
Features of the Chemical Composition and Structure of Birch Phloem Dioxane Lignin: A Comprehensive Study

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. SEM Study
2.3. Isolation of Dioxane Lignin
2.4. NMR Spectroscopy
2.5. Analytical Pyrolysis
2.6. Atmospheric Pressure Photoionization High-Resolution Orbitrap Mass Spectrometry
3. Results and Discussion
3.1. Dioxane Lignin Monomeric Units Identified by Py-GC/MS
3.2. Chemical Composition and Structure of Lignin Analyzed by 2D-NMR
3.3. Molecular Level Characterization by High-Resolution Mass Spectrometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; et al. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Sun, R.C.; Sun, Y.C.; Wen, J.L.; Xu, F. Fractional and structural characterization of organosolv and alkaline lignins from Tamarix austromogoliac. Sci. Res. Essays 2010, 5, 3850–3864. [Google Scholar]
- Lahive, C.W.; Kamer, P.C.J.; Lancefield, C.S.; Deuss, P.J. An Introduction to Model Compounds of Lignin Linking Motifs; Synthesis and Selection Considerations for Reactivity Studies. ChemSusChem 2020, 13, 4238–4265. [Google Scholar] [CrossRef] [PubMed]
- Neiva, D.M.; Rencoret, J.; Marques, G.; Gutiérrez, A.; Gominho, J.; Pereira, H.; del Río, J.C. Lignin from Tree Barks: Chemical Structure and Valorization. ChemSusChem 2020, 13, 4537–4547. [Google Scholar] [CrossRef]
- Branco, D.G.; Santiago, C.; Lourenço, A.; Cabrita, L.; Evtuguin, D.V. Structural Features of Cork Dioxane Lignin from Quercus suber L. J. Agric. Food Chem. 2021, 69, 8555–8564. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.A.; Miranda, I.; Gominho, J.; Pereira, H. Chemical characterization of cork and phloem from Douglas fir outer bark. Holzforschung 2016, 70, 475–483. [Google Scholar] [CrossRef]
- Lourenço, A.; Rencoret, J.; Chematova, C.; Gominho, J.; Gutiérrez, A.; del Río, J.C.; Pereira, H. Lignin composition and structure differs between xylem, phloem and phellem in Quercus suber L. Front Plant Sci. 2016, 7, 1612. [Google Scholar] [CrossRef] [Green Version]
- Dou, J.; Kim, H.; Li, Y.; Padmakshan, D.; Yue, F.; Ralph, J.; Vuorinen, T. Structural characterization of lignins from willow bark and wood. J. Agric. Food Chem. 2018, 66, 7294–7300. [Google Scholar] [CrossRef]
- Kuznetsova, S.A.; Skvortsova, G.P.; Maliar, I.N.; Skurydina, E.S.; Veselova, O.F. Extraction of betulin from birch bark and study of its physico-chemical and pharmacological properties. Russ. J. Bioorgan. Chem. 2014, 40, 742–747. [Google Scholar] [CrossRef]
- Heinämäki, J.; Pirttimaa, M.M.; Alakurtti, S.; Pitkänen, H.P.; Kanerva, H.; Hulkko, J.; Paaver, U.; Aruväli, J.; Yliruusi, J.; Kogermann, K. Suberin Fatty Acids from Outer Birch Bark: Isolation and Physical Material Characterization. J. Nat. Prod. 2017, 80, 916–924. [Google Scholar] [CrossRef]
- Fagerstedt, K.V.; Saranpää, P.; Tapanila, T.; Immanen, J.; Serra, J.A.A.; Nieminen, K. Determining the composition of lignins in different tissues of silver birch. Plants 2015, 4, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Faleva, A.V.; Belesov, A.V.; Kozhevnikov, A.Y.; Falev, D.I.; Chukhchin, D.G.; Novozhilov, E.V. Analysis of the functional group composition of the spruce and birch phloem lignin. Int. J. Biol. Macromol. 2021, 166, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure, Reactions; Walter de Gruyter: Berlin, Germany, 1984. [Google Scholar]
