

Layer-by-Layer Surface Modification of Alendronate-Loaded Polyester Microparticles—Enabling Protein Immobilization


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Methods
2.2.1. PCL and PLA-co-PCL Synthesis Via Ring-Opening Polymerization
2.2.2. Gel Permeation Chromatography
2.2.3. Proton Nuclear Magnetic Resonance
2.2.4. AA-Loaded Polyester Core Preparation
2.2.5. Drug Loading
2.2.6. Differential Scanning Calorimetry
2.2.7. Lbl Coating of Polyester Cores
2.2.8. Dynamic Light Scattering
2.2.9. Scanning Electron Microscopy
2.2.10. Determination of Surface Carboxylic Group Content Via Conductometric Titration
2.2.11. Protein Surface Immobilization
Lysozyme (Lys) Fluorescent Tagging
Protein Coupling on Surface of Core-Shell Microparticles
Fluorescence Assay
3. Results and Discussion
3.1. Physicochemical Properties of Polyesters
3.2. AA-Loaded Core Formulation
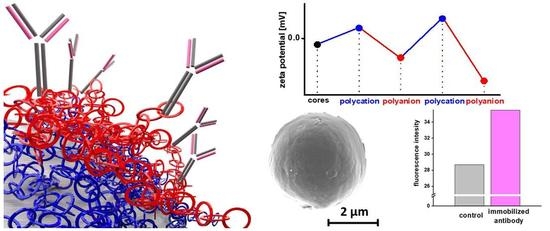
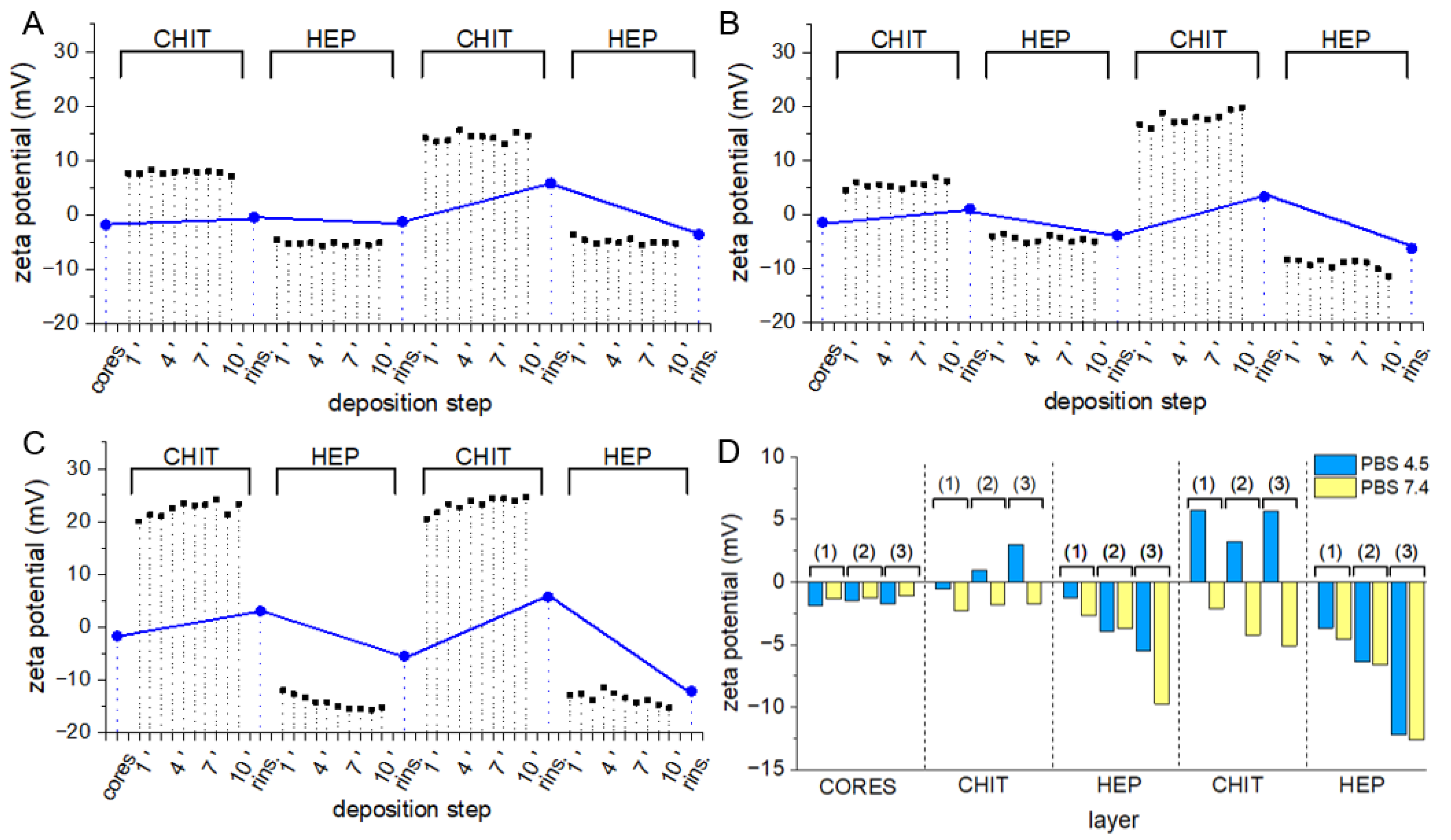
3.3. Layer-by-Layer Coating of AA-Loaded Cores
3.4. Multilayer Crosslinking
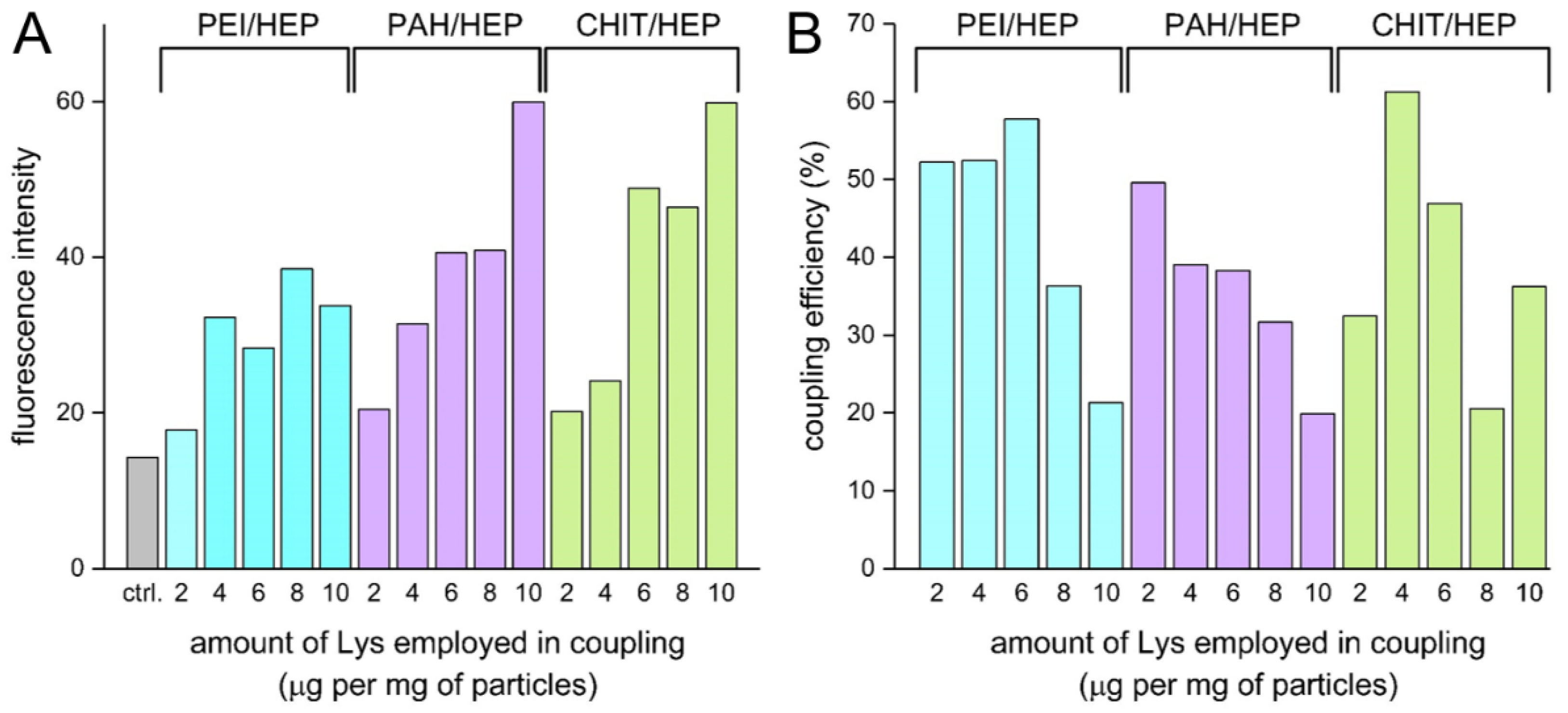
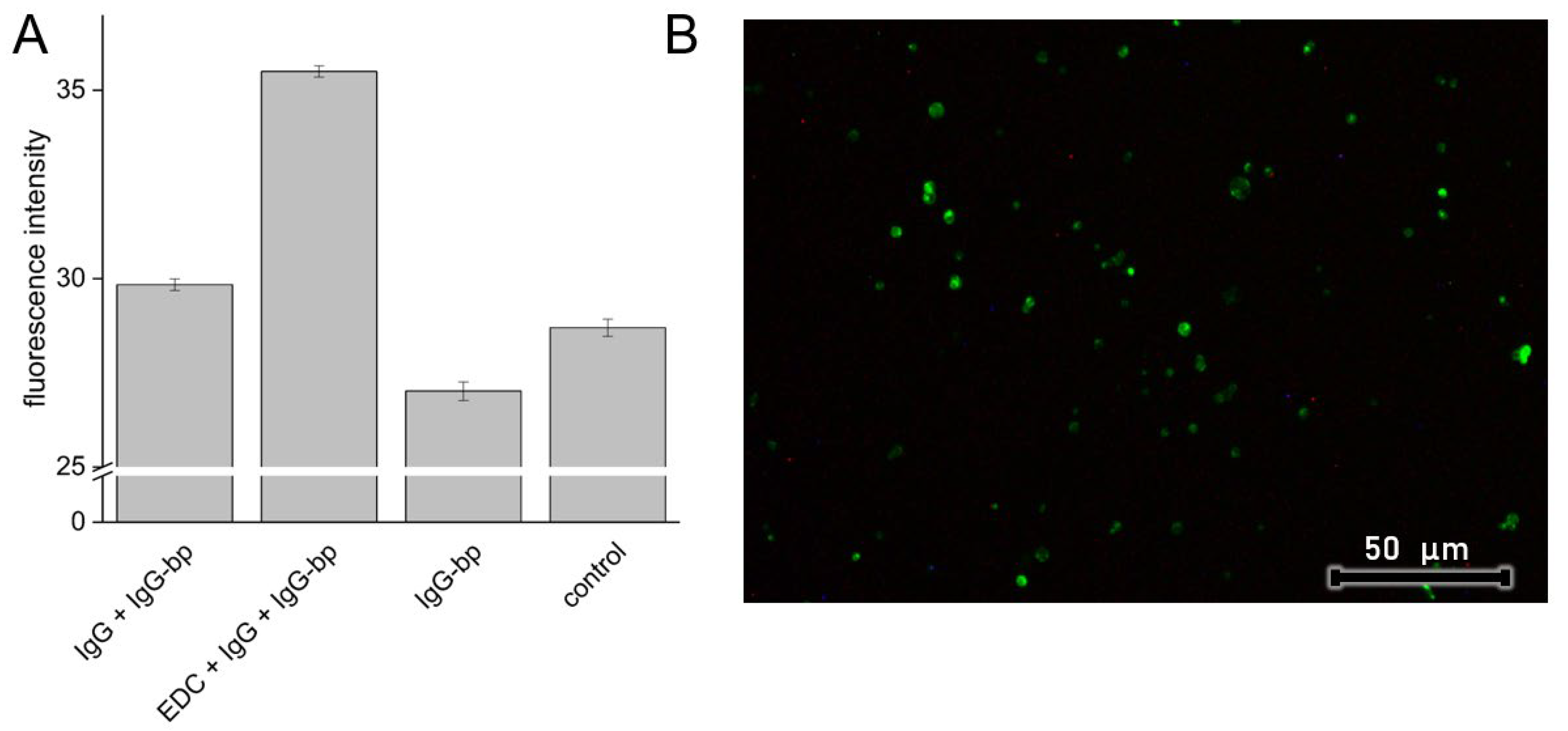
3.5. Model Protein Immobilization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Washington, K.E.; Kularatne, R.N.; Karmegam, V.; Biewer, M.C.; Stefan, M.C. Recent advances in aliphatic polyesters for drug delivery applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1446. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Hu, W.; Liu, Y.; Huang, G.; Sumer, B.D.; Gao, J. Shape-specific polymeric nanomedicine: Emerging opportunities and challenges. Exp. Biol. Med. 2011, 236, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalini, T.; Rossi, F.; Castrovinci, A.; Perale, G. A Perspective on Polylactic Acid-Based Polymers Use for Nanoparticles Synthesis and Applications. Front. Bioeng. Biotechnol. 2019, 7, 259. [Google Scholar] [CrossRef] [PubMed]
