
Development and Validation of HPLC-DAD Method with Pre-Column PMP Derivatization for Monomeric Profile Analysis of Polysaccharides from Agro-Industrial Wastes



Abstract
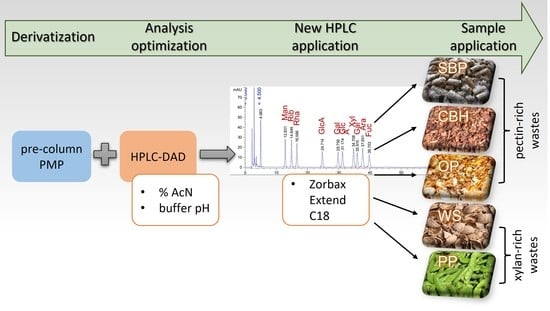
:
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Chemicals
2.3. Preparation of Alcohol-Insoluble Residues
2.4. Complete Acid Hydrolysis
2.5. Determination of Monomeric Composition
2.5.1. Determination of Neutral Sugars by GC-FID
2.5.2. Colorimetric Determination of Uronic Acids
2.5.3. Simultaneous Determination of Neutral and Acidic Sugars by HPLC-DAD
Derivatization Using 3-Methyl-1-Phenyl-2-Pyrazoline-5-One
Optimization of HPLC Mobile-Phase Composition
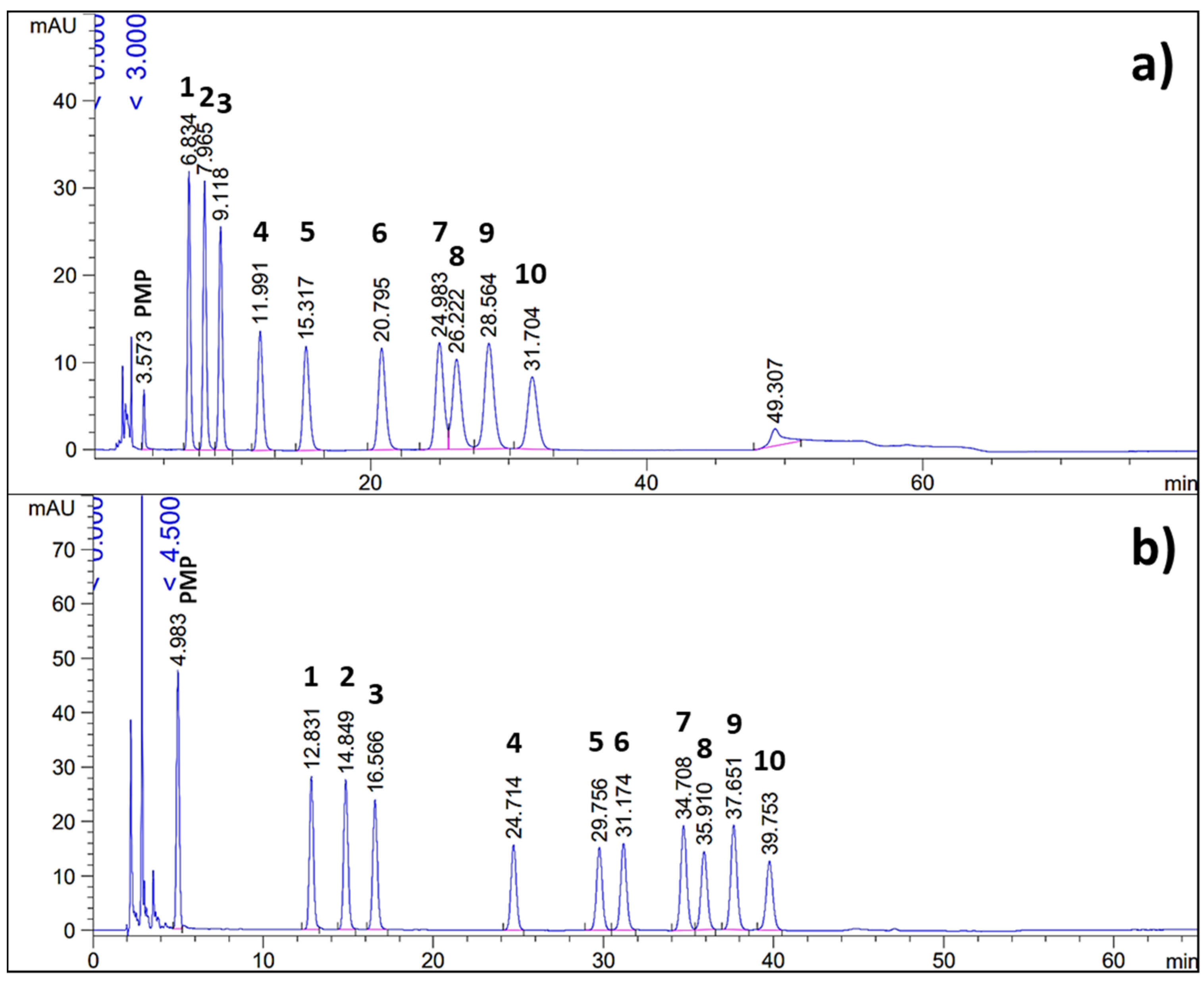
HPLC-DAD Analysis under Optimized Conditions
Validation of Optimized HPLC-DAD Method
2.6. Statistical Analysis
3. Results and Discussion
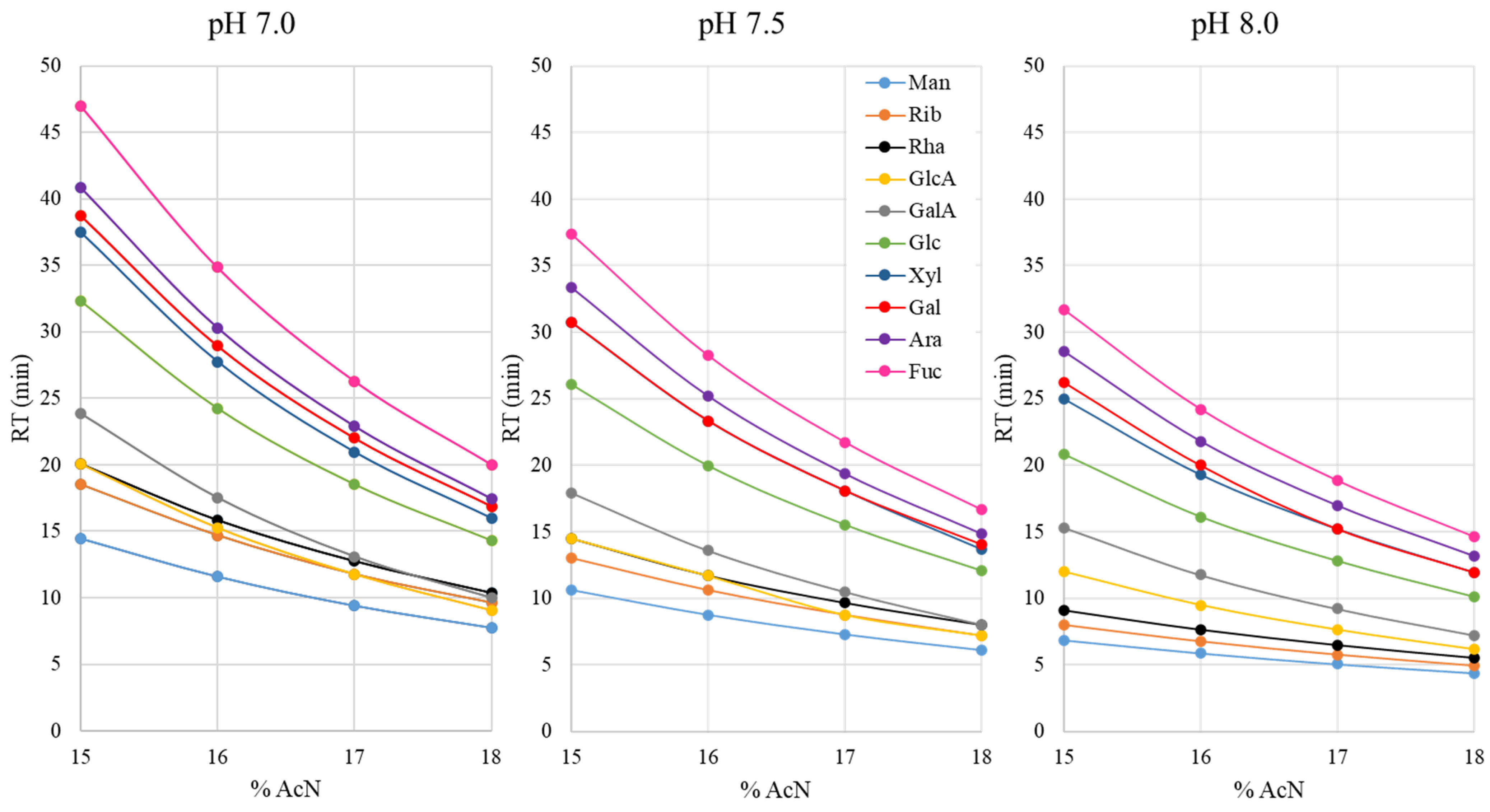
3.1. Influence of the Mobile Phase Composition
3.2. Validation of the Developed HPLC-DAD Method
3.3. Comparative Analysis of the Monomeric Composition of Cell Wall Polysaccharides of Selected Agro-Industrial Wastes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- BeMiller, J.N. Polysaccharides: Occurrence, structures and chemistry. In Carbohydrate Chemistry for Food Scientists, 3rd ed.; AACC International: Saint Paul, MN, USA, 2018; pp. 75–101. [Google Scholar]
- Yang, X.; Li, A.; Li, X.; Sun, L.; Guo, Y. An overview of classifications, properties of food polysaccharides and their links to applications in improving food textures. Trends Food Sci. Technol. 2020, 102, 1–15. [Google Scholar] [CrossRef]
- Liu, D.; Tang, W.; Yin, J.-Y.; Nie, S.-P.; Xie, M.-Y. Monosaccharide composition analysis of polysaccharides from natural sources: Hydrolysis condition and detection method development. Food Hydrocoll. 2021, 116, 106641. [Google Scholar] [CrossRef]
- Harvey, D.J. Derivatization of carbohydrates for analysis by chromatography, electrophoresis and mass spectrometry. J. Chromatogr. B 2011, 879, 1196–1225. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Suzuki, S.; Taga, A. Analysis of carbohydrates as 1-phenyl-3-methyl-5-pyrazolone derivatives by capillary/microchip electrophoresis and capillary electrochromatography. J. Pharm. Biomed. Anal. 2003, 30, 1689–1714. [Google Scholar] [CrossRef]