- del Río, J.C.; Rencoret, J.; Gutierrez, A.; Nieto, L.; Jimenez-Barbero, J.; Martinez, A.T. Structural Characterization of Guaiacyl-rich Lignins in Flax (Linum usitatissimum) Fibers and Shives. J. Agric. Food Chem. 2011, 59, 11088–11099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikovskoi, I.I.; Kosyakov, D.S.; Faleva, A.V.; Shavrina, I.S.; Kozhevnikov, A.Y.; Ul’yanovskii, N.V. Study of the sedge (Cárex) lignin by high-resolution mass spectrometry and NMR spectroscopy. Russ. Chem. Bull. 2020, 69, 2004–2012. [Google Scholar] [CrossRef]
- Evtuguin, D.V.; Neto, C.P.; Silva, A.M.S.; Domingues, P.M.; Amado, F.M.L.; Robert, D.; Faix, O. Comprehensive Study on the Chemical Structure of Dioxane Lignin from Plantation Eucalyptus globulus Wood. J. Agric. Food Chem. 2001, 49, 4252–4261. [Google Scholar] [CrossRef] [PubMed]
- Ralph, J.; Landucci, L.L. NMR of Lignins. In Lignin and Lignans: Advances in Chemistry, 2nd ed.; Heitner, C., Dimmel, D.R., Schmidt, J.A., Eds.; CRC Press (Taylor & Francis Group): Boca Raton, FL, USA, 2010; pp. 137–234. [Google Scholar]
- Faleva, A.V.; Kozhevnikov, A.Y.; Pokryshkin, S.A.; Falev, D.I.; Shestakov, S.L.; Popova, J.A. Structural characteristics of different softwood lignins according to 1D and 2D NMR spectroscopy. J. Wood Chem. Technol. 2020, 40, 178–189. [Google Scholar] [CrossRef]
- Kosyakov, D.S.; Ul’yanovskii, N.V.; Anikeenko, E.A.; Gorbova, N.S. Negative ion mode atmospheric pressure ionization methods in lignin mass spectrometry: A comparative study. Rapid Commun. Mass Spectrom. 2016, 30, 2099–2108. [Google Scholar] [CrossRef]
- Kosyakov, D.S.; Pikovskoi, I.I.; Ul’yanovskii, N.V. Dopant-assisted atmospheric pressure photoionization Orbitrap mass spectrometry—An approach to molecular characterization of lignin oligomers. Anal. Chim. Acta 2021, 1179, 338836. [Google Scholar] [CrossRef]
- Kosyakov, D.S.; Ipatova, E.V.; Krutov, S.M.; Ul’yanovskii, N.V.; Pikovskoi, I.I. Study of Products of the Alkaline Decomposition of Hydrolysis Lignin by Atmospheric Pressure Photoionization High-Resolution Mass Spectrometry. J. Anal. Chem. 2017, 72, 1396–1403. [Google Scholar] [CrossRef]
- Ivakhnov, A.D.; Shavrina, I.S.; Kosyakov, D.S.; Pikovskoi, I.I.; Dumanskii, I.O. Depolymerization of Alkaline Lignin in the Medium of Supercritical 2-Propanol. Russ. J. Appl. Chem. 2020, 93, 99–107. [Google Scholar] [CrossRef]
- Pikovskoi, I.I.; Kosyakov, D.S.; Shavrina, I.S.; Ul’yanovskii, N.V. Study of Nettle (Urtica dióica) Lignin by Atmospheric Pressure Photoionization Orbitrap Mass Spectrometry. J. Anal. Chem. 2019, 74, 1412–1420. [Google Scholar] [CrossRef]
- Obolenskaya, A.V.; Elnitskaya, Z.P.; Leonovich, A.A. Laboratory Work on the Chemistry of Wood and Cellulose: Textbook for High Schools; Ecology: Moscow, Russia, 1991. (In Russian) [Google Scholar]
- Geles, I.S. Woody Biomass and the Basis of Environmentally Acceptable Technologies for Its Chemical-Mechanical Processing; Karelian Scientific Center of the Russian Academy of Sciences: Petrozavodsk, Russia, 2001; p. 382. (In Russian) [Google Scholar]
- Adler, E. Lignin chemistry—Past, present and future. Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar] [CrossRef]
- Pepper, J.M.; Baylis, P.E.T.; Adler, E. The isolation and properties of lignins obtained by the acidolysis of spruce and aspen woods in dioxane-water medium. Can. J. Chem. 1959, 37, 1241–1248. [Google Scholar] [CrossRef]
- Zakis, G.F. The Functional Analysis of Lignins and Their Derivatives; Tappi Press: Atlanta, GA, USA, 1994; 102p. [Google Scholar]
- Ralph, S.A.; Ralph, J.; Landucci, L. NMR Database of Lignin and Cell Wall Model Compounds; US Forest Products Laboratory: Madison, WI, USA, 2004; Available online: https://www.glbrc.org/databases_and_software/nmrdatabase/NMR_DataBase_2009_Complete.pdf (accessed on 1 December 2021).
