
Cross-Linked Gamma Polyglutamic Acid/Human Hair Keratin Electrospun Nanofibrous Scaffolds with Excellent Biocompatibility and Biodegradability



Abstract
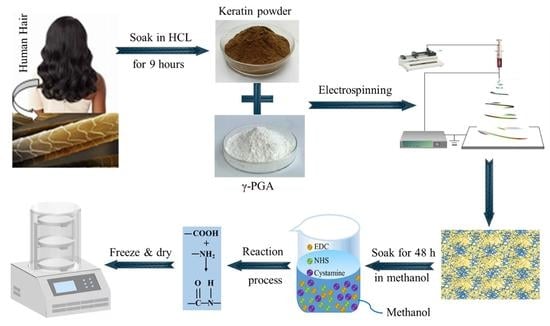
{kind=link}
{kind=link}
{kind=link}
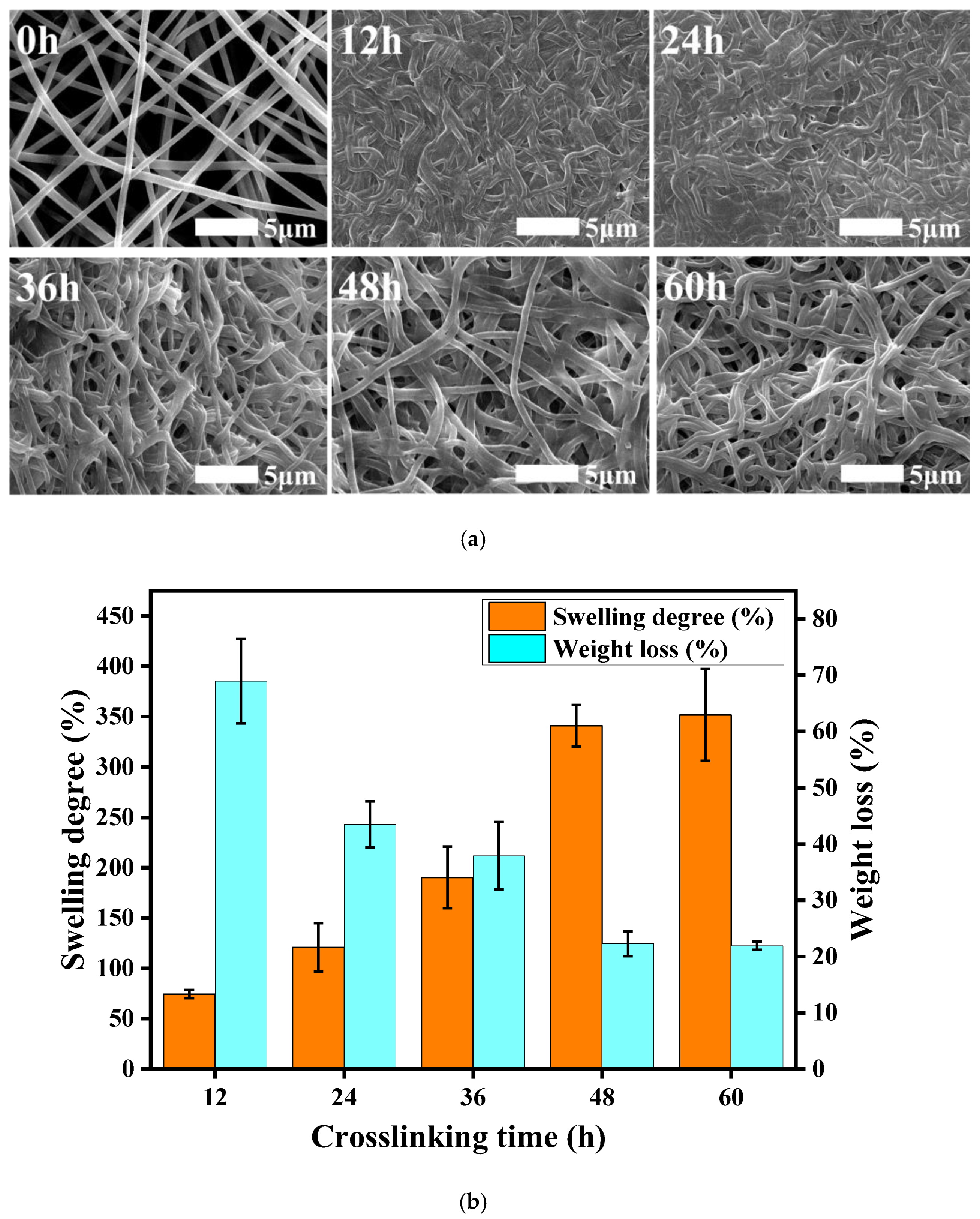{kind=link}
{kind=link}
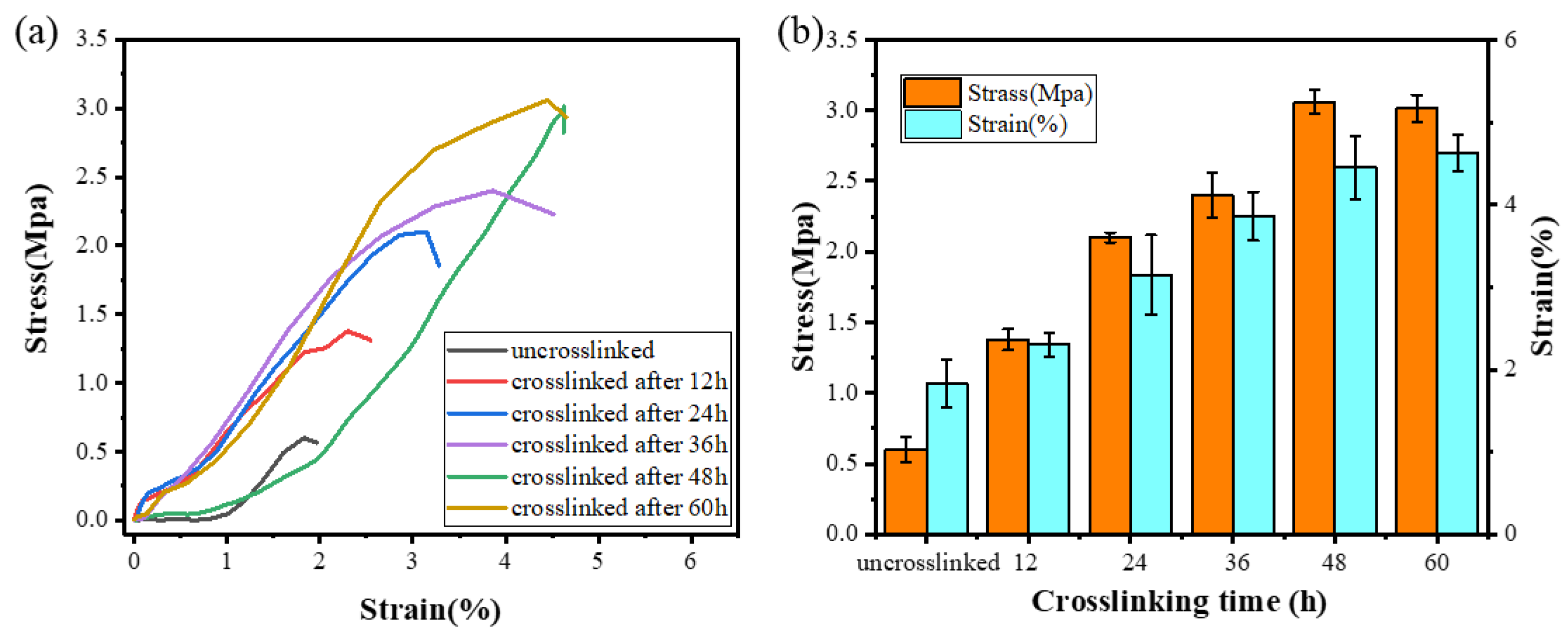{kind=link}
{kind=link}
{kind=link}
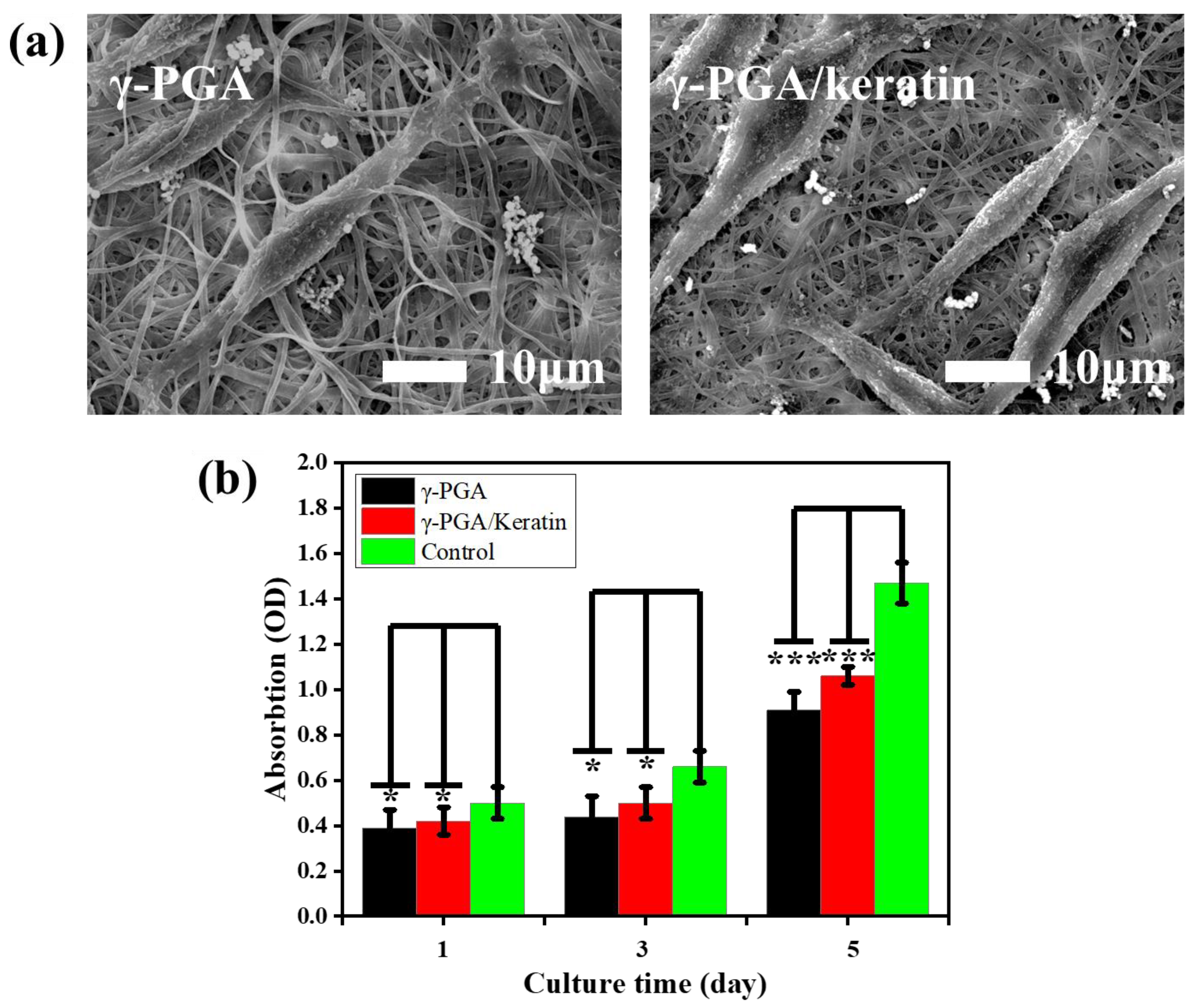{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Regenerated Human Hair Keratin
2.3. Preparation of γ-PGA/Keratin ENSs
2.4. Preparation of Cross-Linked γ-PGA/Keratin ENSs
2.5. Characterization
2.6. Statistical Analysis
3. Results
3.1. Optimization of Process Parameters of γ-PGA/Keratin Nanofibers
3.2. Principle of Cross-Linking
3.3. Resistance to Hydrolysis
3.4. Tensile Properties
3.5. In Vitro Degradation
3.6. Biocompatibility
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Verma, V.; Verma, P.; Ray, P.; Ray, A.R. Preparation of scaffolds from human hair proteins for tissue-engineering applications. Biomed. Mater. 2008, 3, 025007. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Pierschbacher, M.; Clark, R.A. Expression of functional α4β1 integrin by human dermal fibroblasts. J. Investig. Dermatol. 1993, 100, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Hynes, R.O. Lymphoid cells recognize an alternatively spliced segment of fibronectin via the integrin receptor α4β1. Cell 1990, 60, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Teng, Z.; Wang, S.; Xu, Y.; Wu, P.; Zhu, Y.; Wang, C.; Wang, G. Polymeric carbon nitride modified polyacrylonitrile fabrics with efficient self-cleaning and water disinfection under visible light. Chem. Eng. J. 2020, 391, 123506. [Google Scholar] [CrossRef]