- Corradini, C. Liquid Chromatography. In Encyclopedia of Separation Science, Level III: Practical Applications–Carbohydrates; Wilson, I.D., Cooke, M., Poole, C.F., Eds.; Academic Press: Cambridge, MA, USA, 2000; pp. 2224–2235. [Google Scholar]
- Jie, L.; Yuan, Z.; Yu, Z.; Xue-Song, F. Progress in the pretreatment and analysis of carbohydrates in food: An update since 2013. J. Chromatogr. A 2021, 1655, 462496. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wan, Y.; Li, O.; Zhang, X.; Xie, M.; Nie, S.; Yin, J. Two-step hydrolysis method for monosaccharide composition analysis of natural polysaccharides rich in uronic acids. Food Hydrocoll. 2020, 101, 105524. [Google Scholar] [CrossRef]
- Nagel, A.; Sirisakulwat, S.; Carle, R.; Neidhart, S. An acetate–hydroxide gradient for the quantitation of the neutral sugar and uronic acid profile of pectins by HPAEC-PAD without postcolumn pH adjustment. J. Agric. Food Chem. 2020, 62, 2037–2048. [Google Scholar] [CrossRef]
- Lamari, F.N.; Kuhn, R.; Karamanos, N.K. Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometry structure analysis. J. Chromatogr. B 2003, 793, 15–36. [Google Scholar] [CrossRef]
- Ruiz-Matute, A.I.; Hernández-Hernández, O.; Rodríguez-Sánchez, S.; Sanz, M.; Martínez-Castro, I. Derivatization of carbohydrates for GC and GC-MS analyses. J. Chromatogr. B 2011, 879, 1226–1240. [Google Scholar] [CrossRef]
- Bailly, C.; Hecquet, P.-E.; Kouach, M.; Thuru, X.; Goossens, J.-F. Chemical reactivity and uses of 1-phenyl-3-methyl-5-pyrazolone (PMP), also known as edaravone. Bioorgan. Med. Chem. 2020, 28, 115463. [Google Scholar] [CrossRef]
- Honda, S.; Akao, E.; Suzuki, S.; Okuda, M.; Kakehi, K.; Nakamura, J. High-performance liquid chromatography of reducing carbohydrates as strong ultraviolet-absorbing and electrochemically sensitive 1-phenyl-3-methyl-5-pyrazolone derivatives. Anal. Biochem. 1989, 180, 351–357. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, P.; Wang, Z.; Huang, L. An innovative derivatization method for simultaneous determination of uronic acids and neutral and amino sugars in coexisting samples by HPLC-ESI-MS/MS2. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 1754–1771. [Google Scholar] [CrossRef]