- Zikeli, F.; Vinciguerra, V.; D’Annibale, A.; Capitani, D.; Romagnoli, M.; Scarascia Mugnozza, G. Preparation of Lignin Nanoparticles from Wood Waste for Wood Surface Treatment. Nanomaterials 2019, 9, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rencoret, J.; Kim, H.; Evaristo, A.B.; Gutiérrez, A.; Ralph, J.; del Río, J.C. Variability in Lignin Composition and Structure in Cell Walls of Different Parts of Macaúba (Acrocomia aculeata) Palm Fruit. J. Agric. Food Chem. 2017, 66, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Miles-Barrett, D.M.; Neal, A.R.; Hand, C.; Montgomery, J.R.D.; Panovic, I.; Ojo, O.S.; Lancefield, C.S.; Cordes, D.B.; Slawin, A.M.Z.; Lebl, T.; et al. The synthesis and analysis of lignin-bound Hibbert ketone structures in technical lignins. Org. Biomol. Chem. 2016, 14, 10023–10030. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Gellerstedt, G. Quantitative 2D HSQC NMR determination of polymer structures by selecting suitable internal standard references. Magn. Reson. Chem. 2007, 45, 37–45. [Google Scholar] [CrossRef]
- Pikovskoi, I.I.; Ul’yanovskii, N.V.; Gorbova, N.S.; Kosyakov, D.S. Study of Lignin by Atmospheric Pressure Photoionization Orbitrap Mass Spectrometry: Effect of Spectral Resolution. J. Anal. Chem. 2021, 76, 1610–1617. [Google Scholar] [CrossRef]