- Oyane, A.; Uchida, M.; Choong, C.; Triffitt, J.; Jones, J.; Ito, A. Simple surface modification of poly(ε-caprolactone) for apatite deposition from simulated body fluid. Biomaterials 2005, 26, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Chen, Y.R.; Zhang, J.Y.; Jiang, D.; Yuan, F.Z.; Mao, Z.M.; Yang, F.; Jiang, W.B.; Wang, X.; Yu, J.K. Facile Strategy on Hydrophilic Modification of Poly(ε-caprolactone) Scaffolds for Assisting Tissue-Engineered Meniscus Constructs In Vitro. Front. Pharmacol. 2020, 11, 471. [Google Scholar] [CrossRef]
- Lipovka, A.; Rodriguez, R.; Bolbasov, E.; Maryin, P.; Tverdokhlebov, S.; Sheremet, E. Time-stable wetting effect of plasma-treated biodegradable scaffolds functionalized with graphene oxide. Surf. Coat. Technol. 2020, 388, 125560. [Google Scholar] [CrossRef]
- Korzhikov-Vlakh, V.; Averianov, I.; Sinitsyna, E.; Nashchekina, Y.; Polyakov, D.; Guryanov, I.; Lavrentieva, A.; Raddatz, L.; Korzhikova-Vlakh, E.; Scheper, T.; et al. Novel pathway for efficient covalent modification of polyester materials of different design to prepare biomimetic surfaces. Polymers 2018, 10, 1299. [Google Scholar] [CrossRef] [Green Version]
- Morshed, M.N.; Behary, N.; Bouazizi, N.; Guan, J.; Chen, G.; Nierstrasz, V. Surface modification of polyester fabric using plasma-dendrimer for robust immobilization of glucose oxidase enzyme. Sci. Rep. 2019, 9, 15730. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.; Vikulina, A.S. Layer-by-layer assemblies of biopolymers: Build-up, mechanical stability and molecular dynamics. Polymers 2020, 12, 1949. [Google Scholar] [CrossRef]
- Zhao, S.; Caruso, F.; Dahne, L.; Decher, G.; De Geest, B.G.; Fan, J.; Feliu, N.; Gogotsi, Y.; Hammond, P.T.; Hersam, M.C.; et al. The Future of Layer-by-Layer Assembly: A Tribute to ACS Nano Associate Editor Helmuth Mohwald. ACS Nano 2019, 13, 6151–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumorek, M.; Janoušková, O.; Höcherl, A.; Houska, M.; Mázl-Chánová, E.; Kasoju, N.; Cuchalová, L.; Matějka, R.; Kubies, D. Effect of crosslinking chemistry of albumin/heparin multilayers on FGF-2 adsorption and endothelial cell behavior. Appl. Surf. Sci. 2017, 411, 240–250. [Google Scholar] [CrossRef]
- Guzmán, E.; Ritacco, H.A.; Ortega, F.; Rubio, R.G. Growth of polyelectrolyte layers formed by poly(4-styrenesulfonate sodium salt) and two different polycations: New insights from study of adsorption kinetics. J. Phys. Chem. C 2012, 116, 15474–15483. [Google Scholar] [CrossRef]
