
Improving Sustainability of the Griess Reaction by Reagent Stabilization on PDMS Membranes and ZnNPs as Reductor of Nitrates: Application to Different Water Samples

 , and
, and 
Abstract
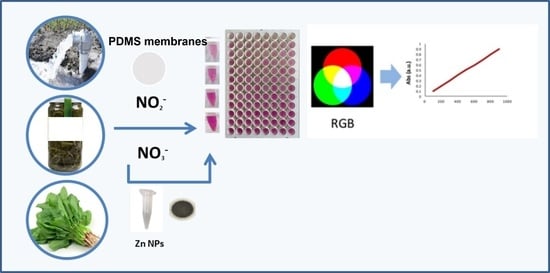
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Apparatus
2.3. Preparation of the Composites
2.3.1. PDMS/TEOS-SiO2NPs-SA-NEDD
2.3.2. PDMS/TEOS-SiO2NPs-SA-NEDD-OMIM PF6
2.3.3. PDMS/SiO2NPs-SA-NEDD-OMIM PF6
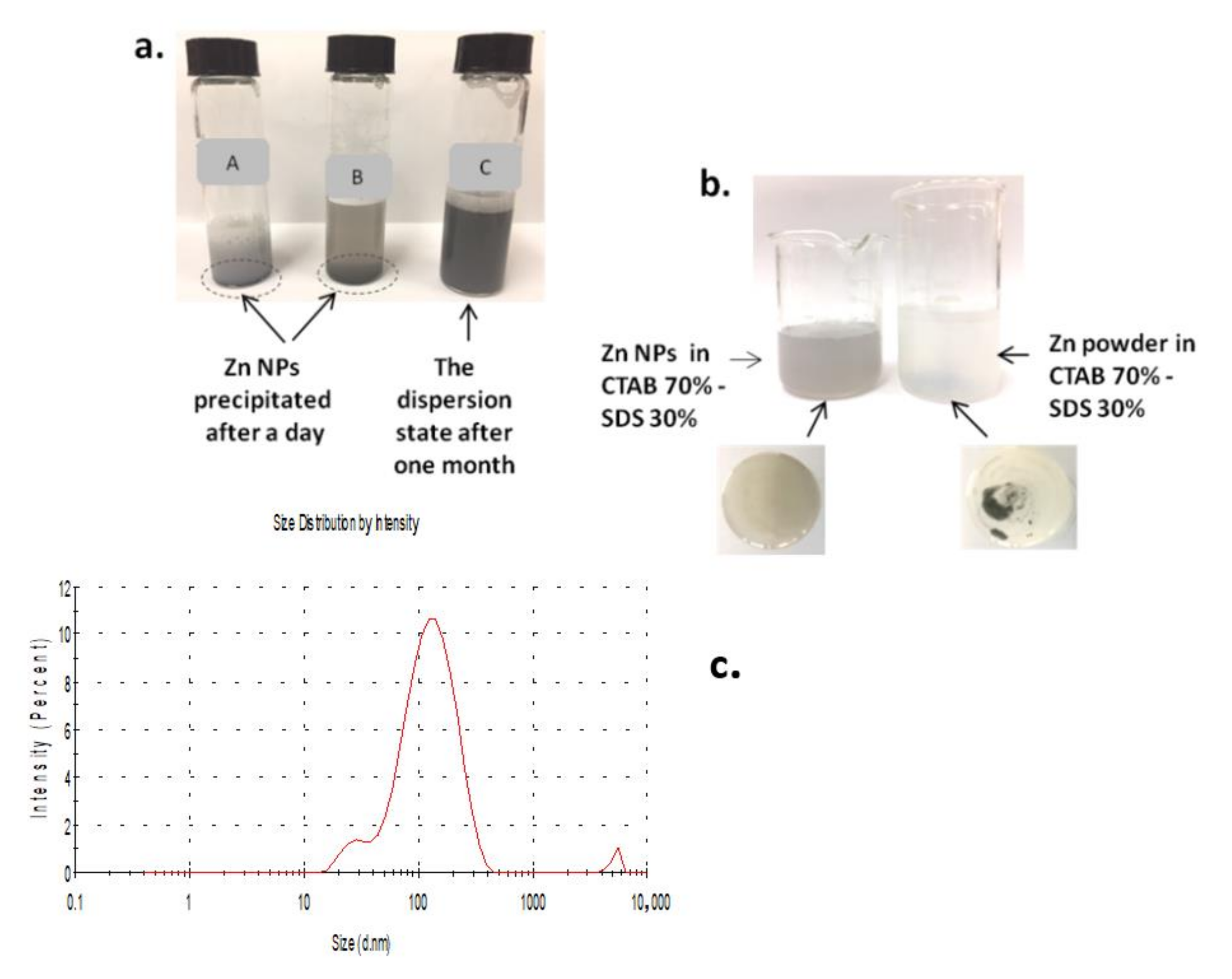
2.3.4. Preparation of ZnNPs Dispersion
2.4. Analytical Response Measurements
2.4.1. Determination of Nitrites or Nitrates Adding the Griess Reagents in Solution
2.4.2. Analytical Response Measurements Adding the Sensor Membrane
2.4.3. Reference Methods: Potentiometric Measurement and UV at 220 nm