- Hou, Z.; Wen, Z.; Wang, D.; Wang, J.; François-Xavier, C.P.; Wintgens, T. Bipolar jet electrospinning bi-functional nanofibrous membrane for simultaneous and sequential filtration of Cd2+ and BPA from water: Competition and synergistic effect. Chem. Eng. J. 2018, 332, 118–130. [Google Scholar] [CrossRef]
- Buivydiene, D.; Todea, A.M.; Asbach, C.; Krugly, E.; Martuzevicius, D.; Kliucininkas, L. Composite micro/nano fibrous air filter by simultaneous melt and solution electrospinning. JAerS 2021, 154, 105754. [Google Scholar] [CrossRef]
- Wu, X.; Li, P.; Cong, L.; Yu, H.; Zhang, D.; Yue, Y.; Xu, H.; Xu, K.; Zheng, X.; Wang, X. Electrospun poly(vinyl alcohol) nanofiber films containing menthol/β-cyclodextrin inclusion complexes for smoke filtration and flavor retention. Colloids Surf. Physicochem. Eng. Asp. 2020, 605, 125378. [Google Scholar] [CrossRef]
- Xu, J.; Liu, C.; Hsu, P.C.; Liu, K.; Cui, Y. Roll-to-Roll Transfer of Electrospun Nanofiber Film for High-Efficiency Transparent Air Filter. Nano Lett. 2016, 16, 1270–1275. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, M.; Quan, Z.; Zhang, H.; Qin, X.; Wang, R.; Yu, J. Polyacrylonitrile/polyimide composite sub-micro fibrous membranes for precise filtration of PM0.26 pollutants. J. Colloid Interface Sci. 2020, 578, 195–206. [Google Scholar] [CrossRef]
- Negm, N.A.; Abubshait, H.A.; Abubshait, S.A.; Abou Kana, M.T.H.; Mohamed, E.A.; Betiha, M.M. Performance of chitosan polymer as platform during sensors fabrication and sensing applications. Int. J. Biol. Macromol. 2020, 165, 402–435. [Google Scholar] [CrossRef]
- Alim, S.; Kafi, A.K.M.; Rajan, J.; Yusoff, M.M. Application of polymerized multiporous nanofiber of SnO2 for designing a bienzyme glucose biosensor based on HRP/GOx. Int. J. Biol. Macromol. 2019, 123, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kang, K.; Wu, J.; Hu, Q.; Harper, D.P.; Du, G.; Wang, S.; Xu, K. Comparative effects of electrospinning ways for fabricating green, sustainable, flexible, porous, nanofibrous cellulose/chitosan carbon mats as anode materials for lithium-ion batteries. J. Mater. Res. Technol. 2021, 11, 50–61. [Google Scholar] [CrossRef]