- Wang, W.; Chen, F.; Wang, Y.; Wang, L.; Fu, H.; Zheng, F.; Beecher, L. Optimization of reactions between reducing sugars and 1-phenyl-3-methyl-5-pyrazolone (PMP) by response surface methodology. Food Chem. 2018, 254, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wu, Y.; Chen, S.W.; Zhu, S.; Yin, H.P.; Wang, M.; Tang, J. Sugar compositional determination of polysaccharides from Dunalliela salina by modified RP-HPLC method of precolumn derivatization with 1-phenyl-3-methyl-5-pyrazolone. Carbohydr. Polym. 2010, 82, 629–635. [Google Scholar] [CrossRef]
- Lv, Y.; Yang, X.; Zhao, Y.; Ruan, Y.; Yang, Y.; Wang, Z. Separation and quantification of component monosaccharides of the tea polysaccharides from Gynostemma pentaphyllum by HPLC with indirect UV detection. Food Chem. 2009, 112, 742–746. [Google Scholar] [CrossRef]
- Li, H.; Long, C.; Zhou, J.; Liu, J.; Wu, X.; Long, M. Rapid analysis of monosaccharides and oligosaccharides in hydrolysates of lignocellulosic biomass by HPLC. Biotechnol. Lett. 2013, 35, 1405–1409. [Google Scholar] [CrossRef]
- Shen, X.; Perreault, H. Characterization of carbohydrates using a combination of derivatization, high-performance liquid chromatography and mass spectrometry. J. Chromatogr. A 1998, 811, 47–59. [Google Scholar] [CrossRef]
- Hu, D.-J.; Cheong, K.-L.; Zhao, J.; Li, S.-P. Chromatography in characterization of polysaccharides from medicinal plants and fungi. J. Sep. Sci. 2013, 36, 1–19. [Google Scholar] [CrossRef]
- Poli, A.; Anzelmo, G.; Fiorentino, G.; Nicolaus, B.; Tommonaro, G.; Di Donato, P. Polysaccharides from wastes of vegetable industrial processing: New opportunities for their eco-friendly re-use. In Biotechnology of Biopolymers; Elnashar, M., Ed.; InTechOpen: London, UK, 2011; Available online: https://www.intechopen.com/chapters/16196 (accessed on 3 October 2021). [CrossRef] [Green Version]
- Reichembach, L.H.; Petkovicz, C.L.O. Pectins from alternative sources and uses beyond sweets and jellies: An overview. Food Hydrocoll. 2021, 118, 106824. [Google Scholar] [CrossRef]