2.4.4. Analytical Response Measurements in 96-Well Multiplate
2.5. Analysis of Real Samples
3. Results and Discussion
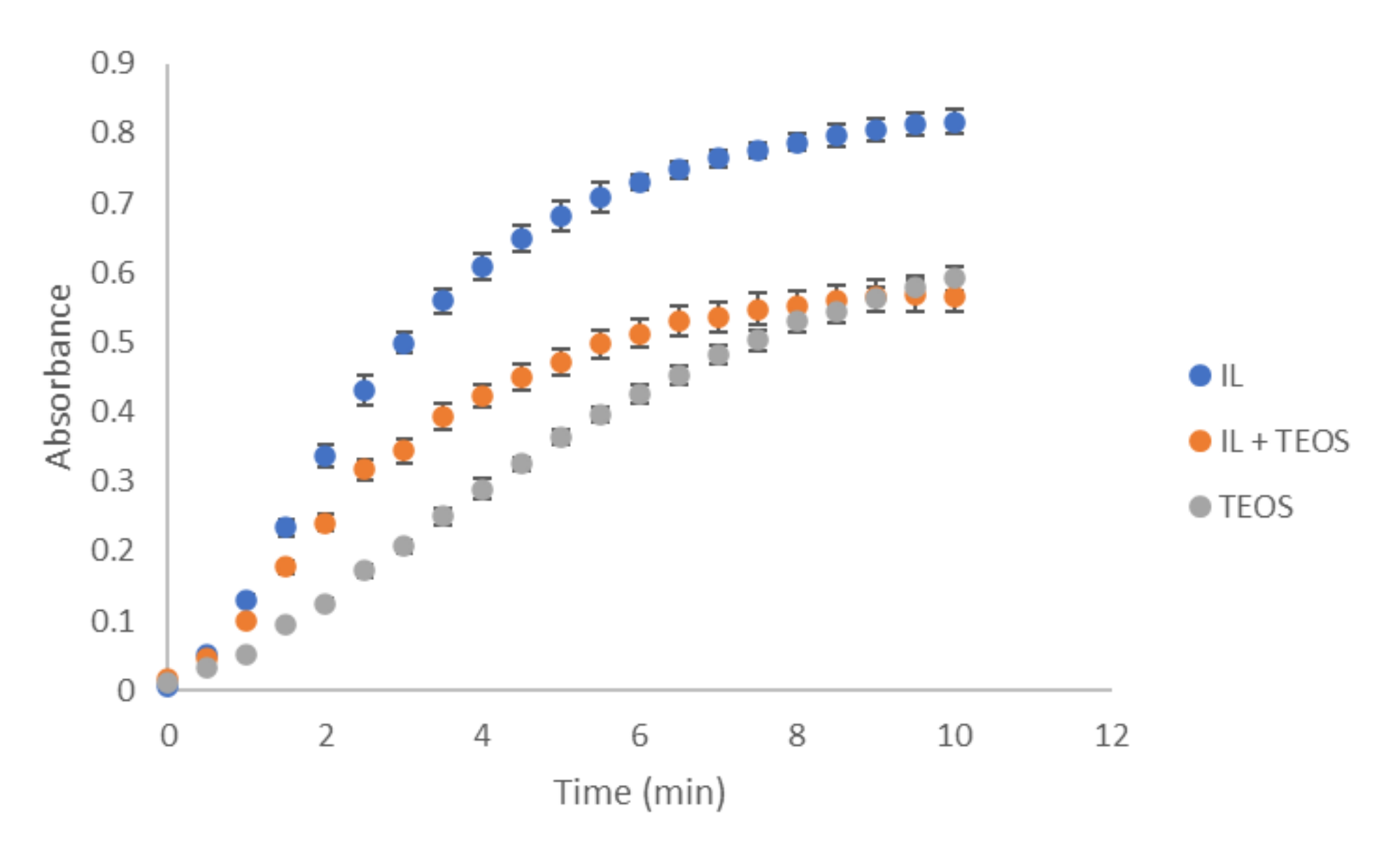
3.1. Study of the Reaction with the Composite
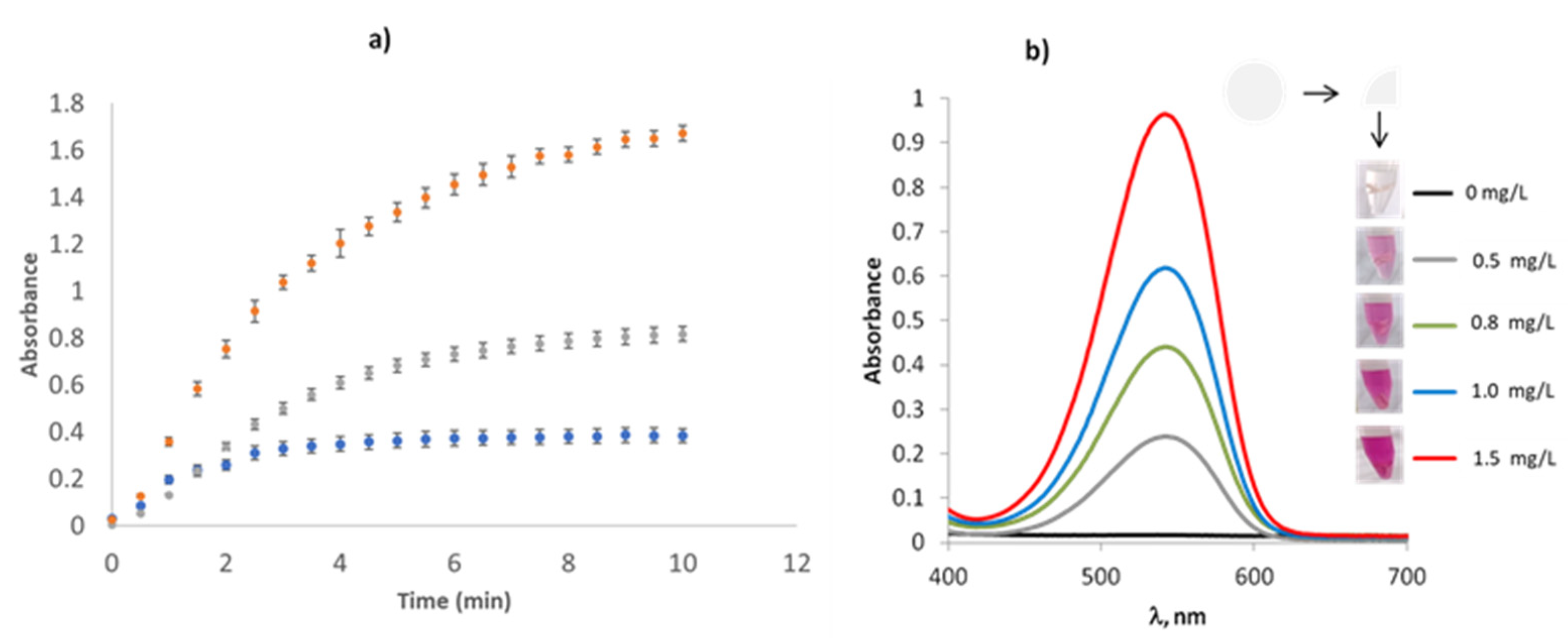
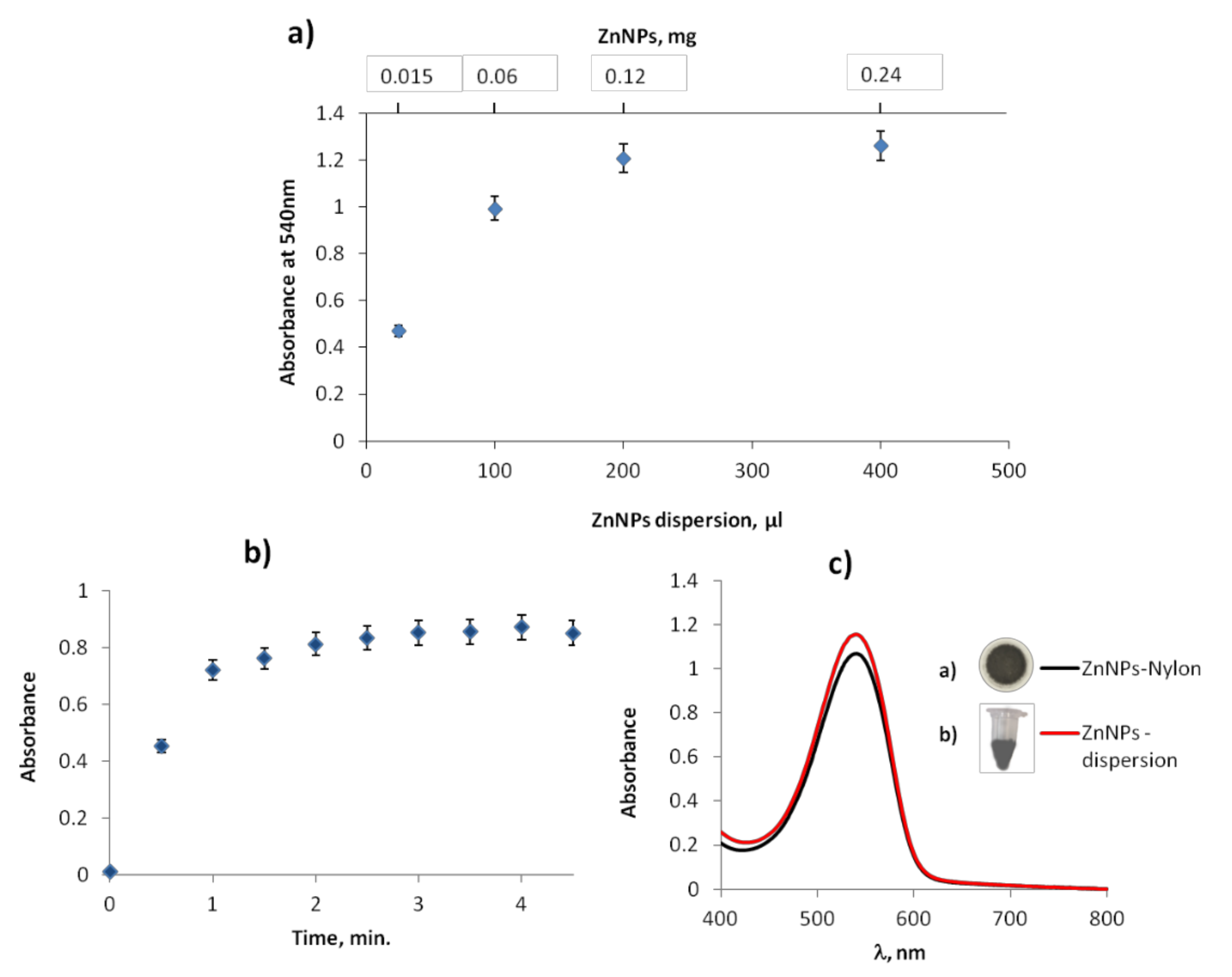
3.2. Study of ZnNPs as Reductor Nitrate Agent
3.3. Transferability to 96 Wells in a Microplate
3.4. Quality Assurance/Quality Control
3.5. Storage Conditions, Stability, and Reusability
3.6. Application to Real Samples
3.7. Comparison with Other Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, M.H.; Jones, R.R.; Brender, J.D.; De Kok, T.M.; Weyer, P.J.; Nolan, B.T.; Villanueva, C.M.; Van Breda, S.G. Drinking Water Nitrate and Human Health: An Updated Review. Int. J. Environ. Res. Public