaris, D.N.; Dorigato, A. Innovative Bio-based Poly(Lactic Acid)/Poly(Alkylene Furanoate)s Fiber Blends for Sustainable Textile Applications. J. Polym. Environ. 2021, 29, 3948–3963. [Google Scholar] [CrossRef]
- Rigotti, D.; Soccio, M.; Dorigato, A.; Gazzano, M.; Siracusa, V.; Fredi, G.; Lotti, N. Novel Biobased Polylactic Acid/Poly(pentamethylene 2,5-furanoate) Blends for Sustainable Food Packaging. ACS Sustain. Chem. Eng. 2021, 9, 13742–13750. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Smyrnioti, D.; Nikolaidis, G.N.; Tsitsimaka, I.; Christodoulou, E.; Bikiaris, D.N.; Charitopoulou, M.A.; Achilias, D.S.; Kapnisti, M.; Papageorgiou, G.Z. Sustainable plastics from biomass: Blends of polyesters based on 2,5-furandicarboxylic acid. Polymers 2020, 12, 225. [Google Scholar] [CrossRef]
- Perin, D.; Fredi, G.; Rigotti, D.; Soccio, M.; Lotti, N.; Dorigato, A. Sustainable textile fibers of bioderived polylactide/poly(pentamethylene 2,5-furanoate) blends. J. Appl. Polym. Sci. 2022, 139, 51740. [Google Scholar] [CrossRef]
- Long, Y.; Zhang, R.; Huang, J.; Wang, J.; Jiang, Y.; Hu, G.H.; Yang, J.; Zhu, J. Tensile Property Balanced and Gas Barrier Improved Poly(lactic acid) by Blending with Biobased Poly(butylene 2,5-furan dicarboxylate). ACS Sustain. Chem. Eng. 2017, 5, 9244–9253. [Google Scholar] [CrossRef]
- Long, Y.; Zhang, R.; Huang, J.; Wang, J.; Zhang, J.; Rayand, N.; Hu, G.H.; Yang, J.; Zhu, J. Retroreflection in binary bio-based PLA/PBF blends. Polymer 2017, 125, 138–143. [Google Scholar] [CrossRef]
- NatureWorks IngeoTM Biopolymer 3052D Technical Data Sheet. NatureWorks 2018, 001, 1–4. Available online: https://www.natureworksllc.com/~/media/Files/NatureWorks/Technical-Documents/Technical-Data-Sheets/TechnicalDataSheet_3052D_injection-molding_pdf.pdf (accessed on 25 October 2022).
- Oliver, W.C.; Pharr, G.M. An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 1992, 7, 1564–1583. [Google Scholar] [CrossRef]
- Tzetzis, D.; Tsongas, K.; Mansour, G. Determination of the mechanical properties of epoxy silica nanocomposites through FEA-supported evaluation of ball indentation test results. Mater. Res. 2017, 20, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Tsongas, K.; Tzetzis, D.; Karantzalis, A.; Banias, G.; Exarchos, D.; Ahmadkhaniha, D.; Zanella, C.; Matikas, T.; Bochtis, D. Microstructural, surface topology and nanomechanical characterization of electrodeposited Ni-P/SiC nanocomposite coatings. Appl. Sci. 2019, 9, 2901. [Google Scholar] [CrossRef] [Green Version]
- Mansour, G.; Tzetzis, D. Nanomechanical Characterization of Hybrid Multiwall Carbon Nanotube and Fumed Silica Epoxy Nanocomposites. Polym.-Plast. Technol. Eng. 2013, 52, 1054–1062. [Google Scholar] [CrossRef]
- Mansour, G.; Tzetzis, D.; Bouzakis, K.D. A nanomechanical approach on the measurement of the elastic properties of epoxy reinforced carbon nanotube nanocomposites. Tribol. Ind. 2013, 35, 190–199. [Google Scholar]