- Elbert, D.L.; Herbert, C.B.; Hubbell, J.A. Thin polymer layers formed by polyelectrolyte multilayer techniques on biological surfaces. Langmuir 1999, 15, 5355–5362. [Google Scholar] [CrossRef]
- Sergeeva, Y.N.; Huang, T.; Felix, O.; Jung, L.; Tropel, P.; Viville, S.; Decher, G. What is really driving cell–surface interactions? Layer-by-layer assembled films may help to answer questions concerning cell attachment and response to biomaterials. Biointerphases 2016, 11, 019009. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, T.; Machová, D.; Janoušková, O.; Musiał, W. Microparticles of Lamivudine—Poly-ε-Caprolactone Conjugate for Drug Delivery via Internalization by Macrophages. Molecules 2019, 24, 723. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2017, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Peterson, K.R.; Cottam, M.A.; Kennedy, A.J.; Hasty, A.H. Macrophage-Targeted Therapeutics for Metabolic Disease. Trends Pharmacol. Sci. 2018, 39, 536–546. [Google Scholar] [CrossRef]
- Sabatino, R.; Antonelli, A.; Battistelli, S.; Schwendener, R.; Magnani, M.; Rossi, L. Macrophage depletion by free bisphosphonates and Zoledronate-loaded red blood cells. PLoS ONE 2014, 9, e101260. [Google Scholar] [CrossRef] [PubMed]
- Pignatello, R.; Sarpietro, M.G.; Castelli, F. Synthesis and Biological Evaluation of a New Polymeric Conjugate and Nanocarrier with Osteotropic Properties. J. Funct. Biomater. 2012, 3, 79–99. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Sela, E.; Rosenzweig, O.; Gao, J.; Epstein, H.; Gati, I.; Reich, R.; Danenberg, H.D.; Golomb, G. Alendronate-loaded nanoparticles deplete monocytes and attenuate restenosis. J. Control. Release 2006, 113, 23–30. [Google Scholar] [CrossRef]
- Ducker, R.E.; Montague, M.T.; Leggett, G.J. A comparative investigation of methods for protein immobilization on self-assembled monolayers using glutaraldehyde, carbodiimide, and anhydride reagents. Biointerphases 2008, 3, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Save, M.; Soum, A. Controlled ring-opening polymerization of lactones and lactide initiated by lanthanum isopropoxide, 2a: Mechanistic studies. Macromol. Chem. Phys. 2002, 203, 2591–2603. [Google Scholar] [CrossRef]
