1. Introduction
Periodontal or gum disease, a prevalent infectious disease worldwide, causes the damage to supporting tissues around teeth and can lead to tooth loss. Periodontal disease can be managed by controlling infections, with mechanical debridement of the tooth surface (such as scaling and root planning, curettage, flap surgery with or without bone graft, etc.), and then the loss tissues would be spontaneously restored. Nevertheless, the regeneration of the lost supporting tissues does not succeed in every case; thus, alternative treatments are needed. Periodontal tissues engineering has gained interest in recent years, as this approach provides complexity to restore lost tooth-supporting structures with inert materials. Attempts for successful tissue regeneration are ultimately dependent on the interplay among the scaffold, cells, and bioactive signals [
1]. Hydrogels, a widely employed scaffold, can be used as an alternative, as they are cytocompatible, possesses distinctive qualities, and are utilized in drug-controlled release [
2,
3].
Hyaluronic acid (HA), a bio-polysaccharide a prominent component in the extracellular matrix of connective tissues and the periodontal ligament matrix, has the potential to achieve beneficial effects in periodontal tissue regeneration [
4,
5,
6]. HA plays a vital role in regulating cell adherence, migration, and differentiation through various binding proteins and cell-surface receptors, such as CD44 [
6]. The binding of HA fragments (below 5000 Da) to CD44 and receptors for HA-mediated motility (RHAMM) could stimulate cell cycle progression, resulting in signal transduction activation and mitogenesis [
7,
8,
9]. However, a limitation of HA administration is its fast turned over rate in the body. HA is digested by hyaluronidase, resulting in short tissue half-lives ranging from hours to days [
10]. Therefore, modification of the HA chain is needed to customize the properties of the resulting material.
Chemical modifications of three targeted functional groups (the glucuronic acid carboxylic acid, the primary and secondary hydroxyl groups, and the N-acetyl group), on an HA chain with reactive molecules (e.g., acrylates, methacrylates, maleimides) could permit crosslinking (including via Michael addition reactions or photoinitiated radical polymerizations) and generate stiffness hydrogels [
4,
11]. Hydrogels with proper mechanical properties would not only stabilize the networking system when placed between two distinct hard-tissue structures, but would also mimic features of the native extra cellular matrix that affects to cell behaviors [
12,
13]. Ibrahim and his team invented a hydrogel system containing both large and small molecular weights of hyaluronic acid. Their findings showed that the amount of HA oligomer had effect on the hydrogel’s properties, such as its swelling ratio and rheological profile. However, greater benefits on the attachment and proliferation of endothelial cells were observed. Further, the HA oligomer had a very mild ability to enhance inflammation [
14]. At this time, there are few studies which have investigated the use of hydrogels containing two different molecular weights of HA as a supporting material for periodontal tissue engineering. Particularly, the effect of this system has not yet been explored in both in vitro and in vivo models. Therefore, there is a clear motivation to investigate the application of a hydrogel system that contains two different molecular weights of HA to support the regeneration of lost periodontal tissues. In this study, the impacts of incorporated short-chain HA (sHA: 3 kDa) suspended in a thiol-ene crosslinking HA hydrogel on the physical properties (e.g., gelation time, mechanical properties, swelling, and degradation profile) and biological properties were investigated.
2. Materials and Methods
2.1. Materials
Sodium hyaluronate (HA, MW 47 kDa and 3 kDa) was purchased from Liuzhou Shengqiang Biotech Co., Ltd. (Guangxi, China). Methacrylic anhydride (Me, MW 154.16 g/mol), dithiothreitol (DTT, molecular weight 153.25 g/mol), carbazole, and crystal violet were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium tetraborate (Na2B4O7), deuterium oxide (D2O), sodium hydroxide (NaOH), absolute ethanol, and absolute methanol were from Merck (Darmstadt, Germany). Dulbecco’s Modified Eagle’s Medium (DMEM)—high glucose, trypsin-EDTA solution, L-glutamine, fetal bovine serum (FBS), and penicillin-streptomycin (10,000 U/mL) were purchased from Gibco (Waltham, MA, USA). Presto Blue TM, a resazurin-based solution, was from Invitrogen (Waltham, MA, USA).