Health 2018, 15, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.; Arcot, J.; Lee, N.A. Nitrate and nitrite quantification from cured meat and vegetables and their estimated dietary intake in Australians. Food Chem. 2009, 115, 334–339. [Google Scholar] [CrossRef]
- DIRECTIVE 98/83/EC. Quality of Water Intended for Human Consumption. In Official Journal of the European Communitie; European Communitie: Brussels, Belgium, 1998.
- EPA Regulation. National Primary Drinking Water Regulations. Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 12 October 2021).
- World Health Organization. Nitrate and Nitrite in Drinking-Water: Background Document for Development of WHO. In Guidelines for Drinking-Water Quality, 4th ed.; World Health Organization: Geneva, Switzerland, 2011; Available online: https://apps.who.int/iris/bitstream/handle/10665/44584/9789241548151_eng.pdf?sequence=1 (accessed on 12 October 2021).
- DIRECTIVE 2006/118/EC. Protection of Groundwater Against Pollution and Deterioration. In Official Journal of the European Union; European Union: Brussels, Belgium, 2006.
- RD 1620/2007 Spanish Regulation, Legal framework for the reuse of treated wastewater. BOE No 294. (2007) 50639–50661. Available online: https://www.iwa-network.org/filemanager-uploads/WQ_Compendium/Database/Selected_guidelines/042_2.pdf (accessed on 12 October 2021).
- COMMISSION REGULATION (EC) No 1881/2006. Setting Maximum Levels Certain Contaminants in Foodstuffs. In Official Journal of the European Union; European Union: Brussels, Belgium, 2006.
