
A New Approach to The Synthesis of Polylactide/Polyacrylonitrile Block Copolymers


Abstract
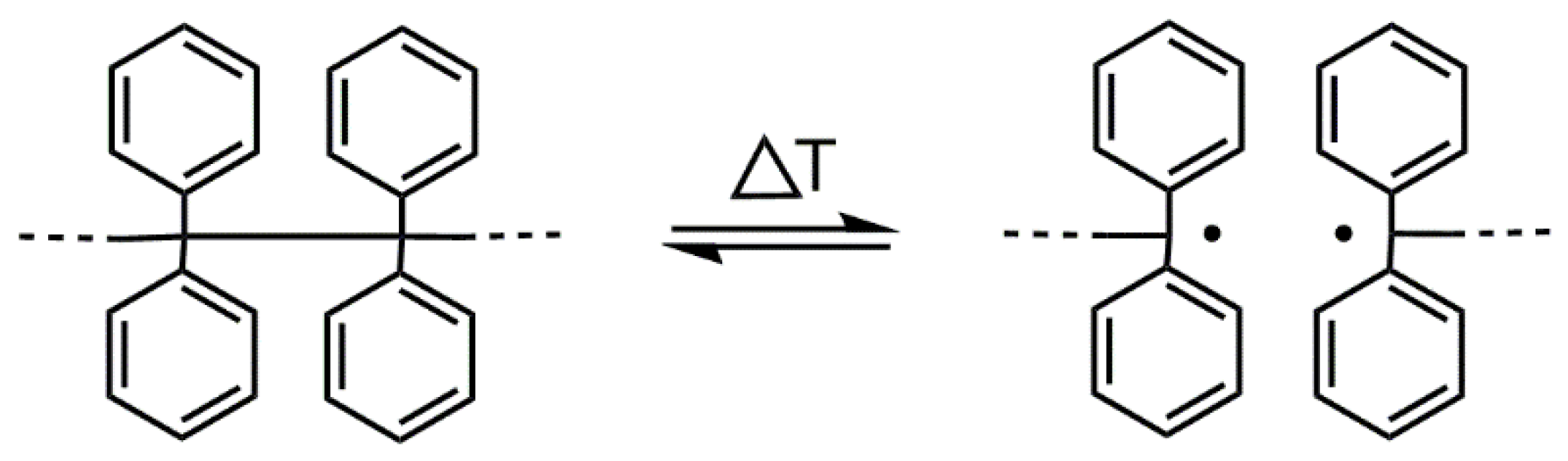
:1. Introduction
2. Materials and Methods
2.1. Materials
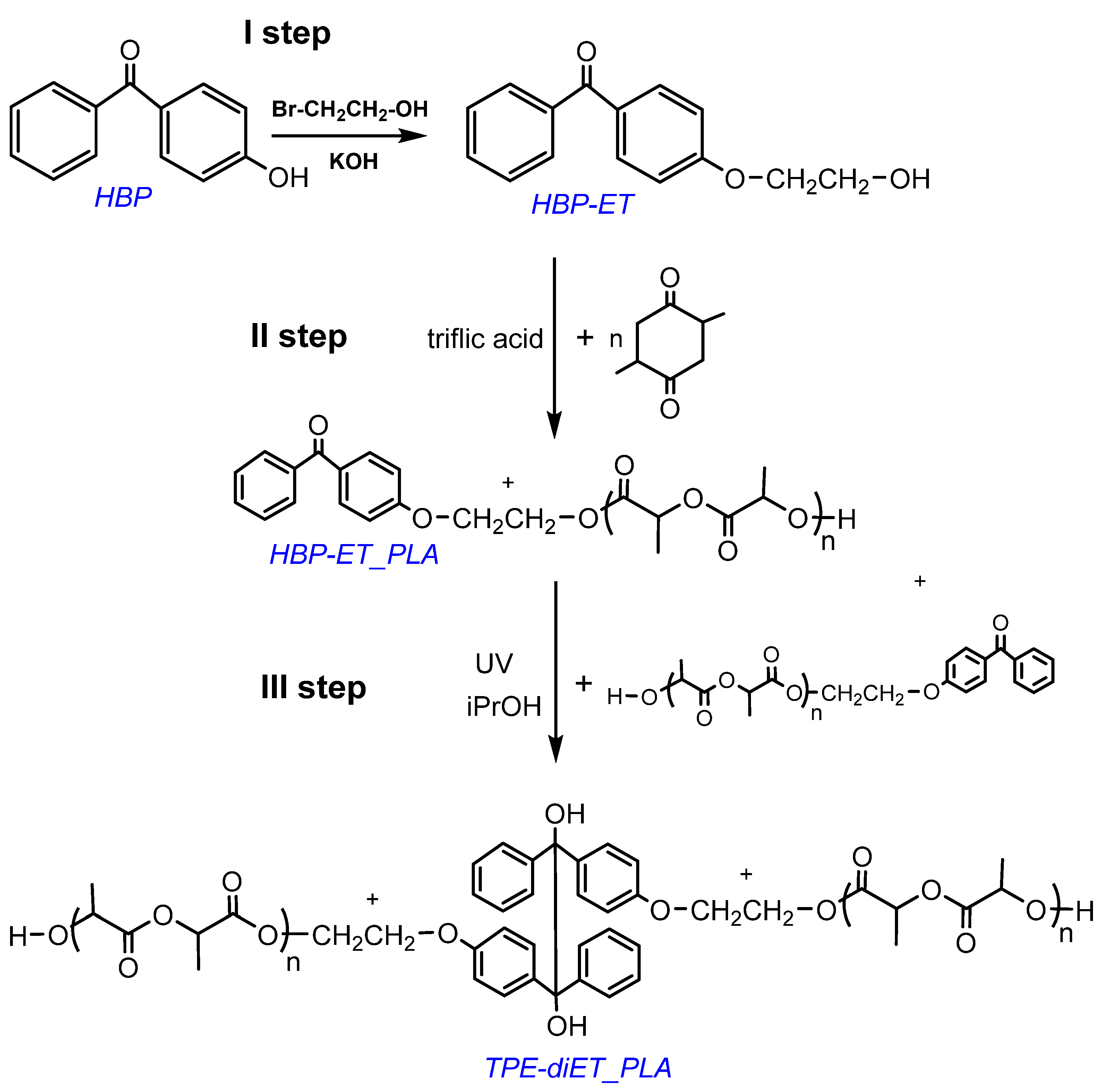
2.2. Synthesis of the HBP Derivative Containing Primary Hydroxyl Groups (HBP-ET)
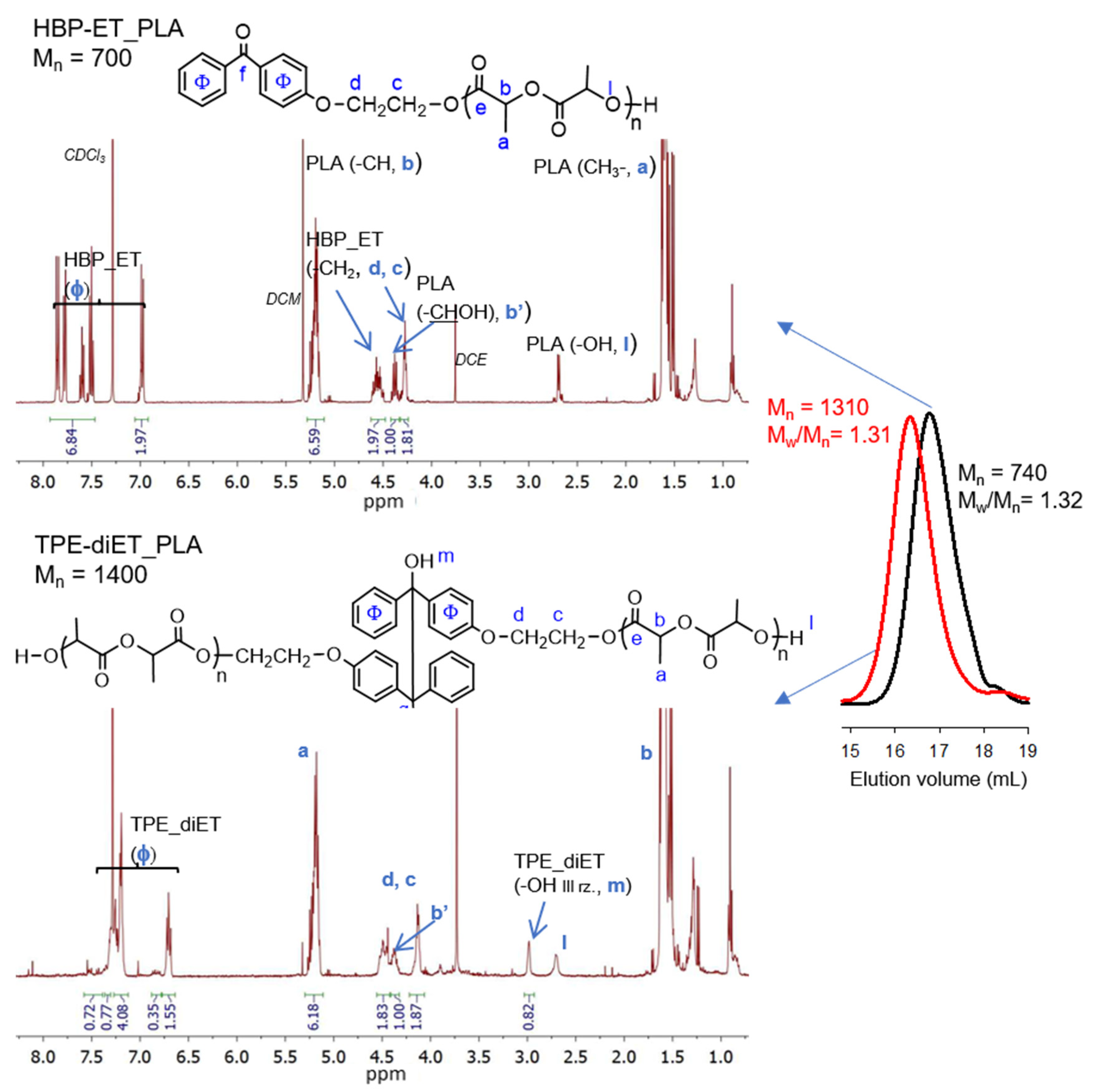
2.3. Synthesis of PLA with HBP-ET Units at One Chain End
2.4. Synthesis of PLA with TPE Units in the Middle of the Chain (TPE-diET_PLA)
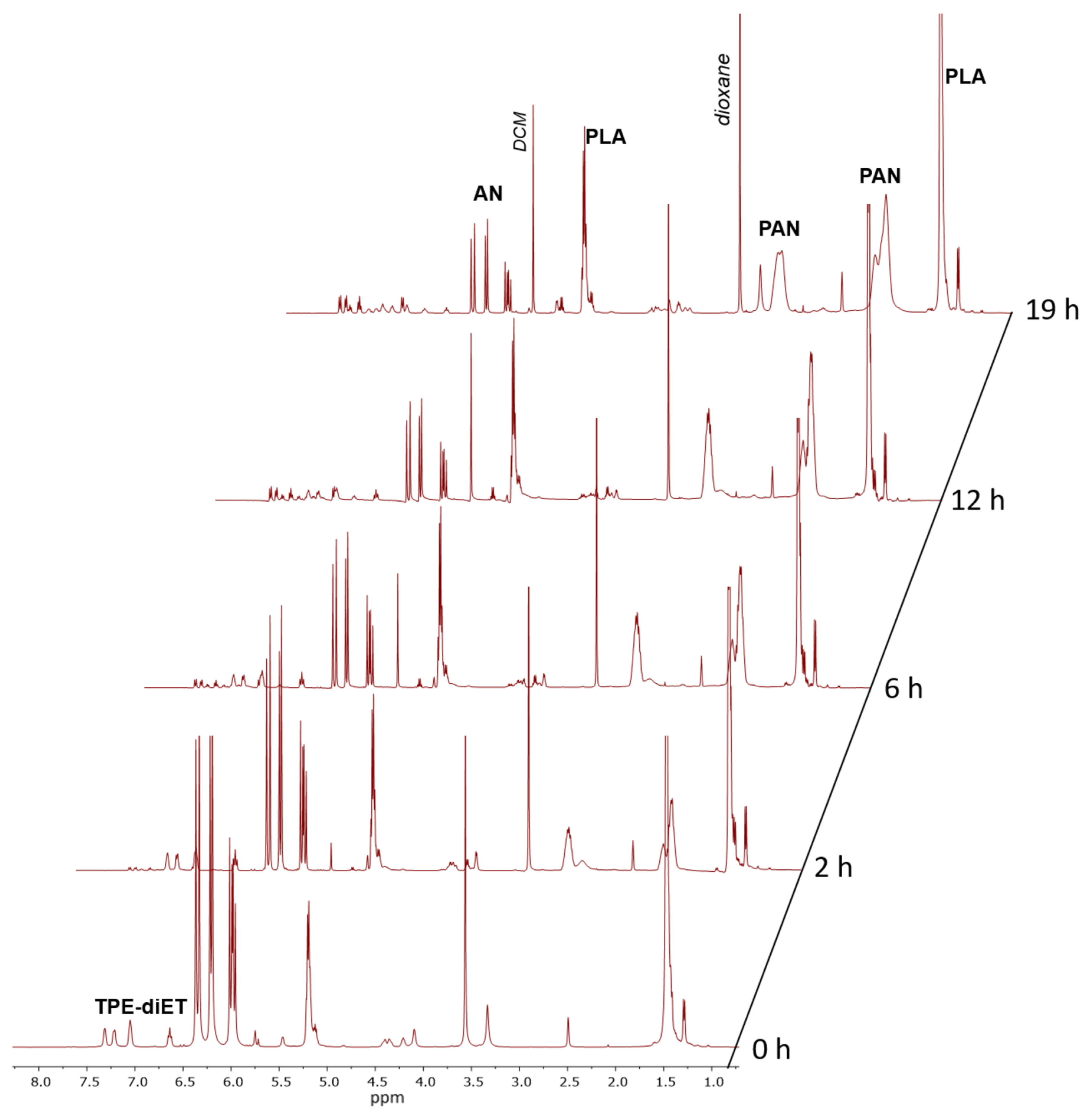
2.5. Polymerization of AN Using TPE-diET_PLA as a Macroiniferter
2.6. Instrumental Methods
3. Results and Discussion
3.1. Synthesis of a PLA Containing TPE Group in the Middle of the Chain
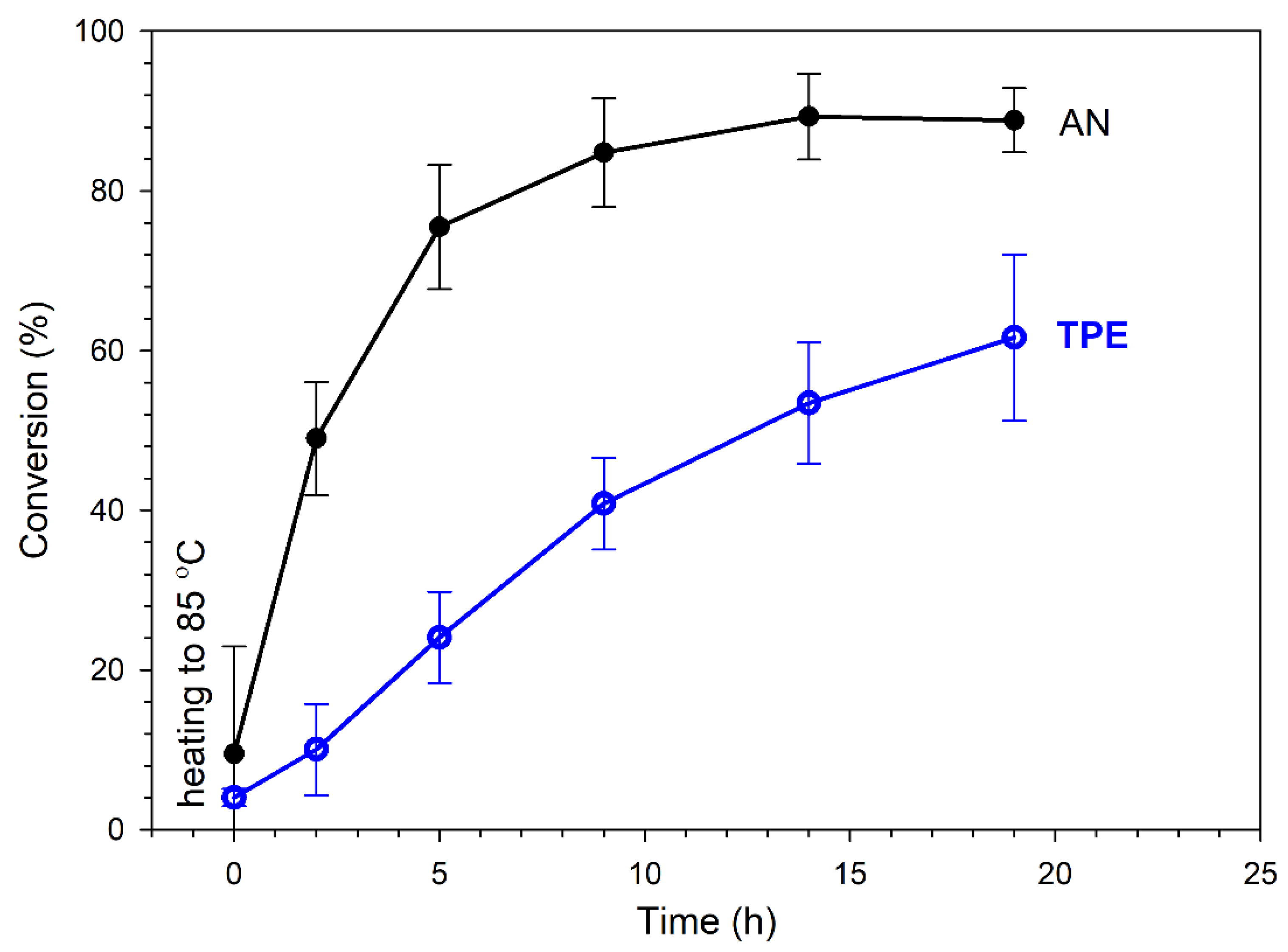
3.2. Polymerization of Acrylonitrile Initiated by TPE-diET_PLA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, V.; Mohanty, A.K.; Misra, M. Perspective on Polylactic Acid (PLA) based Sustainable Materials for Durable Applications: Focus on Toughness and Heat Resistance. ACS Sustain. Chem. Eng. 2016, 4, 2899–2916. [Google Scholar] [CrossRef]