2.2. Polymer Synthesis and Structure Elucidation
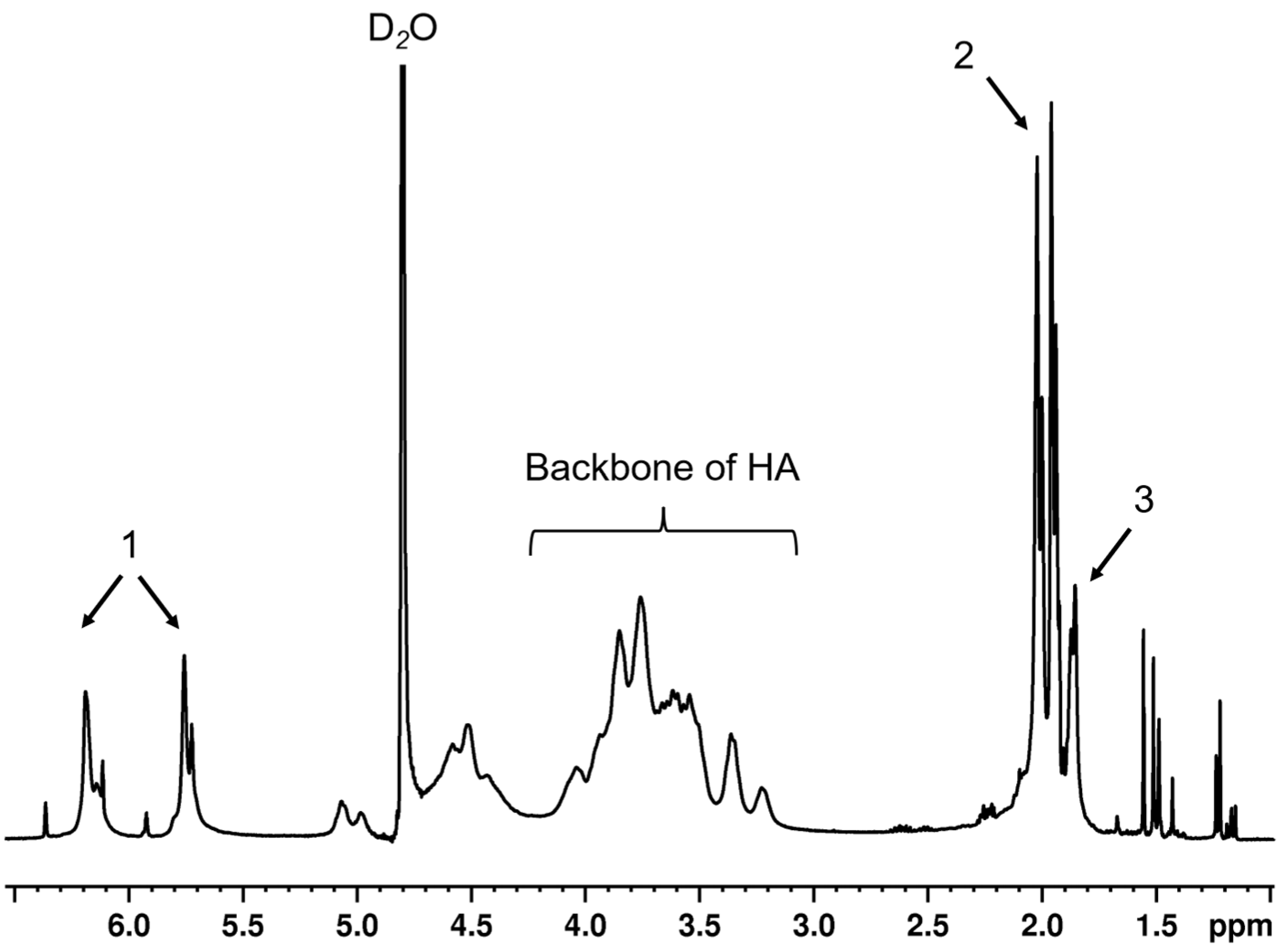
HA (47 kDa) was modified by Me via an esterification reaction, following the previous protocol [
15]. In brief, 10-fold molar excess of Me was added dropwise into 1%
w/
v HA solution in potassium phosphate buffer (PBS) at 0–4 °C, under basic conditions (pH 9–10, adjusted by 5 M NaOH). In this basic solution, the hydroxyl group of HA would be deprotonated, whereas the ester group of Me was cleaved. The reactive moiety of cleaved Me interacts with the deprotonated hydroxyl group of HA and generates MeHA plus methacrylic acid as a by-product. After overnight continuing reaction, the solution was purified by dialysis against ultrapure water for ≥48 h at 4 °C before removal of precipitated residues by centrifugation. The supernatant was collected, flash frozen and lyophilized for 2–3 days, then stored at −20 °C. The lyophilized polymers were dissolved in D
2O before being analyzed with a 400 MHz
1H NMR spectrometer (Bruker, Rheinstetten, Germany). The D
2O peak was calibrated until it represented 4.8 ppm. The degree of modification was calculated by comparing the integrals of the peaks of the protons on methacrylate alkene at 5.8 and 6.2 ppm to the integrals originating from the protons of HA backbones.