- Salehzadeh, H.; Maleki, A.; Rezaee, R.; Shahmoradi, B.; Ponnet, K. The nitrate content of fresh and cooked vegetables and their health-related risks. PLoS ONE 2020, 15, e0227551. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-H.; Yu, L.-J.; Liu, Y.; Lin, L.; Lu, R.-G.; Zhu, J.-P.; He, L.; Lu, Z.-L. Methods for the detection and determination of nitrite and nitrate: A review. Talanta 2017, 165, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, M.K.; Beg, Y.R.; Nishad, G.R. A review on spectroscopic methods for determination of nitrite and nitrate in environmental samples. Talanta 2019, 191, 364–381. [Google Scholar] [CrossRef]
- Griess, P. Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt “Ueber einige Azoverbindungen”. Eur. J. Inorg. Chem. 1879, 12, 426–428. [Google Scholar] [CrossRef] [Green Version]
- Martínez, N.J.; Moliner-Martínez, Y.; Herráez-Hernández, R.; Molins-Legua, C.; Verdu-Andres, J.; Campíns-Falcó, P. Designing solid optical sensors for in situ passive discrimination of volatile amines based on a new one-step hydrophilic PDMS preparation. Sensors Actuators B Chem. 2016, 223, 333–342. [Google Scholar] [CrossRef]
- Bocanegra-Rodríguez, S.; Jornet-Martínez, N.; Molins-Legua, C.; Campíns-Falcó, P. Delivering Inorganic and Organic Reagents and Enzymes from Zein and Developing Optical Sensors. Anal. Chem. 2018, 90, 8501–8508. [Google Scholar] [CrossRef]
- Rattanarat, P.; Dungchai, W.; Cate, D.M.; Siangproh, W.; Volckens, J.; Chailapakul, O.; Henry, C.S. A microfluidic paper-based analytical device for rapid quantification of particulate chromium. Anal. Chim. Acta 2013, 800, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Ortuño, M.; Argente-García, A.; Moliner-Martínez, Y.; Molins-Legua, C.; Campíns-Falcó, P. Polydimethylsiloxane composites containing 1,2-naphtoquinone 4-sulphonate as unique dispositive for estimation of casein in effluents from dairy industries. Anal. Chim. Acta 2015, 873, 31–37. [Google Scholar] [CrossRef]
- Pla-Tolós, J.; Moliner-Martínez, Y.; Molins-Legua, C.; Campíns-Falcó, P. Solid glucose biosensor integrated in a multi-well microplate coupled to a camera-based detector: Application to the multiple analysis of human serum samples. Sensors Actuators B Chem. 2018, 258, 331–341. [Google Scholar] [CrossRef]
- Argente-García, A.; Muñoz-Ortuño, M.; Molins-Legua, C.; Moliner-Martínez, Y.; Campíns-Falcó, P. A solid device based on doped hybrid composites for controlling the dosage of the biocide N-(3-aminopropyl)-N-dodecyl-1,3-propanediamine in industrial formulations. Talanta 2016, 147, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Bhakta Samir, A.; Borba, R.; Taba, M., Jr.; Garcia.C.D.: Carrilho, E. Determination of nitrite in saliva using microfluidic paper-based analytical devices. Analytica Chimica Acta 2014, 809, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballester-Caudet, A.; Hakobyan, L.; Moliner-Martinez, Y.; Molins-Legua, C.; Campíns-Falcó, P. Ionic-liquid doped polymeric composite as passive colorimetric sensor for meat freshness as a use case. Talanta 2021, 223, 121778. [Google Scholar] [CrossRef]
- Han, J.; Tao, W.; Shuo, L.; Zhi-Ping, Z. Fabrication of porous polymer membrane from polysulfone grafted with acid ionic liquid and the catalytic property for inulin hydrolysis. J. Membr. Sci. 2021, 618, 118742. [Google Scholar]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis–Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Wittbrodt, B.T.; Squires, D.A.; Walbeck, J.; Campbell, E.; Campbell, W.; Pearce, J.M. Open-Source Photometric System for Enzymatic Nitrate Quantification. PLoS ONE 2015, 10, e0134989. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.S.; Shabani, A.M.H.; Gentle, B.S.; McKelvie, I.D. Field measurement of nitrate in marine and estuarine waters with a flow analysis system utilizing on-line zinc reduction. Talanta 2011, 84, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hansen, F.; Koroleff, H.P. Determination of nutrients. In Methods Seawater Analysis, 3rd ed.; Grasshoff, K., Ehrhardt, M., Kremling, K., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1999; pp. 159–228. [Google Scholar]