- Pang, X.; Zhuang, X.; Tang, Z.; Chen, X. Polylactic acid (PLA): Research, development and industrialization. Biotechnol. J. 2010, 5, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K. Polylactide (PLA)-based amphiphilic block copolymers: Synthesis, self-assembly, and biomedical applications. Soft Matter 2011, 7, 5096–5108. [Google Scholar] [CrossRef] [Green Version]
- Madhavan Nampoothiri, K.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef] [PubMed]
- Maharana, T.; Pattanaik, S.; Routaray, A.; Nath, N.; Sutar, A.K. Synthesis and characterization of poly(lactic acid) based graft copolymers. React. Funct. Polym. 2015, 93, 47–67. [Google Scholar] [CrossRef]
- Puthumana, M.; Santhana Gopala Krishnan, P.; Nayak, S.K. Chemical modifications of PLA through copolymerization. Int. J. Polym. Anal. Charact. 2020, 25, 634–648. [Google Scholar] [CrossRef]
- Becker, J.M.; Pounder, R.J.; Dove, A.P. Synthesis of poly(lactide)s with modified thermal and mechanical properties. Macromol. Rapid Commun. 2010, 31, 1923–1937. [Google Scholar] [CrossRef]
- Yildirim, I.; Weber, C.; Schubert, U.S. Old meets new: Combination of PLA and RDRP to obtain sophisticated macromolecular architectures. Prog. Polym. Sci. 2018, 76, 111–150. [Google Scholar] [CrossRef]
- Otsu, T. Iniferter concept and living radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2121–2136. [Google Scholar] [CrossRef]
- Otsu, T.; Matsumoto, A.; Tazaki, T. Radical polymerization of methyl methacrylate with some 1,2-disubstituted tetraphenylethanes as thermal iniferters. Polym. Bull. 1987, 17, 323–330. [Google Scholar] [CrossRef]
- Yao, X.Q.; Hou, X.J.; Wu, G.S.; Xu, Y.Y.; Xiang, H.W.; Jiao, H.; Li, Y.W. Estimation of C-C bond dissociation enthalpies of large aromatic hydrocarbon compounds using DFT methods. J. Phys. Chem. A 2002, 106, 7184–7189. [Google Scholar] [CrossRef]
- Braun, D.; Becker, K.H. Aromatische Pinakole als Polymerisationsinitiatoren. Die Angew. Makromol. Chemie 1969, 6, 186–189. [Google Scholar] [CrossRef]
- Braun, D.; Becker, K.H. Kinetik der Polymerisationsauslösung mit aromatischen pinakolen. Die Makromol. Chemie 1971, 147, 91–99. [Google Scholar] [CrossRef]