2.3. Short-Chain HA Loading and Hydrogel Formation
Short-chain hyaluronic acid (3 kDa HA or sHA) was physically added at various concentrations (
Table 1) into 3%
w/
v of methacrylated HA (MeHA) solution. Dithiothreitol (DTT) was added into the pre-gel solution at a molar ratio of thiol:ene = 2:1, then pH was adjusted in the range of 9–11 to form a gel.
2.4. Gel Point Determination
Gelation time of hydrogels was detected by inverted tube methods and oscillation time sweep. First, the tube containing 0.5 mL of mixed pre-gel solution, prepared as previously described, was inverted. The gelation time was defined as the time that the liquid transformed to gel (each sample showed no flow within 20 s during tube inversion). The gelation time was confirmed by oscillation time sweep mode using HAAKETM MARSTM rheometer (Thermo Scientific, Karlsruhe, Germany). The pre-gel solution was placed between 35 mm of titanium parallel plate and rotor, with 1% strain and a frequency of 1 Hz at 25 °C. The gelation time was decided from where the storage modulus (G′) was equal to the loss modulus (G″) (cross-over point), which represents liquid–solid transition.
2.5. Mechanical Property Analysis
HAAKE
TM MARS
TM rheometer (Thermo Scientific, Karlsruhe, Germany) was used to analyze the rheological properties of the hydrogels. One milliliter samples were placed between the 35 mm of titanium parallel plate and rotor. After that, samples were analyzed under oscillation amplitude sweep mode (1–1000% strain and a frequency of 1 Hz at 25 °C). The mechanical profile of each formulation was reported, as well as the yield point value, which was plotted from the relationship between tan δ and percentage of strain. Tan δ was calculated following this equation:
2.6. Swelling and Degradation Behavior
The gel disks (1 mm thickness, 8 mm diameter) were punched from the silicone mold before transferred to centrifuge tubes, weighed, then immersed in 3 mL of PBS for 48 h. At each time point, the swollen gels were weighed again. The swelling ratio was reported, which was calculated from the following equation:
where µ
t refers to the weight of the swollen gel at each time point, and µ
0 is the initial weight of the gel sample.
The degradation study was performed in PBS for 21 days. At each time point, the resting gels were washed with distilled water before being lyophilized for 2 days and weighed. The degradation ratio was calculated as the below equation:
where µ
0 and µ
d are the initial weight of gel sample and the weight of dry gel sample at each time point, respectively [
16].
2.7. Micromorphology Analysis
The punched gels (1 mm thickness, 8 mm diameter) were frozen in liquid nitrogen for 5–30 min before being lyophilized for 48 h. Sputter films were coated on the materials for Scanning Electron Microscope (SEM (IT-500HR), JEOL, Tokyo, Japan). Surfaces and internal sections of each lyophilized hydrogel were observed at 3 random locations per sample. The pore size was measured by Image-Analysis J 1.45S software.