- Jiang, G.; Nowakowski, D.J.; Bridgwater, A.V. Effect of the temperature on the composition of lignin pyrolysis products. Energy Fuel 2010, 24, 4470–4475. [Google Scholar] [CrossRef]
- Lourenco, A.; Neiva, D.M.; Gominho, J.; Marques, A.V.; Pereira, H. Characterization of lignin in heartwood, sapwood and bark from Tectona grandis using Py–GC–MS/FID. Wood Sci. Technol. 2015, 49, 159–175. [Google Scholar] [CrossRef]
- Stojakowska, A.; Malarz, J. Bioactive phenolics from in vitro cultures of Lactuca aculeata Boiss. et Kotschy. Phytochem. Lett. 2017, 19, 7–11. [Google Scholar] [CrossRef]
- Prozil, S.O.; Evtuguin, D.V.; Silva, A.M.S.; Lopes, L.P.C. Structural Characterization of Lignin from Grape Stalks (Vitis vinifera L.). J. Agric. Food Chem. 2014, 62, 5420–5428. [Google Scholar] [CrossRef] [PubMed]
- Liimatainen, J.; Karonen, M.; Sinkkonen, J.; Helander, M.; Salminen, J.-P. Characterization of phenolic compounds from inner bark of Betula pendula. Holzforschung 2012, 66, 171–181. [Google Scholar] [CrossRef]
- Ralph, J.; MacKay, J.J.; Hatfield, R.D.; O’Malley, D.M.; Whetten, R.W.; Sederoff, R.R. Abnormal lignin in a loblolly pine mutant. Science 1997, 277, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyy, V.A.; Karmanov, A.P.; Kocheva, L.S.; Nekrasova, P.S.; Kaneva, M.V.; Lobov, A.N.; Spirikhin, L.V. Comparative study of chemical and topological structure of macromolecules of lignins of birch (Betula verrucosa) and apple (Malus domestica) wood. Int. J. Biol. Macromol. 