- Grigora, M.E.; Terzopoulou, Z.; Tsongas, K.; Bikiaris, D.N.; Tzetzis, D. Physicochemical Characterization and Finite Element Analysis-Assisted Mechanical Behavior of Polylactic Acid-Montmorillonite 3D Printed Nanocomposites. Nanomaterials 2022, 12, 2641. [Google Scholar] [CrossRef]
- Mansour, G.; Zoumaki, M.; Tsongas, K.; Tzetzis, D. Microstructural and finite element analysis—Assisted nanomechanical characterization of maize starch nanocomposite films. Mater. Res. 2021, 24, e20200409. [Google Scholar] [CrossRef]
- Peng, S.; Wu, L.; Li, B.G.; Dubois, P. Hydrolytic and compost degradation of biobased PBSF and PBAF copolyesters with 40–60 mol% BF unit. Polym. Degrad. Stab. 2017, 146, 223–228. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Papageorgiou, D.G.; Exarhopoulos, S.; Papageorgiou, M.; Bikiaris, D.N. Evaluation of polyesters from renewable resources as alternatives to the current fossil-based polymers. Phase transitions of poly(butylene 2,5-furan-dicarboxylate). Polymer 2014, 55, 3846–3858. [Google Scholar] [CrossRef]
- Ma, J.; Yu, X.; Xu, J.; Pang, Y. Synthesis and crystallinity of poly(butylene 2,5-furandicarboxylate). Polymer 2012, 53, 4145–4151. [Google Scholar] [CrossRef]
- Papadopoulos, L.; Xanthopoulou, E.; Nikolaidis, G.N.; Zamboulis, A.; Achilias, D.S.; Papageorgiou, G.Z.; Bikiaris, D.N. Towards high molecular weight furan-based polyesters: Solid state polymerization study of bio-based poly(propylene furanoate) and poly(butylene furanoate). Materials 2020, 13, 4880. [Google Scholar] [CrossRef]
- Papadopoulos, L.; Terzopoulou, Z.; Vlachopoulos, A.; Klonos, P.A.; Kyritsis, A.; Tzetzis, D.; Papageorgiou, G.Z.; Bikiaris, D. Synthesis and characterization of novel polymer/clay nanocomposites based on poly (butylene 2,5-furan dicarboxylate). Appl. Clay Sci. 2020, 190, 105588. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, J.; Xie, W.; Chen, P.H.; Gazzano, M.; Scandola, M.; Gross, R.A. Poly(butylene 2,5-furan dicarboxylate), a biobased alternative to PBT: Synthesis, physical properties, and crystal structure. Macromolecules 2013, 46, 796–804. [Google Scholar] [CrossRef]
- Nikolic, M.S.; Djonlagic, J. Synthesis and characterization of biodegradable poly(butylene succinate-co-butylene adipate)s. Polym. Degrad. Stab. 2001, 74, 263–270. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Doi, Y. Temperature-induced polymorphic crystals of poly(butylene adipate). Macromol. Chem. Phys. 2002, 203, 2369–2374. [Google Scholar] [CrossRef]
- Siafaka, P.I.; Barmbalexis, P.; Bikiaris, D.N. Novel electrospun nanofibrous matrices prepared from poly(lactic acid)/poly(butylene adipate) blends for controlled release formulations of an anti-rheumatoid agent. Eur. J. Pharm. Sci. 2016, 88, 12–25. [Google Scholar] [CrossRef]
- Farias da Silva, J.M.; Soares, B.G. Epoxidized cardanol-based prepolymer as promising biobased compatibilizing agent for PLA/PBAT blends. Polym. Test. 2021, 93, 106889. [Google Scholar] [CrossRef]