- Santibáñez, L.; Henríquez, C.; Corro-Tejeda, R.; Bernal, S.; Armijo, B.; Salazar, O. Xylooligosaccharides from lignocellulosic biomass: A comprehensive review. Carbohydr. Polym. 2021, 251, 117118. [Google Scholar] [CrossRef]
- Rao, L.V.; Goli, J.K.; Gentela, J.; Koti, S. Bioconversion of lignocellulosic biomass to xylitol: An overview. Bioresour. Technol. 2016, 2013, 299–310. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Li, J.; Yan, L.; Li, S.; Ye, X.; Liu, D.; Ding, T.; Linhardt, R.J.; Orfila, C.; et al. Extraction and characterization of RG-I enriched pectic polysaccharides from mandarin citrus peel. Food Hydrocoll. 2018, 79, 579–586. [Google Scholar] [CrossRef]
- Zhang, F.; Ran, C.; Zheng, J.; Ding, Y.; Chen, G. Polysaccharides obtained from bamboo shoots (Chimonobambusa quadrangularis) processing by-products: New insight into ethanol precipitation and characterization. Int. J. Biol. Macromol. 2018, 112, 951–960. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kishimura, H.; Kinoshita, Y.; Saburi, W.; Kumagai, Y.; Yasui, H.; Ojima, T. Enzymatic production of xylooligosaccharides from red algae dulse (Palmaria sp.) wasted in Japan. Process Biochem. 2019, 82, 117–122. [Google Scholar] [CrossRef]
- Wu, S.; Lan, Y.; Wu, Z.; Peng, Y.; Chen, S.; Huang, Z.; Xu, L.; Gelbič, I.; Guan, X.; Zhang, L.; et al. Pretreatment of spent mushroom substrate for enhancing the conversion of fermentable sugar. Bioresour. Technol. 2013, 148, 596–600. [Google Scholar] [CrossRef]
- Wu, H.; Li, H.; Xue, Y.; Luo, G.; Gan, L.; Liu, J.; Mao, L.; Long, M. High efficiency co-production of ferulic acid and xylooligosaccharides from wheat bran by recombinant xylanase and feruloyl esterase. Biochem. Eng. J. 2017, 120, 41–48. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Chen, F.; Zheng, F. Comparison of determination of sugar-PMP derivatives by two different stationary phases and two HPLC detectors: C18 vs. amide columns and DAD vs. ELSD. J. Food Compos. Anal. 2021, 96, 103715. [Google Scholar] [CrossRef]
- Englyst, H.N.; Cummings, J.H. Improved method of measurement of dietary fiber as non-starch polysaccharides in plant foods. J. Assoc. Off. Anal. Chem. 1988, 71, 808–814. [Google Scholar] [CrossRef]