- Al Deeb, S.K.; Hamdan, I.I.; Al Najjar, S.M. Spectroscopic and HPLC methods for the determination of alendronate in tablets and urine. Talanta 2004, 64, 695–702. [Google Scholar] [CrossRef]
- Callejas-Fernández, J.; Ramos, J.; Sanz, O.; Forcada, J.; Ortega-Vinuesa, J.L.; Martín-Molina, A.; Rodríguez-Valverde, M.A.; Tirado-Miranda, M.; Schmitt, A.; Sierra-Martin, B.; et al. Experimental Techniques Used for the Characterization of Soft Nanoparticles. In Soft Nanoparticles for Biomedical Applications; Royal Society of Chemistry: London, UK, 2014; Volume 2014-January, ISBN 9781782625216. [Google Scholar]
- Miladi, K.; Sfar, S.; Fessi, H.; Elaissari, A. Encapsulation of alendronate sodium by nanoprecipitation and double emulsion: From preparation to in vitro studies. Ind. Crops Prod. 2015, 72, 24–33. [Google Scholar] [CrossRef]
- Koulouktsi, C.; Nanaki, S.; Barmpalexis, P.; Kostoglou, M.; Bikiaris, D. Preparation and characterization of Alendronate depot microspheres based on novel poly(-ε-caprolactone)/Vitamin E TPGS copolymers. Int. J. Pharm. X 2019, 1, 100014. [Google Scholar] [CrossRef]
- Matrali, S.S.H.; Ghag, A.K. Feedback-controlled release of alendronate from composite microparticles. J. Funct. Biomater. 2020, 11, 46. [Google Scholar] [CrossRef]
- Mohamed, F.; Van Der Walle, C.F. PLGA microcapsules with novel dimpled surfaces for pulmonary delivery of DNA. Int. J. Pharm. 2006, 311, 97–107. [Google Scholar] [CrossRef]
- Li, M.; Rouaud, O.; Poncelet, D. Microencapsulation by solvent evaporation: State of the art for process engineering approaches. Int. J. Pharm. 2008, 363, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.C.; Caldas, M.; Pattekari, P.; Fontes Ribeiro, C.; Ribeiro, A.J.; Lvov, Y.; Veiga, F. Layer-by-Layer Coated Drug-Core Nanoparticles as Versatile Delivery Platforms. In Design and Development of New Nanocarriers; William Andrew Publishing: Norwich, NY, USA, 2018; ISBN 9780128136270. [Google Scholar]