- Madsen, B. Utilization of flow injection with hydrazine reduction and photometric detection for the determination of nitrate in rain-water. Anal. Chim. Acta 1981, 124, 437–441. [Google Scholar] [CrossRef]
- Wang, S.; Lin, K.; Chen, N.; Yuan, D.; Ma, J. Automated determination of nitrate plus nitrite in aqueous samples with flow injection analysis using vanadium (III) chloride as reductant. Talanta 2016, 146, 744–748. [Google Scholar] [CrossRef]
- Lin, K.; Li, P.; Ma, J.; Yuan, D. An automatic reserve flow injection method using vanadium (III) reduction for simultaneous determination of nitrite and nitrate in estuarine and coastal waters. Talanta 2019, 195, 613–618. [Google Scholar] [CrossRef]
- Martinez-Cisneros, C.; da Rocha, Z.; Seabra, A.; Valdés, F.; Alonso-Chamarro, J. Highly integrated autonomous lab-on-a-chip device for on-line and in situ determination of environmental chemical parameters. Lab Chip 2018, 18, 1884–1890. [Google Scholar] [CrossRef]
- Jayawardane, B.M.; Wei, S.; McKelvie, I.; Kolev, S. Microfluidic Paper-Based Analytical Device for the Determination of Nitrite and Nitrate. Anal. Chem. 2014, 86, 7274–7279. [Google Scholar] [CrossRef]
- Martínez-Aviño, A.; Molins-Legua, C.; Pilar, C.-F. Scaling the Analytical Information Given by Several Types of Colorimetric and Spectroscopic Instruments Including Smartphones: Rules for Their Use and Establishing Figures of Merit of Solid Chemosensors. Anal. Chem. 2021, 93, 6043–6052. [Google Scholar] [CrossRef]
- Rice, L.S.; Baird, E.W.; Eaton, R.B.; Clesceri, A.D. Standard Methods for the Examination of Water and Wastewater (Méto-dos Normalizados para Análisis de Aguas Potables y Residuales), 17th ed.; Díaz de Santos: Madrid, Spain, 2012. [Google Scholar]
- Zhou, Y.; Schattka, A.J.H.; Antonietti, M. Room-Temperature Ionic Liquids as Template to Monolithic Mesoporous Silica with Wormlike Pores via a Sol−Gel Nanocasting Technique. Nano Lett. 2004, 4, 477–481. [Google Scholar] [CrossRef]
- Sasikumar, B.; Arthanareeswaran, G.; Ismail, A. Recent progress in ionic liquid membranes for gas separation. J. Mol. Liq. 2018, 266, 330–341. [Google Scholar] [CrossRef]
- Friess, K.; Jansen, J.C.; Bazzarelli, F.; Izák, P.; Jarmarová, V.; Kačírková, M.; Schauer, J.; Clarizia, G.; Bernardo, P. High ionic liquid content polymeric gel membranes: Correlation of membrane structure with gas and vapour transport properties. J. Membr. Sci. 2012, 415-416, 801–809. [Google Scholar] [CrossRef]
- Horowitz, A.I.; Panzer, M.J. Poly(dimethylsiloxane)-Supported Ionogels with a High Ionic Liquid Loading. Angew. Chem. Int. Ed. 2014, 53, 9780–9783. [Google Scholar] [CrossRef] [PubMed]
- Prajapat, G.; Gupta, R.; Bhojak, N. Thermal, Spectroscopic and Antimicrobial Properties of Novel Nickel(II) Complexes with Sulfanilamide and Sulfamerazine Drugs. Chem. Sci. Int. J. 2018, 24, 1–13. [Google Scholar] [CrossRef]
- Bocanegra-Rodríguez, S.; Jornet-Martínez, N.; Molins-Legua, C.; Campíns-Falcó, P. New Reusable Solid Biosensor with Covalent Immobilization of the Horseradish Peroxidase Enzyme: In Situ Liberation Studies of Hydrogen Peroxide by Portable Chemiluminescent Determination. ACS Omega 2020, 5, 2419–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refat, M.S.; Adam, A.M.A.; Sharshar, T.; Saad, H.A.; Eldaroti, H.H. Utility of positron annihilation lifetime technique for the assessment of spectroscopic data of some charge-transfer complexes derived from N-(1-Naphthyl)ethylenediamine dihydrochloride. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2014, 122, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Merino, L. Development and Validation of a Method for Determination of Residual Nitrite/Nitrate in Foodstuffs and Water After Zinc Reduction. Food Anal. Methods 2009, 2, 212–220. [Google Scholar] [CrossRef]