- Hoebler, C.; Barry, J.L.; David, A.; Delort-Laval, J. Rapid acid hydrolysis of plant cell wall polysaccharides and simplified quantitative determination of their neutral monosaccharides by gas-liquid chromatography. J. Agric. Food Chem. 1989, 37, 360–367. [Google Scholar] [CrossRef]
- Blakeney, A.B.; Rarris, P.J.; Henry, R.J.; Stone, B.A. A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydr. Res. 1983, 113, 291–299. [Google Scholar] [CrossRef]
- Blumenkrantz, N.; Asboe-Hansen, G. New method for quantitative determination of uronic acids. Anal. Biochem. 1973, 54, 484–489. [Google Scholar] [CrossRef]
- Ahmed, A.E.R.; Labavitch, M. A simplified method for accurate determination of cell wall uronide content. J. Food Biochem. 1977, 1, 361–365. [Google Scholar] [CrossRef]
- Thibault, J.-F. Automatisation du dosage des substances pectiques par la méthode au méta-hydroxydiphenyl. LWT-Food Sci. Technol. 1979, 12, 247–251. [Google Scholar]
- Filisetti-Cozzi, T.M.; Carpita, N.C. Measurement of uronic acids without interference from neutral sugars. Anal. Biochem. 1991, 197, 157–162. [Google Scholar] [CrossRef]
- Sun, X.; Wang, H.; Han, X.; Chen, S.; Zhu, S.; Dai, J. Fingerprint analysis of polysaccharides from different Ganoderma by HPLC combined with chemometrics methods. Carbohydr. Polym. 2014, 114, 432–439. [Google Scholar] [CrossRef]
- Guillon, F.; Auffret, A.; Robertson, J.A.; Thibault, J.-F.; Barry, J.-L. Relationships between physical characteristics of sugar beet fibre and its fermentability by human faecal flora. Carbohydr. Polym. 1998, 37, 185–197. [Google Scholar] [CrossRef]
- Kühnel, S.; Schols, H.A.; Gruppen, H. Aiming for the complete utilization of sugar-beet pulp: Examination of the effects of mild acid and hydrothermal pretreatment followed by enzymatic digestion. Biotechnol. Biofuels 2011, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Bonvehí, J.S.; Benería, M.A. Composition of dietary fibre in cocoa husk. Zeitschrift für Lebensmitteluntersuchung und-Forschung A 1998, 207, 105–109. [Google Scholar] [CrossRef]