- Btedzki, A.; Braun, D.; Menzel, W.; Titzschkau, K. Polymerizationsauslosung mit subsstituierten Ethanen. Makromol. Chemie 1983, 294, 287–294. [Google Scholar] [CrossRef]
- Błedzki, A.; Braun, D.; Titzschkau, K. Polymerisationsauslösung mit substituierten ethanen, 6. Polymerisation von methylmethacrylat mit verschiedenen tetraphenylethanen. Die Makromol. Chem. 1983, 184, 745–754. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. A Novel Polyurethane Macroinitiator for Free Radical Polymerization. Eur. Polym. J. 1994, 30, 1351–1355. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. Tetraphenylethane Iniferters. II. Toluene Diisocyanate-Based Polyurethane Iniferter for “living Radical Polymerization of Acrylonitrile. J. Polym. Sci. Part A Polym. Chem. 1996, 34, 1723–1731. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. Tetraphenylethane iniferters. 3. “living” radical polymerization of methyl methacrylate using toluene-diisocyanate-based polyurethane iniferter. J. Macromol. Sci. Part A Pure Appl. Chem. 1996, A33, 417–437. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. “Living” radical polymerization of styrene using tetraphenylethane-based polyurethane iniferter. Polym. Int. 1997, 43, 13–21. [Google Scholar] [CrossRef]
- Tharanikkarasu, K.; Radhakrishnan, G. Tetraphenylethane iniferters: Polyurethane-polystyrene multiblock copolymers through “living” radical polymerization. J. Appl. Polym. Sci. 1997, 66, 1551–1560. [Google Scholar] [CrossRef]
- Zhou, G.; Ma, C.; Zhang, G. Synthesis of polyurethane-g-poly(ethylene glycol) copolymers by macroiniferter and their protein resistance. Polym. Chem. 2011, 2, 1409–1414. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhang, Z.; Zhang, Y.; Tuo, X. Synthesis and simultaneous self-assembly of multiblock fluorinated polyurethane in iniferter polymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2161–2170. [Google Scholar] [CrossRef]
- Grabowski, M.; Kost, B.; Gostyński, B.; Bednarek, M. Can tetraphenylethane (TPE) “iniferter” group be introduced into the polymer chain by coupling TPE diol with diisocyanate? Polymer (Guildf). 2022, 246, 124738. [Google Scholar] [CrossRef]