- Trybała, A.; Szyk-Warszyńska, L.; Warszyński, P. The effect of anchoring PEI layer on the build-up of polyelectrolyte multilayer films at homogeneous and heterogeneous surfaces. Colloids Surf. A Physicochem. Eng. Asp. 2009, 343, 127–132. [Google Scholar] [CrossRef]
- Pop-Georgievski, O.; Kubies, D.; Zemek, J.; Neykova, N.; Demianchuk, R.; Chánová, E.M.; Šlouf, M.; Houska, M.; Rypácek, F. Self-assembled anchor layers/polysaccharide coatings on titanium surfaces: A study of functionalization and stability. Beilstein J. Nanotechnol. 2015, 6, 617–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lew, J.H.; Matar, O.K.; Müller, E.A.; Maung, M.T.M.; Luckham, P.F. Adsorption of Hydrolysed Polyacrylamide onto Calcium Carbonate. Polymers 2022, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Gemeinhar, R.A. Understanding the adsorption mechanism of chitosan onto poly (lactide-co-glycolide) particles. Eur. J. Pharm. Biopharm. 2008, 70, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Ji, J.; Barbosa, M.A.; Fonseca, C.; Shen, J. Constructing thromboresistant surface on biomedical stainless steel via layer-by-layer deposition anticoagulant. Biomaterials 2003, 24, 4699–4705. [Google Scholar] [CrossRef]
- Mao, Z.; Ma, L.; Zhou, J.; Gao, C.; Shen, J. Bioactive thin film of acidic fibroblast growth factor fabricated by layer-by-layer assembly. Bioconjug. Chem. 2005, 16, 1316–1322. [Google Scholar] [CrossRef]
- Barany, S. Polymer adsorption and electrokinetic potential of dispersed particles in weak and strong electric fields. Adv. Colloid Interface Sci. 2015, 222, 58–69. [Google Scholar] [CrossRef]
- Fang, F.; Satulovsky, J.; Szleifer, I. Kinetics of protein adsorption and desorption on surfaces with grafted polymers. Biophys. J. 2005, 89, 1516–1533. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Frischknecht, A.L.; Winey, K.I.; Riggleman, R.A. Effect of surface properties and polymer chain length on polymer adsorption in solution. J. Chem. Phys. 2021, 155, 034701. [Google Scholar] [CrossRef]