- Murray, E.; Nesterenko, E.P.; McCaul, M.; Morrin, A.; Diamond, D.; Moore, B. A colorimetric method for use within portable test kits for nitrate determination in various water matrices. Anal. Methods 2017, 9, 680–687. [Google Scholar] [CrossRef]
- Hak, C.R.C.; Fatanah, D.N.E.; Abdullah, Y.; Sulaiman, M.Y.M. The Effect of Surfactants on the Stability of TiO2 Aqueous Suspension. Int. J. Curr. Res. Sci. Eng. Technol. 2018, 1, 172–178. [Google Scholar] [CrossRef]
- Mal, A.; Bag, S.; Ghosh, S.; Moulik, S.P. Physicochemistry of CTAB-SDS interacted catanionic micelle-vesicle forming system: An extended exploration. Colloids Surfaces A Physicochem. Eng. Asp. 2018, 553, 633–644. [Google Scholar] [CrossRef]
- Tomašić, V.; Štefanić, I.; Filipović-Vinceković, N. Adsorption, association and precipitation in hexadecyltrimethylammonium bromide/sodium dodecyl sulfate mixtures. Colloid Polym. Sci. 1999, 277, 153–163. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Regulation | ||
|---|---|---|---|
| Drinking water | 50 mg L−1 | 0.5 mgL−1 | Council Directive 98/83/EC of 3 November 1998 EPA—United States Environmental Protection Agency WHO—World Health Organization |
| Groundwater | 50 mg L−1 | - | Directive 2006/118/EC of the European Parliament and of the Council of 12 December 2006 |
| Sewage treatment plant | 25 mg L−1 | - | Spanish regulation, Royal Decree 1620/2007 of 7 December |
| Fresh spinach | 2500–3000 mg kg−1 | - | Commission Regulation (EC) No 1881/2006 of 19 December 2006 |
| Preserved, deep-frozen, or frozen spinach | 2000 mg kg−1 | - | Commission Regulation (EC) No 1881/2006 of 19 December 2006 |
| Linearity (y = a + bx) (mg L−1) | Precision RSD (%) | LOD | |||||
|---|---|---|---|---|---|---|---|
| a ± sa | b ± sb | R2 | Linear Interval (mg·L−1) | Intraday (n = 3) | Interday (n = 3) | (mg·L−1) | |
| NITRITE | |||||||
| (a) Solution | 0.007 ± 0.003 | 0.661 ± 0.004 | 0.99 | 0.02–1.5 | 0.12 | 0.9 | 0.005 |
| (b) Sensor | 0.0035 ± 0.009 | 0.551 ± 0.007 | 0.99 | 0.04–2.7 | 0.4 | 1.6 | 0.01 |
| (b) RGB (green) | 0.062 ± 0.013 | 0.550 ± 0.004 | 0.99 | 0.09–1.3 | 1.1 | 7.2 | 0.03 |
| (b) RGB (green) multiplate | 0.079 ± 0.013 | 0.56 ± 0.08 | 0.99 | 0.02–2.7 | 1.3 | 4.3 | 0.01 |
| NITRATE | |||||||
| (a) Solution | 0.028 ± 0.013 | 0.056 ± 0.001 | 0.99 | 0.3–30 | 0.2 | 4.3 | 0.1 |
| (b) Sensor | 0.034 ± 0.007 | 0.0409 ± 0.005 | 0.99 | 1.6–30 | 0.7 | 5.8 | 0.5 |
| (c) Sensor | 0.031 ± 0.008 | 0.038 ± 0.0004 | 0.99 | 1.6–30 | 0.2 | 6.2 | 0.5 |
| (b) RGB (green) | 0.114 ± 0.013 | 0.052 ± 0.003 | 0.99 | 2.8–25 | 1.1 | 7.2 | 0.8 |
| (b) RGB (green) Microplate | 0.04 ± 0.013 | 0.0532 ± 0.004 | 0.97 | 0.7–20 | 0.8 | 7.0 | 0.2 |
| Samples | Recovery (%) | |||
|---|---|---|---|---|
| Proposed Method | Comparative Method | |||
| Irrigation water | Chanel | 69.2 ± 0.9 | 67.13 ± 0.2 (a) | 97.2 ± 0.2 |
| Well | 21.42 ± 0.05 | 21.6 ± 0.1 (a) | 100.5 ± 0.5 | |
| Well | 2.79 ± 0.01 | ---- | ---- | |
| Canned vegetables | Chard | 1103 ± 60 | 1210 ± 90 (b) | 103.9 ± 0.4 |
| Spinach | 810 ± 40 | - | 107.3 ± 0.2 | |
| Fresh vegetables | Chard | 1600 ± 100 | 1700 ± 70 (b) | 93.0 ± 0.4 |
| Spinach | 990 ± 40 | 980 ± 70 (b) | 104.0 ± 0.5 | |
| Drinking water (*) Tap water (*) | M84 M85 M86 M87 M88 M89 M90 M81 M91 | 10.6 2.8 <LOD 1.8 2.0 2.0 2.2 7.0 6.0 | ||
| River water (*) Lake water (*) | M65 M1 | 74 204 | ||
| Analyte/ Sample | Procedure | Evaluation | Reference | ||||
|---|---|---|---|---|---|---|---|
| Analytical Parameters | Green Points | Economical Points | |||||
| Analytical Time/ Robustez | Figures of Merits LODs/ Dynamic Range | Footprint Kg CO2/100 Samples | Waste | Reagent Consumption Personal Time Instrument | |||