2019, 128, 40–48. [Google Scholar] [CrossRef] [PubMed]
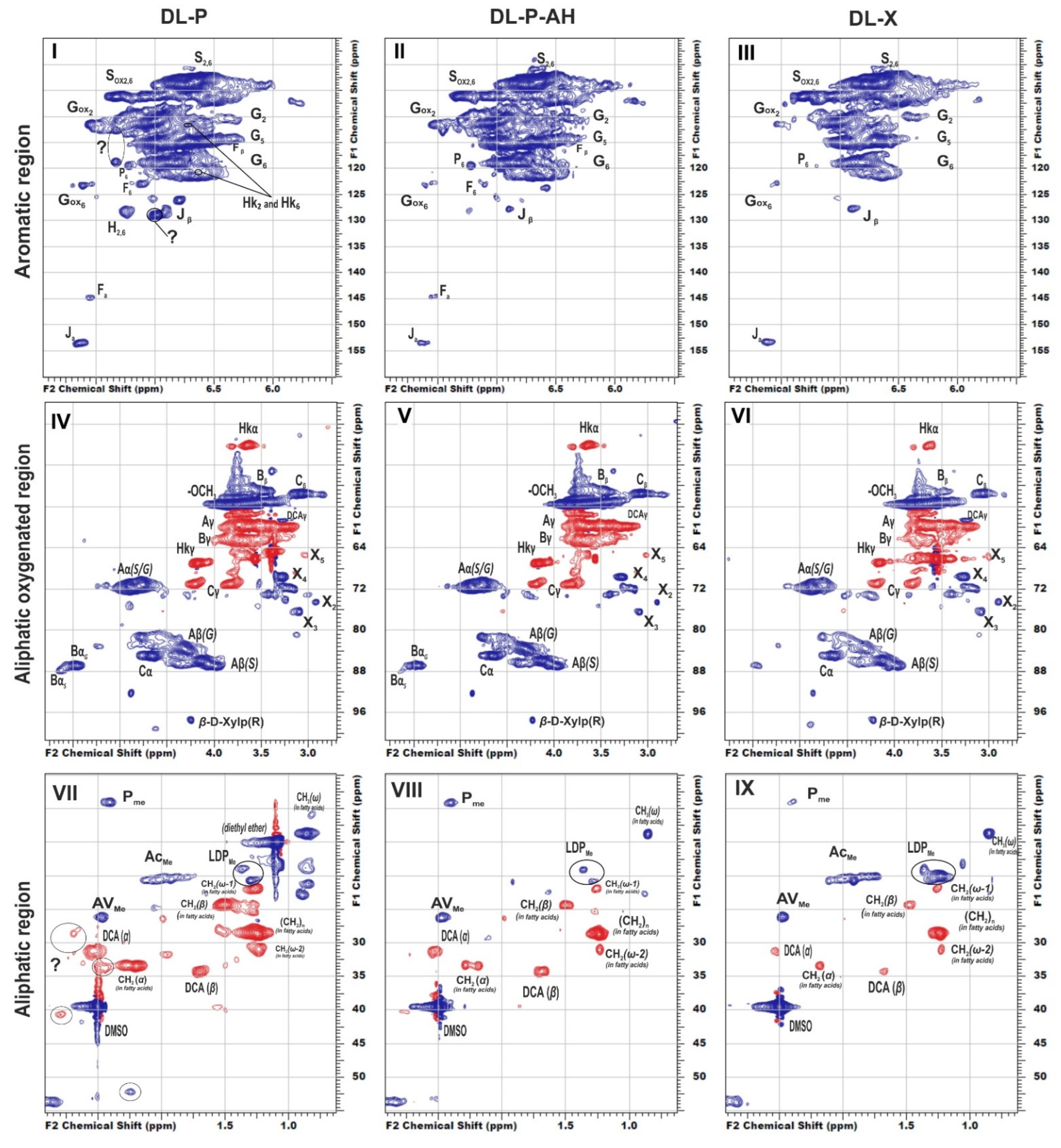



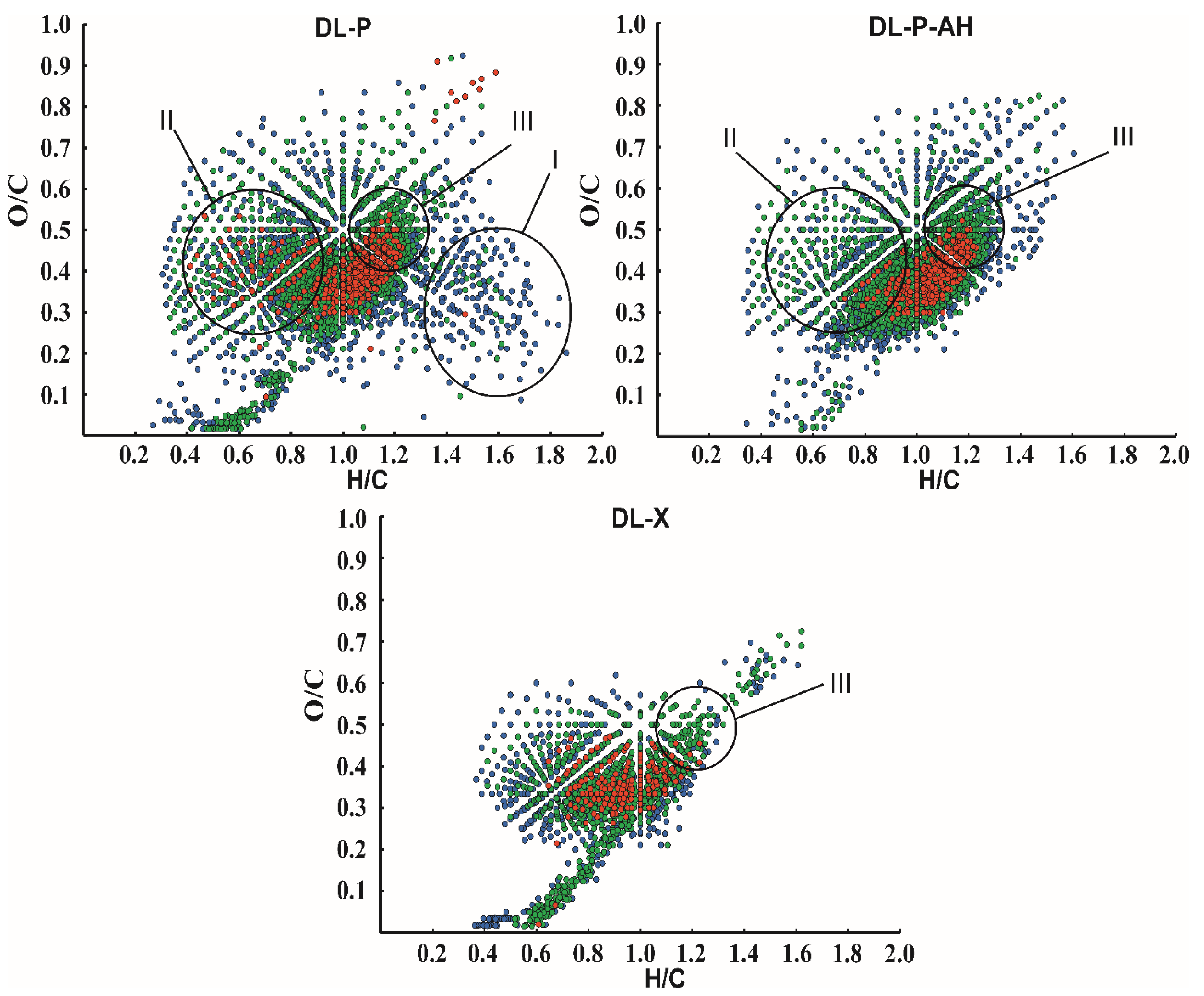

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Content, % (Recalculated to Dry Material) | |
|---|---|---|
| Experimental Data | Literature Data [25] | |
| Total extractives | 16.7 | 14.6 |
| Klason lignin after Soxhlet extraction | 28.5 | - |
| Substances extracted with 1% NaOH | 21.2 | 23.8 |
| Klason lignin after Soxhlet and 1% NaOH extractions | 22.5 | 19.5 |
| Parameter | Lignin Sample | ||
|---|---|---|---|
| DL-P | DL-P-AH | DL-X | |
| Mw, Da | 4350 | 4400 | 4400 |
| Mn, Da | 2200 | 1500 | 1700 |
| PDI | 1.97 | 2.93 | 2.59 |
| OHphen, % | 4.4 | 3.8 | 2.8 |
| OHaliph, % | 7.9 | 7.8 | 7.4 |
| COOH, % | 0.33 | 0.33 | 0.18 |
| Structure Type | DL-P | DL-P-AH | DL-X |
|---|---|---|---|
| S | 38 | 40 | 63 |
| G | 52 | 54 | 33 |
| H | 3 | 2 | 1 |
| C | 6 | 4 | 3 |
| S/G | 0.73 | 0.74 | 1.91 |
| Structure | DL-P | DL-P-AH | DL-X |
|---|---|---|---|
| β-O-4′ (β-aryl ether), including: | 35.9 | 34.3 | 42.5 |
| between G- and S-type units, AS | 20.2 | 19.1 | 27.8 |
| between G-type units; AG) | 10.5 | 10.1 | 8.0 |