- Wang, L.; Feng, L.F.; Gu, X.P.; Zhang, C.L. Influences of the Viscosity Ratio and Processing Conditions on the Formation of Highly Oriented Ribbons in Polymer Blends by Tape Extrusion. Ind. Eng. Chem. Res. 2015, 54, 11080–11086. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, Q.; Zhang, Q.; Ye, C.; Zhou, G. A series of furan-aromatic polyesters synthesized via direct esterification method based on renewable resources. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1026–1036. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Kasmi, N.; Bikiaris, D.N.; Papageorgiou, D.G.; Floudas, G.; Papageorgiou, G.Z. Sustainable Polymers from Renewable Resources: Polymer Blends of Furan-Based Polyesters. Macromol. Mater. Eng. 2018, 303, 1800153. [Google Scholar] [CrossRef]
- Xu, J.; Reiter, G.; Alamo, R.G. Concepts of nucleation in polymer crystallization. Crystals 2021, 11, 304. [Google Scholar] [CrossRef]
- Mitra, M.K.; Muthukumar, M. Theory of spinodal decomposition assisted crystallization in binary mixtures. J. Chem. Phys. 2010, 132, 184908. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Disorder-to-order phase transition and multiple melting behavior of poly(L-lactide) investigated by simultaneous measurements of WAXD and DSC. Macromolecules 2008, 41, 1352–1357. [Google Scholar] [CrossRef]
- Su, S.; Duhme, M.; Kopitzky, R. Uncompatibilized pbat/pla blends: Manufacturability, miscibility and properties. Materials 2020, 13, 4897. [Google Scholar] [CrossRef]
- Liu, J.; Mei, Y.; Xia, R. A new wetting mechanism based upon triple contact line pinning. Langmuir 2011, 27, 196–200. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, R.; Sousa, A.; Long, Y.; Ying, W.B.; Wang, J.; Zhu, J. Bio-based poly(butylene 2,5-furandicarboxylate)-b-poly(ethylene glycol) copolymers with adjustable degradation rate and mechanical properties: Synthesis and characterization. Eur. Polym. J. 2018, 106, 42–52. [Google Scholar] [CrossRef]
- Nova Institute Biodegradable Polymers in Various Environments. Available online: https://renewable-carbon.eu/publications/product/biodegradable-polymers-in-various-environments-−-graphic-pdf/ (accessed on 8 June 2022).
- Dong, Y.; Wang, J.; Yang, Y.; Wang, Q.; Zhang, X.; Hu, H.; Zhu, J. Bio-based poly(butylene diglycolate-co-furandicarboxylate) copolyesters with balanced mechanical, barrier and biodegradable properties: A prospective substitute for PBAT. Polym. Degrad. Stab. 2022, 202, 110010. [Google Scholar] [CrossRef]
- Kasmi, N.; Terzopoulou, Z.; Chebbi, Y.; Dieden, R.; Habibi, Y.; Bikiaris, D.N. Tuning thermal properties and biodegradability of poly(isosorbide azelate) by compositional control through copolymerization with 2,5-furandicarboxylic acid. Polym. Degrad. Stab. 2022, 195, 109804. [Google Scholar] [CrossRef]
- Marten, E.; Müller, R.J.; Deckwer, W.D. Studies on the enzymatic hydrolysis of polyesters. II. Aliphatic-aromatic copolyesters. Polym. Degrad. Stab. 2005, 88, 371–381. [Google Scholar] [CrossRef]