2.8. Short-Chain HA Released Kinetics
The release kinetics of 3 kDa HA-loaded hydrogels were evaluated in PBS (pH 7.0) for 48 h. The hydrogels (8 mm thickness, 13 mm diameter) were transferred to dialysis bags (12–14 kDa (MWCO), Spectra/Por
® 4, Spectrum Laboratories, New Brighton, MN, USA), immersed in 100 mL of PBS, and incubated at 37 °C. One milliliter of the solutions were sampled at each time point and replaced with an equal amount of fresh medium. Then, 100 µL of the collected samples were digested with 50 µL of 100 U hyaluronidase and incubated overnight at 37 °C before measuring the amount of released uronic acid by carbazole assay. Briefly, 100 µL of sodium tetraborate in sulfuric acid was added into each tested sample and heated at 100 ± 5 °C for 10 min before being cooled down at room temperature for 15 min. Then, 0.25% carbazole in 50 µL absolute ethanol was added into each tube and heated at 100 ± 5 °C for 10 min. Following this, 100 µL of final solution was transferred to a 96-well plate, after being cooled down at room temperature for 15 min, and analyzed by microplate reader (CLARIOstar
®, BMG LABTECH, Rotenberg, Germany) at a wavelength of 550 nm. The standard curve of D-glucuronic acid was used to convert the measured absorbances to the concentration of released uronic acid [
16].
2.9. Cell Cytocompatibility
PDLs were extracted and cultured as the previous protocol [
17]. The isolation and culture protocols were approved by the Ethic Committee, Faculty of Dentistry, Chulalongkorn University (No. HREC-DCU 2021-084). Cells at passage 4–14 were used in the in vitro cell studies.
Indirect cytotoxicity assay was performed following ISO 10993-1:2009 (ISO, 2009a) with slight modification, and the hydrogel samples were described as ISO 10993-5:2009 (ISO, 2012) [
18,
19]. In brief, pre-gel solution was prepared under sterile conditions. After that, the gel was transferred to a sterile silicone mold. The gel was incubated at 37 °C overnight to ensure complete hydrogel formation. Afterwards, the hydrogels were punched into a disk shape with calculated surface area of 3.14 cm
2. Each hydrogel disc was immersed in 1 mL of serum-free DMEM containing penicillin/streptomycin for 24 h at 37 °C. On the same day, 1 × 10
4 of PDLs were seeded into each well of a 96-well plate and incubated at 37 °C in a humidified atmosphere supplemented with 5% CO
2 for 24 h. The following day, the sample extracts (prepared at different concentrations of 12.5%, 25%, 50%, and 100%) were used to replace the cell culture media and incubated at 37 °C for 24 h. In this assay, the extract vehicle, serum-free DMEM, was used as a control. The morphology of the cells was observed the following day by microscopy (Nikon Eclipse TS2, Nikon, Tokyo, Japan). Then, 50 μL of 0.5 mg/mL MTT solution was prepared and added into each well after the medium was removed, and the cells were washed once with PBS. After incubation for 30 min at 37 °C, 100 μL of DMSO replaced the MTT solution to the wells for formazan extraction before being measured by a microplate reader at 570 nm [
20].
2.10. Proliferation Assay
Sterile MeHA polymers were dissolved in complete DMEM media (DMEM mixing with 10% FBS, 1% L-glutamine, and 1% penicillin–streptomycin) as a pre-gel solution, before being combined with sHA at each concentration in order to form a gel coated layer on 96-well plates. Five thousand cells per 100 µL were seeded on the top of the gel layer and cultured in 5% CO
2 incubator at 37 °C for 1, 3, 5, and 7 days. At each time-point, the morphology of cells was observed by using phase contrast imaging with microscope (Nikon Eclipse TS2, Nikon, Tokyo, Japan). Additionally, 10% resazurin-based solution, a cell viability indicator, was reacted with the cells at 37 °C for 1 h, before being analyzed with a microplate reader at excitation/emission of 560 ± 15/590 ± 15 nm [
21].