- Sinner, M.; Puls, J.; Dietrichs, H. Carbohydrate composition of nut shells and some other agricultural residues. Starch 1979, 31, 267–269. [Google Scholar] [CrossRef]
- Altun, T.; Pehlivan, E. Removal of Cr(VI) from aqueous solutions by modified walnut shells. Food Chem. 2012, 132, 693–700. [Google Scholar] [CrossRef]
- Mateos-Aparicio, I.; Redondo-Cuenca, A.; Villanueva-Suárez, M.-J.; Zapata-Revilla, M.-A.; Tenorio-Sanz, M.-D. Pea pod, broad bean pod and okara, potential sources of functional compounds. LWT–Food Sci. Technol. 2010, 43, 1467–1470. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Monosaccharide (PMP–Sugar Derivative) | Linear Equation | R2 | Concentration Range (µg/mL) | LOD | LOQ |
|---|---|---|---|---|---|
| (µg/mL) | |||||
| Mannose | y = 0.9037x + 0.0031 | 0.9999 | 10–400 (0.025–1) * | 1.65 | 5.01 |
| Rhamnose | y = 0.8679x + 0.0005 | 0.9999 | 10–400 (0.025–1) * | 1.17 | 3.55 |
| Glucuronic acid | y = 0.8068x − 0.0015 | 0.9996 | 10–400 (0.025–1) * | 6.05 | 18.32 |
| Galacturonic acid | y = 0.8275x − 0.0009 | 0.9996 | 10–400 (0.025–1) * | 3.04 | 9.21 |
| Glucose | y = 0.8535x − 0.0031 | 0.9998 | 10–400 (0.025–1) * | 3.79 | 11.47 |
| Xylose | y = 1.0185x − 0.0029 | 0.9998 | 10–400 (0.025–1) * | 2.78 | 8.43 |
| Galactose | y = 0.9861x − 0.0065 | 0.9998 | 10–400 (0.025–1) * | 4.52 | 13.71 |
| Arabinose | y = 1.1865x − 0.0049 | 0.9998 | 10–400 (0.025–1) * | 3.85 | 11.67 |
| Fucose | y = 0.873x − 0.0095 | 0.9995 | 10–400 (0.025–1) * | 4.83 | 14.64 |
| Method Precision (%RSD) | |||||
|---|---|---|---|---|---|
| Repeatability (Intraday Precision) | Intermediate Precision (Interday Precision) | ||||
| RT | A | RT | A | ||
| Mannose | Absolute | 0.06–0.22 | 0.09–0.36 | 2.72–2.91 | 2.72–3.10 |
| Relative | 0.01–0.03 | 0.02–0.32 | 0.11–0.13 | 0.18–1.63 | |
| Ribose | Absolute | 0.07–0.21 | 0.07–0.45 | 2.84–3.02 | 3.02–4.26 |
| Relative | - | - | - | - | |
| Rhamnose | Absolute | 0.09–0.36 | 0.09–0.55 | 2.58–2.76 | 3.33–3.81 |
| Relative | 0.01–0.03 | 0.09–0.55 | 0.26–0.26 | 0.34–0.97 | |
| Glucuronic acid | Absolute | 0.09–0.31 | 0.04–0.50 | 0.69–0.89 | 3.50–4.04 |
| Relative | 0.02–0.22 | 0.02–0.44 | 2.10–2.17 | 0.32–0.66 | |
| Galacturonic acid | Absolute | 0.08–0.43 | 0.11–1.05 | 0.30–0.45 | 2.66–4.70 |