- Muñiz, M.K. 3.14 The Pinacol Rearrangement. Compr. Org. Synth. Second Ed. 2014, 3, 741–756. [Google Scholar]
- Zalibera, M.; Nesvadba, P.; Gescheidt, G. Reaction of benzopinacol with non-ionic bases: Reversing the pinacol coupling. Org. Lett. 2013, 15, 4627–4629. [Google Scholar] [CrossRef]
- Tanimoto, S.; Toshimitsu, A. Synthesis of Ultraviolet Absorbers Having 2-Hydroxybenzophenone Moiety as the Functional Group. Bull. Inst. Chem. Res., Kyoto Univ. 1992, 69, 560–570. [Google Scholar]
- Baśko, M.; Kubisa, P. Cationic polymerization of L,L-lactide. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2650–2658. [Google Scholar] [CrossRef]
- Dashan, I.; Karaca, D.; Aydogan, B.; Temel, G. Preparation of single chain nanoparticles via photoinduced radical coupling process. Eur. Polym. J. 2019, 113, 183–191. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPE_PLA | Polymerization No. | AN/LA mol Ratio in Feed (wt. ratio) | Conversion of AN, % 2 | PLA-PAN-PLA Copolymers 3 | |||
|---|---|---|---|---|---|---|---|
| Mn 1 g/mol | TPE/LA | AN/LA 1 | TPE/LA 1 | Mn1 (SEC) 4 g/mol | |||
| 1400 | 0.157 | 1 | 4.2 (1) | 73 | 11.1 | 0.153 | 5130 (4250) |
| 2200 | 0.084 | 2 | 1.7 (0.5) | 65 | 2.5 | 0.049 | 5760 (4580) |
| 3 | 3.2 (1) | 69 | 3.45 | 0.060 | 5670 (4780) | ||
| 4 | 7.0 (2) | 71 | 9.9 | 0.071 | 9370 (8500) | ||
| 3300 | 0.054 | 5 | 3.5 (1) | 62 81_4 days 89_7 days | 2.3 N.d. 5 ” | 0.050 N.d. 5 ” | 6070 (5850) Bimodal SEC “ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, M.; Kost, B.; Kubisa, P.; Bednarek, M. A New Approach to The Synthesis of Polylactide/Polyacrylonitrile Block Copolymers. Polymers 2022, 14, 1529. https://doi.org/10.3390/polym14081529
Grabowski M, Kost B, Kubisa P, Bednarek M. A New Approach to The Synthesis of Polylactide/Polyacrylonitrile Block Copolymers. Polymers. 2022; 14(8):1529. https://doi.org/10.3390/polym14081529
Chicago/Turabian StyleGrabowski, Mateusz, Bartłomiej Kost, Przemysław Kubisa, and Melania Bednarek. 2022. "A New Approach to The Synthesis of Polylactide/Polyacrylonitrile Block Copolymers" Polymers 14, no. 8: 1529. https://doi.org/10.3390/polym14081529