- Liu, T.; Yao, T.; Li, L.; Zhu, L.; Wang, J.; Li, F.; Wang, H. Embedding amorphous lithium vanadate into carbon nanofibers by electrospinning as a high-performance anode material for lithium-ion batteries. J. Colloid Interface Sci. 2020, 580, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Chen, J.; He, N.; Yu, K.; Liang, C. Controllable SnO2/ZnO@PPy hollow nanotubes prepared by electrospinning technology used as anode for lithium ion battery. JPCS 2021, 150, 109861. [Google Scholar] [CrossRef]
- Zhou, W.; Gong, X.; Li, Y.; Si, Y.; Zhang, S.; Yu, J.; Ding, B. Waterborne electrospinning of fluorine-free stretchable nanofiber membranes with waterproof and breathable capabilities for protective textiles. J. Colloid Interface Sci. 2021, 602, 105–114. [Google Scholar] [CrossRef]
- Zhou, W.; Gong, X.; Li, Y.; Si, Y.; Zhang, S.; Yu, J.; Ding, B. Environmentally friendly waterborne polyurethane nanofibrous membranes by emulsion electrospinning for waterproof and breathable textiles. Chem. Eng. J. 2022, 427, 130925. [Google Scholar] [CrossRef]
- Qi, K.; Zhou, Y.; Ou, K.; Dai, Y.; You, X.; Wang, H.; He, J.; Qin, X.; Wang, R. Weavable and stretchable piezoresistive carbon nanotubes-embedded nanofiber sensing yarns for highly sensitive and multimodal wearable textile sensor. Carbon 2020, 170, 464–476. [Google Scholar] [CrossRef]
- Veeramuthu, L.; Cho, C.-J.; Venkatesan, M.; Kumar, G.R.; Hsu, H.-Y.; Zhuo, B.-X.; Kau, L.-J.; Chung, M.-A.; Lee, W.-Y.; Kuo, C.-C. Muscle fibers inspired electrospun nanostructures reinforced conductive fibers for smart wearable optoelectronics and energy generators. Nano Energy 2022, 101, 107592. [Google Scholar] [CrossRef]