| (water) | Nitrate Reduction— column Zn granules (0.15–0.40 mm) 8 cm 3 min. Derivatization Griess—solution Analytical signal— Absorbance | FIA in lab or in situ (40 samples/h) | 0.006 mg L−1 0.04–0.3 mg L−1 | 0.25 | High amount of waste (Continuous flow) | Reagents: *** Personal: ** Instrument: *** | [24] |
(water and food samples) | Nitrate Reduction—Zn powder (0.1 g/sample)-5 min volume 100 mL Derivatization Griess—10 min Analytical signal—Absorbance | Batch | 3 to 5 mg Kg−1 | 5.99 | High volumes used | Reagents: *** Personal: *** Instrument: ** | [40] |
| (water) | Nitrate Reduction—Zn powder (150 µm) (25 mg/sample) 10 min Derivatization Griess—solution Anaytical signal—Absorbance | Batch | 0.5 mg L−1 0.5–45 mg L−1 | 0.23 | 10 mL sample/1 min reagent | Reagents: ** Personal: *** Instrument: ** | [41] |
| (a)(b) (Synthetic, tap, pond, and mineral water) | Inkjet printing with AKD Zn suspension prepared by mixing 500 mg of Zn dust (<10 µm); 1 mg/sample (75 s) Derivatization Griess—µPAD paper support (3–7 min) Analytical signal—scanned imagens processed | In situ Microfluidic Sensor Stable for 30 days Stored in vacuum at ≤−20 °C) | (a) 0.04/(b) 1.2 mg L−1 0.5–6.9 mg L−1 (a) 3.1–62 mg L−1 (b) | 9.45 In the fabrication of the sensor/scanner | Low consumption of reagents (µL) | Reagents: * Personal: * Instrument: ** | [30] |
| (a)/(b) | Nitrate Reduction—Dispersion of ZnNPs (0.12 mg/sample) 3 min Derivatization Griess—5 min, solution Analytical signal—processed imagens (RGB) or Absorbance | In situ Reagents supported in PDMS Stable for more than 6 moths | (a) 0.01/(b) 0.5 mg L−1 0.04–2.5 mg L−1 (a) 1.6–30 mg L−1 (b) | 0.028 | Low consumption of reagents (µL) | Reagents: * Personal: * Instrument: * | Proposed method |
| Reference methods | |||||||
| Electrode Nitrate 10 mL sample 10 mL of buffer (Al3SO4)3 17 mg; Ag2SO4 34.3 mg; H3BO4 18.6 mg, H2SO3H 25.4 mg/per sample) Analytical signal—Electrochemical | In situ in the lab Interferences: Cl−, CO3−2, NO2−, CN−, S2−, Br−, I−, ClO3−, ClO4− | 0.6 mg L−1 0.6–620 mg L−1 | 1.8 × 10−4 | High volumes | Reagent: ** Time/sample: 3 min Instrumentation: *** | [32] | |
| Absorbance measurement at 220 nm and 275 nm Analytical signal—Absorbance | In situ (probes) In the lab (batch or FIA) Interferences: soaps, NO2−, Cr6+ | 0.1 mg L−1 0.3 a 30 mg L−1 | 0.025 | Reagent: Time/sample: 1 min Instrumentation: *** | [32] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakobyan, L.; Monforte-Gómez, B.; Moliner-Martínez, Y.; Molins-Legua, C.; Campíns-Falcó, P. Improving Sustainability of the Griess Reaction by Reagent Stabilization on PDMS Membranes and ZnNPs as Reductor of Nitrates: Application to Different Water Samples. Polymers 2022, 14, 464. https://doi.org/10.3390/polym14030464
Hakobyan L, Monforte-Gómez B, Moliner-Martínez Y, Molins-Legua C, Campíns-Falcó P. Improving Sustainability of the Griess Reaction by Reagent Stabilization on PDMS Membranes and ZnNPs as Reductor of Nitrates: Application to Different Water Samples. Polymers. 2022; 14(3):464. https://doi.org/10.3390/polym14030464
Chicago/Turabian StyleHakobyan, Lusine, Belén Monforte-Gómez, Yolanda Moliner-Martínez, Carmen Molins-Legua, and Pilar Campíns-Falcó. 2022. "Improving Sustainability of the Griess Reaction by Reagent Stabilization on PDMS Membranes and ZnNPs as Reductor of Nitrates: Application to Different Water Samples" Polymers 14, no. 3: 464. https://doi.org/10.3390/polym14030464
APA StyleHakobyan, L., Monforte-Gómez, B., Moliner-Martínez, Y., Molins-Legua, C., & Campíns-Falcó, P. (2022). Improving Sustainability of the Griess Reaction by Reagent Stabilization on PDMS Membranes and ZnNPs as Reductor of Nitrates: Application to Different Water Samples. Polymers, 14(3), 464. https://doi.org/10.3390/polym14030464