- Kreke, M.R.; Badami, A.S.; Brady, J.B.; Michael Akers, R.; Goldstein, A.S. Modulation of protein adsorption and cell adhesion by poly(allylamine hydrochloride) heparin films. Biomaterials 2005, 26, 2975–2981. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, P.; Diamanti, E.; Py-Daniel, K.R.; Cáceres-Vélez, P.R.; Martinelli, C.; Politakos, N.; Escobar, A.; Muzi-Falconi, M.; Azevedo, R.; Moya, S.E. Exploring the pH Sensitivity of Poly(allylamine) Phosphate Supramolecular Nanocarriers for Intracellular siRNA Delivery. ACS Appl. Mater. Interfaces 2017, 9, 38242–38254. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Besseling, N.A.M. Formation of polyelectrolyte multilayers: Ionic strengths and growth regimes. Soft Matter 2016, 12, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boddohi, S.; Killingsworth, C.E.; Kipper, M.J. Polyelectrolyte multilayer assembly as a function of pH and ionic strength using the polysaccharides chitosan and heparin. Biomacromolecules 2008, 9, 2021–2028. [Google Scholar] [CrossRef]
- Sun, L.; Xiong, X.; Zou, Q.; Ouyang, P.; Burkhardt, C.; Krastev, R. Design of intelligent chitosan/heparin hollow microcapsules for drug delivery. J. Appl. Polym. Sci. 2017, 134, 44425. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [CrossRef]
- Welch, N.G.; Scoble, J.A.; Muir, B.W.; Pigram, P.J. Orientation and characterization of immobilized antibodies for improved immunoassays (Review). Biointerphases 2017, 12, 02D301. [Google Scholar] [CrossRef] [Green Version]
- Risse, F.; Gedig, E.T.; Gutmann, J.S. Carbodiimide-mediated immobilization of acidic biomolecules on reversed-charge zwitterionic sensor chip surfaces. Anal. Bioanal. Chem. 2018, 410, 4109–4122. [Google Scholar] [CrossRef]
- Hagiya, K.; Miyagawa, A.; Nagatomo, S.; Nakatani, K. Direct Quantification of Proteins Modified on a Polystyrene Microparticle Surface Based on ζ Potential Change. Anal. Chem. 2022, 94, 6304–6310. [Google Scholar] [CrossRef]
- Cortez, C.; Tomaskovic-Crook, E.; Johnston, A.P.R.; Radt, B.; Cody, S.H.; Scott, A.M.; Nice, E.C.; Heath, J.K.; Caruso, F. Targeting and uptake of multilayered particles to colorectal cancer cells. Adv. Mater. 2006, 18, 1998–2003. [Google Scholar] [CrossRef]