- Busolo, T.; Szewczyk, P.K.; Nair, M.; Stachewicz, U.; Kar-Narayan, S. Triboelectric Yarns with Electrospun Functional Polymer Coatings for Highly Durable and Washable Smart Textile Applications. ACS Appl. Mater. Inter. 2021, 13, 16876–16886. [Google Scholar] [CrossRef]
- Tao, F.; Cheng, Y.; Tao, H.; Jin, L.; Wan, Z.; Dai, F.; Xiang, W.; Deng, H. Carboxymethyl chitosan/sodium alginate-based micron-fibers fabricated by emulsion electrospinning for periosteal tissue engineering. Mater. Design 2020, 194, 108849. [Google Scholar] [CrossRef]
- Juncos Bombin, A.D.; Dunne, N.J.; McCarthy, H.O. Electrospinning of natural polymers for the production of nanofibres for wound healing applications. Mater. Sci. Eng. C 2020, 114, 110994. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Khani, M.-M.; Rabbani, S.; Mashaghi, A.; Noorizadeh, F.; Faridi-Majidi, R.; Ghanbari, H. Development of poly(mannitol sebacate)/poly(lactic acid) nanofibrous scaffolds with potential applications in tissue engineering. Mater. Sci. Eng. C 2020, 110, 110626. [Google Scholar] [CrossRef]
- Liu, X.; Chen, M.; Luo, J.; Zhao, H.; Zhou, X.; Gu, Q.; Yang, H.; Zhu, X.; Cui, W.; Shi, Q. Immunopolarization-regulated 3D printed-electrospun fibrous scaffolds for bone regeneration. Biomaterials 2021, 276, 121037. [Google Scholar] [CrossRef] [PubMed]
- Keirouz, A.; Fortunato, G.; Zhang, M.; Callanan, A.; Radacsi, N. Nozzle-free electrospinning of Polyvinylpyrrolidone/Poly(glycerol sebacate) fibrous scaffolds for skin tissue engineering applications. Med. Eng. Phys. 2019, 71, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Singhal, P. Preparation and characterization of poly (E-CAPROLACTONE) nano fibers by electrospinning technique for tissue enginerring applications. Mater. Today Proc. 2021, 37, 2997–3001. [Google Scholar] [CrossRef]