| α-O-4 (Phenylcoumarane) | 4.7 | 4.3 | 1.4 |
| α-O-γ’ (Resinol) | 8.1 | 7.4 | 7.3 |
| S | 43.5 | 46.1 | 73.3 |
| G | 54.5 | 53.9 | 26.7 |
| H | 2.0 | 0.0 | 0.0 |
| S/G | 0.87 | 0.86 | 2.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faleva, A.V.; Pikovskoi, I.I.; Pokryshkin, S.A.; Chukhchin, D.G.; Kosyakov, D.S. Features of the Chemical Composition and Structure of Birch Phloem Dioxane Lignin: A Comprehensive Study. Polymers 2022, 14, 964. https://doi.org/10.3390/polym14050964
Faleva AV, Pikovskoi II, Pokryshkin SA, Chukhchin DG, Kosyakov DS. Features of the Chemical Composition and Structure of Birch Phloem Dioxane Lignin: A Comprehensive Study. Polymers. 2022; 14(5):964. https://doi.org/10.3390/polym14050964
Chicago/Turabian StyleFaleva, Anna V., Ilya I. Pikovskoi, Sergey A. Pokryshkin, Dmitry G. Chukhchin, and Dmitry S. Kosyakov. 2022. "Features of the Chemical Composition and Structure of Birch Phloem Dioxane Lignin: A Comprehensive Study" Polymers 14, no. 5: 964. https://doi.org/10.3390/polym14050964
APA StyleFaleva, A. V., Pikovskoi, I. I., Pokryshkin, S. A., Chukhchin, D. G., & Kosyakov, D. S. (2022). Features of the Chemical Composition and Structure of Birch Phloem Dioxane Lignin: A Comprehensive Study. Polymers, 14(5), 964. https://doi.org/10.3390/polym14050964