2.11. Transwell Migration Assay
Twenty-four-well size transwell inserts with an 8.0 µm pore polycarbonate membrane and 0.33 cm
2 effective growth area (Corning
®, Corning, NY, USA) were used to perform cell migration experiments. The lower wells were layered with hydrogel and topped up with serum-free media. PDLs were trypsinized and re-suspended in serum-free media, then seeded in each insert at a concentration of 30,000 cells/well. Each seeded insert was placed into each lower well and incubated at 37 °C for 24 h in order to prevent the effects of cell migration. After the maturity period, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with acetic acid:methanol at the ratio of 1:3 for 5 min, then stained with 0.7% crystal violet and washed with PBS. A cotton swab was used to remove non-migrated cells from the upper surface of the membrane. The migrated cells were observed by microscope (Nikon Eclipse TS2, Nikon, Tokyo, Japan). Image-Analysis J 1.45S software was used to count the number of migrated cells from five randomly chosen fields (×40) [
22].
2.12. Animal Study
Male Wistar rats (250–300 g) were obtained from the Nomura Siam International Co. Ltd. All the in vivo studies were approved by the Faculty of Pharmaceutical Sciences Animal Care and Use Committee, Chulalongkorn University, protocol number 1933004. The housing and experimental procedures complied with the guidelines of the Animals for Scientific Purposes Act, B.E. 2558 (A.D. 2015), Thailand. Prior to the beginning of in vivo experiments, animals were quarantined/acclimated for 7 days to adjust to the standardized laboratory temperature (22 ± 2 °C), humidity (40–60%), and light–dark cycle (12:12 h).
In total, 36 animals were used (calculated using G*Power program version 3.1.9.4) and were randomly divided into three groups with
n = 12 per group: Control, MeHA, and 1.5%
w/
v sHA. The randomization was generated from the random function (Rand) in Excel program. On day 0, the rats were anesthetized using isoflurane (1–3%) with 100% oxygen. The depth of anesthesia and vital signs of the animals were monitored every 5 min. Subcutaneous injection of tramadol was used for peri-operative and post-operative analgesia. Moreover, moist feed was provided at the floor cage level to promote the eating of the rats after the procedure. The oral wounds were created by 3-mm diameter punch biopsy on the anterior palate in the mucoperiosteum of the midline of the hard palate. The soft tissue was removed by sharp dissection. Cotton gauze was placed over the wound until hemostasis was achieved. All procedures were performed using an aseptic technique. The treatment gels were applied to the wound. Every 1, 3, 5, and 7 days after treatment, 3 rats from each group were euthanized by CO
2 asphyxiation, following the open thorax (American Veterinary Medical Association (AVMA) Guidelines for Euthanasia (2013 edition). The hard palate samples were collected for histopathological studies. The wound sizes were measured. For the histological analysis, the sample sections were stained with Hematoxylin and Eosin (H&E) staining and observed by Apotome.2 apparatus (Carl Zeiss, Jena, Germany) (×400) [
23].
2.13. Statistical Analysis
All results were reported as mean ± standard deviation (SD), resulting from three or more replications. Statistical significance between different groups was analyzed using one-way analysis of variance followed by Tukey’s post hoc test using GraphPad Prism Software (V.5) with a p-value < 0.05.
4. Discussion
There are numerous clinical HA products that have been approved by the Food and Drug administration for use as medical devices for dental diseases (e.g., Gengigel, Flex Barrier) [
26,
27]. These products are in gel form, that provides weak mechanical properties and stability, resulting in increased frequency of use. Since the treatment of periodontal disease is long-term management, reducing frequency of use could improve patients’ compliance. In this work, we chose to fabricate the scaffolding of hydrogels from a modified HA chain (MeHA), since this system can be digested and supply HA itself. Moreover, the mechanical properties of this hydrogel can be adjusted by degree of modification or crosslinking system. Another attractive characteristic of this hydrogel system is that it represents itself as an
in situ gel system, a feature of drug delivery systems that are in liquid forms before administration; once it reaches the target areas, it transforms into a gel [
28]. This feature is very useful as it could prevent the deposition of food residues into the depth or irregular shapes of periodontal wounds.