- Fortunato, G.M.; Da Ros, F.; Bisconti, S.; De Acutis, A.; Biagini, F.; Lapomarda, A.; Magliaro, C.; De Maria, C.; Montemurro, F.; Bizzotto, D.; et al. Electrospun Structures Made of a Hydrolyzed Keratin-Based Biomaterial for Development of in vitro Tissue Models. Front. Bioeng. Biotechnol. 2019, 7, 174. [Google Scholar] [CrossRef]
- Zhao, X.; Lui, Y.S.; Choo, C.K.C.; Sow, W.T.; Huang, C.L.; Ng, K.W.; Tan, L.P.; Loo, J.S.C. Calcium phosphate coated Keratin–PCL scaffolds for potential bone tissue regeneration. Mater. Sci. Eng. C 2015, 49, 746–753. [Google Scholar] [CrossRef]
- Li, P.; Wang, Y.; Jin, X.; Dou, J.; Han, X.; Wan, X.; Yuan, J.; Shen, J. Catalytic Generation of Nitric Oxide from Poly(ε-caprolactone)/Phosphobetainized Keratin Mats for a Vascular Tissue Engineering Scaffold. Langmuir 2020, 36, 4396–4404. [Google Scholar] [CrossRef]
- Sawadkar, P.; Mohanakrishnan, J.; Rajasekar, P.; Rahmani, B.; Kohli, N.; Bozec, L.; García-Gareta, E. A Synergistic Relationship between Polycaprolactone and Natural Polymers Enhances the Physical Properties and Biological Activity of Scaffolds. ACS Appl. Mater. Inter. 2020, 12, 13587–13597. [Google Scholar] [CrossRef]
- Mohammadalizadeh, Z.; Bahremandi-Toloue, E.; Karbasi, S. Synthetic-based blended electrospun scaffolds in tissue engineering applications. J. Mater. Sci. 2022, 57, 4020–4079. [Google Scholar] [CrossRef]
- Kunioka, M.; Choi, H.J.; Kunioka, M. Preparation conditions and swelling equilibria of biodegradable hydrogels prepared from microbial poly(γ-glutamic acid) and poly(ε-lysine). J. Polym. Environ. 1996, 4, 123–129. [Google Scholar] [CrossRef]
- Tsao, C.T.; Chang, C.H.; Lin, Y.Y.; Wu, M.F.; Wang, J.L.; Han, J.L.; Hsieh, K.H. Antibacterial activity and biocompatibility of a chitosan–γ-poly(glutamic acid) polyelectrolyte complex hydrogel. Carbohydr. Res. 2010, 345, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.W.; Peng, S.F.; Su, C.J.; Mi, F.L.; Chen, H.L.; Wei, M.C.; Lin, H.J.; Sung, H.W. The use of biodegradable polymeric nanoparticles in combination with a low-pressure gene gun for transdermal DNA delivery. Biomaterials 2008, 29, 742–751. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Mishra, R.; Rana, D. Strategies for effective oral insulin delivery with modified chitosan nanoparticles: A review. Prog. Polym. Sci. 2012, 37, 1457–1475. [Google Scholar] [CrossRef]
- Bajaj, I.; Singhal, R. Poly (glutamic acid)—An emerging biopolymer of commercial interest. Bioresour. Technol. 2011, 102, 5551–5561. [Google Scholar] [CrossRef]
- Castro, F.; Pinto, M.L.; Pereira, C.L.; Serre, K.; Barbosa, M.A.; Vermaelen, K.; Gärtner, F.; Gonçalves, R.M.; De Wever, O.; Oliveira, M.J. Chitosan/γ-PGA nanoparticles-based immunotherapy as adjuvant to radiotherapy in breast cancer. Biomaterials 2020, 257, 120218. [Google Scholar] [CrossRef]
- Díez-Pascual, A.M.; Díez-Vicente, A.L. Multifunctional poly(glycolic acid-co-propylene fumarate) electrospun fibers reinforced with graphene oxide and hydroxyapatite nanorods. J. Mater. Chem. B 2017, 5, 4084–4096. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Arimoto, M.; Takeuchi, K.; Fujii, T. A rapid extraction procedure of human hair proteins and identification of phosphorylated species. Biol. Pharm. Bull. 2002, 25, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Klinkhammer, K.; Matsusaki, M.; Mller, M.; Klee, D.; Akashi, M. Disulfide-Crosslinked Electrospun Poly(γ-glutamic acid) Nonwovens as Reduction-Responsive Scaffolds. Macromol. Biosci. 2009, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, X.; Shen, M.; Guo, R.; Banyai, I.; Shi, X. Fabrication and morphology control of electrospun poly(γ-glutamic acid) nanofibers for biomedical applications. Colloids Surf. B Biointerfaces 2012, 89, 254–264. [Google Scholar] [CrossRef]