- Masereel, B.; Dinguizli, M.; Bouzin, C.; Moniotte, N.; Feron, O.; Gallez, B.; Vander Borght, T.; Michiels, C.; Lucas, S. Antibody immobilization on gold nanoparticles coated layer-by-layer with polyelectrolytes. J. Nanoparticle Res. 2011, 13, 1573–1580. [Google Scholar] [CrossRef]
- Alkekhia, D.; Hammond, P.T.; Shukla, A. Layer-by-Layer Biomaterials for Drug Delivery. Annu. Rev. Biomed. Eng. 2020, 22, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Gao, W.; Li, Z.; Zhang, Y.; Xiao, L.; Xiao, Y. Current Development of Nano-Drug Delivery to Target Macrophages. Biomedicines 2022, 10, 1203. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyester | Mcalc. [kDa] | Mn [kDa] a | Mw [kDa] a | PDI a | Monomer ratio b | Melting point [°C] c |
|---|---|---|---|---|---|---|
| PCL | 7.472 d | 7.373 | 11.923 | 1.66 | - | 58.4 |
| PLA-co-PCL | 7.453 d | 7.966 | 11.010 | 1.39 | 1:6.07 | 50.1 |
| PLGA | 7.500 e | - | - | - | 1:1 | 44.6 |
| Core Material | Hydrodynamic Diameter [nm] a | PDI a | ζ Potential [mV] a | Drug Loading b |
|---|---|---|---|---|
| PCL | 2310 ± 112 | 0.26 ± 0.01 | −14.6 ± 0.28 | 28% ± 3% |
| PLA-co-PCL | 2367 ± 98 | 0.53 ± 0.06 | −23.0 ± 0.24 | 39% ± 9% |
| PLGA | 2543 ± 110 | 0.14 ± 0.03 | −31.2 ± 0.45 | 90% ± 2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbaniak, T.; Musiał, W. Layer-by-Layer Surface Modification of Alendronate-Loaded Polyester Microparticles—Enabling Protein Immobilization. Polymers 2022, 14, 4943. https://doi.org/10.3390/polym14224943
Urbaniak T, Musiał W. Layer-by-Layer Surface Modification of Alendronate-Loaded Polyester Microparticles—Enabling Protein Immobilization. Polymers. 2022; 14(22):4943. https://doi.org/10.3390/polym14224943
Chicago/Turabian StyleUrbaniak, Tomasz, and Witold Musiał. 2022. "Layer-by-Layer Surface Modification of Alendronate-Loaded Polyester Microparticles—Enabling Protein Immobilization" Polymers 14, no. 22: 4943. https://doi.org/10.3390/polym14224943
APA StyleUrbaniak, T., & Musiał, W. (2022). Layer-by-Layer Surface Modification of Alendronate-Loaded Polyester Microparticles—Enabling Protein Immobilization. Polymers, 14(22), 4943. https://doi.org/10.3390/polym14224943