HA fragments have been found mostly in tissue inflammation areas [
29,
30]. On the other hand, it has been reported that HA fragments could probably enhance the regeneration of damaged tissues [
31]. A previous study has revealed that a hydrogel containing two different sizes of HA (long-chain vs. fragmented HA), could lower the inflammatory effect of HA fragments, and improve the attachment and proliferation of endothelial cells. They also found that HA fragments showed some interference with the physical properties of hydrogels (swelling profiles, mechanical properties, crosslinking density) [
14]. Trakiatkul and team, illustrated the suitability of a MeHA-based hydrogel for loading polysaccharides and proteins, such as mannitol and BSA. They found no significant changes to the physical properties of this hydrogel system when carrying mannitol/BSA [
32]. Accordingly, we decided to explore the possibility of directly incorporating HA fragments (or sHA) into a MeHA injectable hydrogel, and we sought to investigate the impact of sHA content on the gel’s physical properties, in addition to their biological effects (e.g., biocompatibility, migratory).
One of the huge challenges in developing an
in situ gel system is to obtain an appropriate gelation time. Having a short gelation time is essential for the prompt attachment of the gel to the wound or pockets in oral cavities and to minimize the loss of intra-pocket carriers, further, enhancing patients’ compliance [
33]. In order to observe the effects of incorporated sHA on gelation time, the concentration of MeHA and the crosslinking molar ratios (thiol:ene) were fixed at 3% and 2:1, based on our prior work [
34]. The content of the incorporated sHA were divided into three concentrations, 0.5%, 1.0%, and 1.5%
w/
v (referring to the concentration of commercial products containing HA, which are in the range of 0.2–1.0%
w/
w) [
27,
35]. Our prior work reported the typical gelation time of a hydrogel fabricated from MeHA with 40–60% modifications, which was in the range of 15–30 min [
34]. The increase in modification degree minimizes gelation time, due to the increasing of crosslinking moiety (vinyl groups) [
36]. In this study, we adjusted the degree of modification of MeHA into the range of 80–100% in order to minimize the gel onset time. From the results, we could achieve gelation time within a few minutes. Additional sHA seemed to have effects on the gelation time, since the gel time of the formulations containing 1.0 and 1.5%
w/
v of sHA were less than others, which was related to their pre-gel viscosity, which seemed to be lower than the 0.5%
w/
v and non-sHA formulation. Existing HA fragments in an HA macromers solution has been reported to interrupt the interpolymeric interaction of HA solutions [
33]. Therefore, a higher content of sHA in a MeHA solution may interfere with the networking system between MeHA macromer chains dissolved in aqueous solutions, resulting in the decrease of viscosity that allowed a faster crosslinking reaction and showed an earlier sol to gel transition. Nonetheless, adding sHA at this range of concentration did not have a significant impact on the gelation time.
Rheological analysis quantitatively estimated the elastic responses of a MeHA-based hydrogel by simulating an external strain ranking from 1–1000% to the gel. By increasing the degree of modification of MeHA, we could obtain the storage modulus (G′) of approximately 1000–1500 Pa; that was even higher than that of a previously developed MeHA hydrogel with 40% degree of modification; the G′ was ~400 Pa [
16]. Though sHA did not have an effect on G′, it seemed to have interference with the yield point of hydrogels, which represents the maximum tolerance of the material to given strain. Increasing amounts of sHA reduces the yield point of hydrogels; we believed that this event was related to the possible causes of the high solid contents in the formulation. We observed a correlation between the yield point and microstructure of the hydrogels. A high percentage of sHA incorporation causes a weaker gel structure; additionally, the presence of small pore size might be the effect of entanglement between polymers which formed an impermanent interaction across networks [
37]. Another possible reason is the disturbance of sHA to the crosslinking system. Overall, there was no significant difference among all hydrogels, they could tolerate to external strains of 150–300% strain, which could be survivable when surrounded by distributed strain during mastication [
38].