- Qi, R.; Jones, D.L.; Liu, Q.; Liu, Q.; Li, Z.; Yan, C. Field test on the biodegradation of poly(butylene adipate-co-terephthalate) based mulch films in soil. Polym. Test. 2021, 93, 107009. [Google Scholar] [CrossRef]
- Han, Y.; Shi, J.; Mao, L.; Wang, Z.; Zhang, L. Improvement of Compatibility and Mechanical Performances of PLA/PBAT Composites with Epoxidized Soybean Oil as Compatibilizer. Ind. Eng. Chem. Res. 2020, 59, 21779–21790. [Google Scholar] [CrossRef]
- Saeidlou, S.; Huneault, M.A.; Li, H.; Park, C.B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [η] (dL/g) | Mn (g/mol) | PDI |
|---|---|---|---|
| PBAd oligo | 0.26 | 3600 | 2.18 |
| PBAd | 0.47 | 15,400 | 2.28 |
| PBF oligo | 0.06 | N.D. | N.D. |
| PBF | 0.6 | N.D. | N.D. |
| PBF-PBAd 75 25 | 0.54 | 23,600 | 2.59 |
| PBF-PBAd 50 50 | 0.41 | 9900 | 2.25 |
| PBF-PBAd 25 75 | 0.63 | 15,790 | 2.25 |
| PBF-PBAd Feed Ratio (mol%) | Ratio Calculated with NMR (mol%) | LBF | LBAd | R |
|---|---|---|---|---|
| PBF-PBAd 75 25 | 74–26 | 2.0 | 1.1 | 1.41 |
| PBF-PBAd 50 50 | 60–40 | 1.7 | 1.4 | 1.31 |
| PBF-PBAd 25 75 | 31–69 | 1.4 | 3.7 | 1.00 |
| Property | Elastic Modulus | FEA Elastic Modulus | FEA Ultimate Stress | Nanoindentation Hardness |
|---|---|---|---|---|
| Units | MPa | MPa | MPa | MPa |
| PLA | 3572 ± 260 | 3600 | 60 | 142 ± 7.4 |
| PBF | 774 ± 43 | 1080 | N.D. | 39.2 ± 3.1 |
| PBF PLA | 2260 ± 107 | 1873 | 30.569 | 125.5 ± 11.4 |
| PBF-PBAd 75 25 | 217.7 ± 4.51 | 574 | 29.679 | 14.5 ± 0.7 |
| PBF-PBAd 75 25 PLA | 1262 ± 38 | 1204 | 27.512 | 72.9 ± 0.4 |
| PBF-PBAd 50 50 | 35 ± 1 | 383 | 23.743 | 2.6 ± 0.08 |
| PBF-PBAd 50 50 PLA | 1004 ± 16.4 | 1070 | 24.455 | 66.7 ± 2.8 |
| PBF-PBAd 25 75 | 31 ± 0.43 | 255 | 18.994 | 1.9 ± 0.08 |
| PBF-PBAd 25 75 PLA | 561 ± 19.3 | 617 | 18.342 | 37.7 ± 1.5 |
| PBAd | 506.03 ± 57 | 502 | 20.570 | 29.72 ± 5.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terzopoulou, Z.; Zamboulis, A.; Papadopoulos, L.; Grigora, M.-E.; Tsongas, K.; Tzetzis, D.; Bikiaris, D.N.; Papageorgiou, G.Z. Blending PLA with Polyesters Based on 2,5-Furan Dicarboxylic Acid: Evaluation of Physicochemical and Nanomechanical Properties. Polymers 2022, 14, 4725. https://doi.org/10.3390/polym14214725
Terzopoulou Z, Zamboulis A, Papadopoulos L, Grigora M-E, Tsongas K, Tzetzis D, Bikiaris DN, Papageorgiou GZ. Blending PLA with Polyesters Based on 2,5-Furan Dicarboxylic Acid: Evaluation of Physicochemical and Nanomechanical Properties. Polymers. 2022; 14(21):4725. https://doi.org/10.3390/polym14214725
Chicago/Turabian StyleTerzopoulou, Zoi, Alexandra Zamboulis, Lazaros Papadopoulos, Maria-Eirini Grigora, Konstantinos Tsongas, Dimitrios Tzetzis, Dimitrios N. Bikiaris, and George Z. Papageorgiou. 2022. "Blending PLA with Polyesters Based on 2,5-Furan Dicarboxylic Acid: Evaluation of Physicochemical and Nanomechanical Properties" Polymers 14, no. 21: 4725. https://doi.org/10.3390/polym14214725