| Relative | 0.02–0.31 | 0.08–0.88 | 2.54–2.60 | 0.45–1.04 | |
| Glucose | Absolute | 0.08–0.19 | 0.06–1.32 | 0.85–1.00 | 3.23–4.54 |
| Relative | 0.02–0.25 | 0.08–0.93 | 1.97–2.01 | 0.58–0.68 | |
| Xylose | Absolute | 0.07–0.22 | 0.06–0.73 | 0.66–0.80 | 2.57–4.73 |
| Relative | 0.02–0.26 | 0.04–0.32 | 2.16–2.22 | 0.48–0.68 | |
| Galactose | Absolute | 0.07–0.15 | 0.02–1.15 | 0.38–0.51 | 1.60–5.82 |
| Relative | 0.02–0.26 | 0.04–0.72 | 2.45–2.50 | 1.23–1.65 | |
| Arabinose | Absolute | 0.07–0.15 | 0.04–1.00 | 0.35–0.47 | 2.84–5.39 |
| Relative | 0.03–0.25 | 0.14–0.59 | 2.49–2.54 | 0.79–1.25 | |
| Fucose | Absolute | 0.06–0.16 | 0.03–1.27 | 0.35–0.48 | 2.58–6.09 |
| Relative | 0.03–0.25 | 0.14–0.85 | 2.48–2.53 | 0.50–1.91 | |
| Monosaccharide (PMP–Sugar Derivative | Recovery (%) at 25/100 µg/mL | RSD(%) | Recovery (%) at 50/200 µg/mL | RSD(%) | Recovery (%) at 75/300 µg/mL | RSD(%) |
|---|---|---|---|---|---|---|
| Mannose | 102.06 | 1.13 | 106.44 | 0.24 | 104.95 | 0.28 |
| Rhamnose | 101.66 | 1.29 | 107.40 | 0.25 | 109.61 | 0.43 |
| Glucuronic acid | 99.15 | 0.17 | 104.42 | 1.01 | 103.57 | 0.95 |
| Galacturonic acid | 103.99 | 0.89 | 107.64 | 0.70 | 107.34 | 0.76 |
| Glucose | 99.40 | 0.00 | 103.30 | 0.33 | 100.97 | 0.29 |
| Xylose | 105.51 | 1.09 | 107.15 | 0.08 | 104.19 | 0.29 |
| Galactose | 99.95 | 2.44 | 101.43 | 0.01 | 100.53 | 0.47 |
| Arabinose | 101.32 | 1.62 | 108.92 | 0.10 | 108.73 | 0.41 |
| Fucose | 94.76 | 1.72 | 104.23 | 2.69 | 103.56 | 0.94 |
| Monomer(% dmb) | SBP | WS | CBH | OP | PP | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| HPLC | GC + MHDP | HPLC | GC + MHDP | HPLC | GC + MHDP | HPLC | GC + MHDP | HPLC | GC + MHDP | |
| Man | 1.16 ± 0.02 a | 1.19 ± 0.04 a | n.d. | n.d. | 2.40 ± 0.00 a | 2.69 ± 0.06 a | 1.21 ± 0.05 | 1.33 ± 0.03 | 0.40 ± 0.00 | 0.64 ± 0.06 |
| Rha | 1.97 ± 0.06 | 1.26 ± 0.09 | 0.46 ± 0.03 | 0.41 ± 0.02 | 2.07 ± 0.02 | 1.33 ± 0.04 | 0.86 ± 0.05 | 0.58 ± 0.01 | 0.52 ± 0.00 a | 0.49 ± 0.03 a |
| GlcA | n.d. | n.d. | 2.10 ± 0.09 | n.d. | 1.23 ± 0.02 a | 1.33 ± 0.15 a | n.d. | n.d. | 0.58 ± 0.04 | n.d. |
| GalA | 15.29 ± 0.19 | 20.52 ± 0.54 | 1.96 ± 0.33 | 5.07 ± 0.14 | 7.95 ± 0.15 | 13.21 ± 0.26 | 19.74 ± 0.68 | 24.16 ± 0.56 | 4.24 ± 0.10 | 9.49 ± 0.40 |