In order to evaluate the long-term retention of incorporated sHA within the MeHA hydrogel, a swelling and degradation study, and in vitro release study should be performed. We found that a high content of sHA increased the gel swelling and degradation ratio. This phenomenon was related to a previous study, which reported that the effect of having HA oligomers in an HA hydrogel could increase the swelling ratio and cause low crosslinking density [
14]. We also found an association between the release profile and swelling behavior of the hydrogels. The highest released amount of glucuronic acid was found in the 1.0%
w/
v of sHA hydrogel, it also had the highest swelling ratio and fastest weight loss, which could refer to the early degradation of the hydrogel formulation. On the other hand, the formulation containing 1.5%
w/
v of sHA could slow release the amount of glucuronic acid, which associated to the smaller pore size of itself, compared to the other formulation. All in all, the addition of sHA at these concentrations did not have much interference on the hydrogels’ physical properties.
To assess the wound healing properties of MeHA hydrogels loaded with sHA, we firstly performed in vitro cell studies, including a proliferation assay and migration test with PDLs, because establishing a PDL cell population in a periodontal defect is an important requirement for the remodeling and rebuilding of the periodontium [
24]. All hydrogels were proved to be biocompatible with PDLs. We executed the proliferation test by culturing PDLs on hydrogels to demonstrate their practical use dimension, wherein the cell would directly contact to the surface of the hydrogels. We found that PDLS appeared to have poor attachment on the surface of hydrogels, forming a spheroidal shape, which impacts their proliferation behavior. Two factors known to influence this phenomenon are the material stiffness and receptor binding affinity of the cell and modified hyaluronan. Material stiffness plays a vital role in controlling cell behavior. Recently, a study reported that the hardness of culture material (ranking from 6–135 kPa) had effects on the alteration of PDLs’ proliferation rate. PDLs cultured on soft material (6 kPa) showed an inferior proliferation rate [
12,
39]. Therefore, it can be presumed that the elastic modulus of the hydrogels (around 1–1.5 kPa) would not be strong enough to support cell proliferation. Another related reason is the binding affinity of the cell and modified HA. The hydroxyl groups on the HA chain are used as one of the binding sites that could associate to CD44 [
40]. Kwon and coworkers disclosed the relationship between the level of HA modification and binding affinity to CD44. A high level of HA modification had poor binding affinity to CD44 due to the unavailable binding site [
41]. Since the investigated MeHA had very high degree of modification, this could cause the deficiency of HA-CD44 binding which resulted in poor cell-surface attachment.
For the transwell migration test, we found that the hydrogel containing 0.5%
w/
v of sHA presented the best performance in inducing migration. We hypothesized that some amount of sHA would have a chemo-attractive effect. This assumption is in agreement with an earlier study, which found that HA promotes the migration of human meniscus cells in a concentration-dependent manner [
42]. On the contrary, the result of this study showed that the increased amount of sHA resulted in the decrease of migrated cells. The possible reason for this is that the release of sHA from the highest content of sHA hydrogel formulation was hindered by the small pore size of the hydrogels. The microstructure study mentioned above found that the pore size of the gels was reduced as a consequence of an increase in the content of sHA in the network.
As the hydrogel formulation containing 1.5%
w/
v of sHA appeared to have a prolonged release effect of sHA, it was selected for further study of its impacts on tissue regeneration in an in vivo model, while the base MeHA gel was utilized as a blank. Small defects were simulated on the rats’ upper palates by punching, which damaged the epidermis layers (keratin layer and keratinized stratified squamous epithelium) [
25]. Our findings showed that applying hydrogels to the wound site reduced the treatment consuming time. In histological section, our hydrogels could induce epithelium cells originating from the three basal layers of the oral squamous epithelium which migrated and repaired the injury caused by the shallower depth of the wound [
43]. Our work has proved the possibility of forming two different sizes of HA and clarified the interference with the physical properties of the hydrogel, which did not show any significant changes. Our study illustrated that our hydrogels were biocompatible with tissues in the oral cavity, in both in vitro and in vivo models. The ability to shorten the healing time of the hydrogels could promote the chance to use this approach as a medical device to support the regeneration of damaged tissues related to dental diseases; however, further studies are needed.




 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}