- Yang, C. Enhanced physicochemical properties of collagen by using EDC/NHS-crosslinking. Bull. Mater. Sci. 2012, 35, 913–918. [Google Scholar] [CrossRef]
- Kim, S.J.; Ise, H.; Goto, M.; Komura, K.; Cho, C.S.; Akaike, T. Gene delivery system based on highly specific recognition of surface-vimentin with N-acetylglucosamine immobilized polyethylenimine. Biomaterials 2011, 32, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, V.V.; Strikovsky, A.G.; Verteletskaya, T.E. Amide I absorption band: Description of the urethane group association scheme in polyether urethane elastomers. Polymer 1993, 34, 938–941. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, M.; Liu, Y.; Chen, Z.; Hu, X.; Zhang, T.; Zhu, X.; He, X.; Yang, B. Cross-Linked Gamma Polyglutamic Acid/Human Hair Keratin Electrospun Nanofibrous Scaffolds with Excellent Biocompatibility and Biodegradability. Polymers 2022, 14, 5505. https://doi.org/10.3390/polym14245505
Hao M, Liu Y, Chen Z, Hu X, Zhang T, Zhu X, He X, Yang B. Cross-Linked Gamma Polyglutamic Acid/Human Hair Keratin Electrospun Nanofibrous Scaffolds with Excellent Biocompatibility and Biodegradability. Polymers. 2022; 14(24):5505. https://doi.org/10.3390/polym14245505
Chicago/Turabian StyleHao, Ming, Yanbo Liu, Zhijun Chen, Xiaodong Hu, Tianyi Zhang, Xinyu Zhu, Xingyu He, and Bo Yang. 2022. "Cross-Linked Gamma Polyglutamic Acid/Human Hair Keratin Electrospun Nanofibrous Scaffolds with Excellent Biocompatibility and Biodegradability" Polymers 14, no. 24: 5505. https://doi.org/10.3390/polym14245505
APA StyleHao, M., Liu, Y., Chen, Z., Hu, X., Zhang, T., Zhu, X., He, X., & Yang, B. (2022). Cross-Linked Gamma Polyglutamic Acid/Human Hair Keratin Electrospun Nanofibrous Scaffolds with Excellent Biocompatibility and Biodegradability. Polymers, 14(24), 5505. https://doi.org/10.3390/polym14245505