| Glc | 21.85 ± 1.10 a | 20.85 ± 1.29 a | 25.09 ± 0.53 a | 22.97 ± 1.18 a | 16.13 ± 0.11 | 17.99 ± 0.58 | 25.30 ± 0.51 | 23.60 ± 0.64 | 19.41 ± 0.42 a | 20.92 ± 1.07 a |
| Xyl | 1.21 ± 0.06 | 1.57 ± 0.01 | 19.96 ± 0.73 a | 22.39 ± 0.68 a | 1.34 ± 0.02 | 1.80 ± 0.04 | 1.64 ± 0.03 | 2.06 ± 0.04 | 12.92 ± 0.05 a | 12.71 ± 0.86 a |
| Gal | 5.82 ± 0.35 | 5.24 ± 0.13 | 1.16 ± 0.23 | 1.08 ± 0.01 | 3.10 ± 0.00 | 3.54 ± 0.11 | 2.29 ± 0.19 a | 2.37 ± 0.12 a | 2.43 ± 0.33 a | 3.06 ± 0.19 a |
| Ara | 17.76 ± 0.78 a | 17.95 ± 0.58 a | 0.39 ± 0.07 | 0.54 ± 0.02 | 1.22 ± 0.00 | 1.62 ± 0.05 | 0.61 ± 0.03 | 0.76 ± 0.03 | 0.88 ± 0.17 | 1.39 ± 0.12 |
| Fuc | 0.15 ± 0.02 a | 0.18 ± 0.01 a | n.d. | n.d. | 0.16 ± 0.00 | 0.18 ± 0.01 | 0.24 ± 0.00 a | 0.25 ± 0.01 a | n.d. | 0.13 ± 0.01 |
| TOTAL | 65.22 ± 2.15 | 68.76 ± 1.64 | 52.76 ± 4.21 | 52.76 ± 1.73 | 35.59 ± 0.02 | 43.71 ± 0.96 | 51.76 ± 0.58 | 55.10 ± 0.85 | 40.73 ± 0.15 | 48.82 ± 1.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojvodić Cebin, A.; Komes, D.; Ralet, M.-C. Development and Validation of HPLC-DAD Method with Pre-Column PMP Derivatization for Monomeric Profile Analysis of Polysaccharides from Agro-Industrial Wastes. Polymers 2022, 14, 544. https://doi.org/10.3390/polym14030544
Vojvodić Cebin A, Komes D, Ralet M-C. Development and Validation of HPLC-DAD Method with Pre-Column PMP Derivatization for Monomeric Profile Analysis of Polysaccharides from Agro-Industrial Wastes. Polymers. 2022; 14(3):544. https://doi.org/10.3390/polym14030544
Chicago/Turabian StyleVojvodić Cebin, Aleksandra, Draženka Komes, and Marie-Christine Ralet. 2022. "Development and Validation of HPLC-DAD Method with Pre-Column PMP Derivatization for Monomeric Profile Analysis of Polysaccharides from Agro-Industrial Wastes" Polymers 14, no. 3: 544. https://doi.org/10.3390/polym14030544
APA StyleVojvodić Cebin, A., Komes, D., & Ralet, M.-C. (2022). Development and Validation of HPLC-DAD Method with Pre-Column PMP Derivatization for Monomeric Profile Analysis of Polysaccharides from Agro-Industrial Wastes. Polymers, 14(3), 544. https://doi.org/10.3390/polym14030544