
A Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
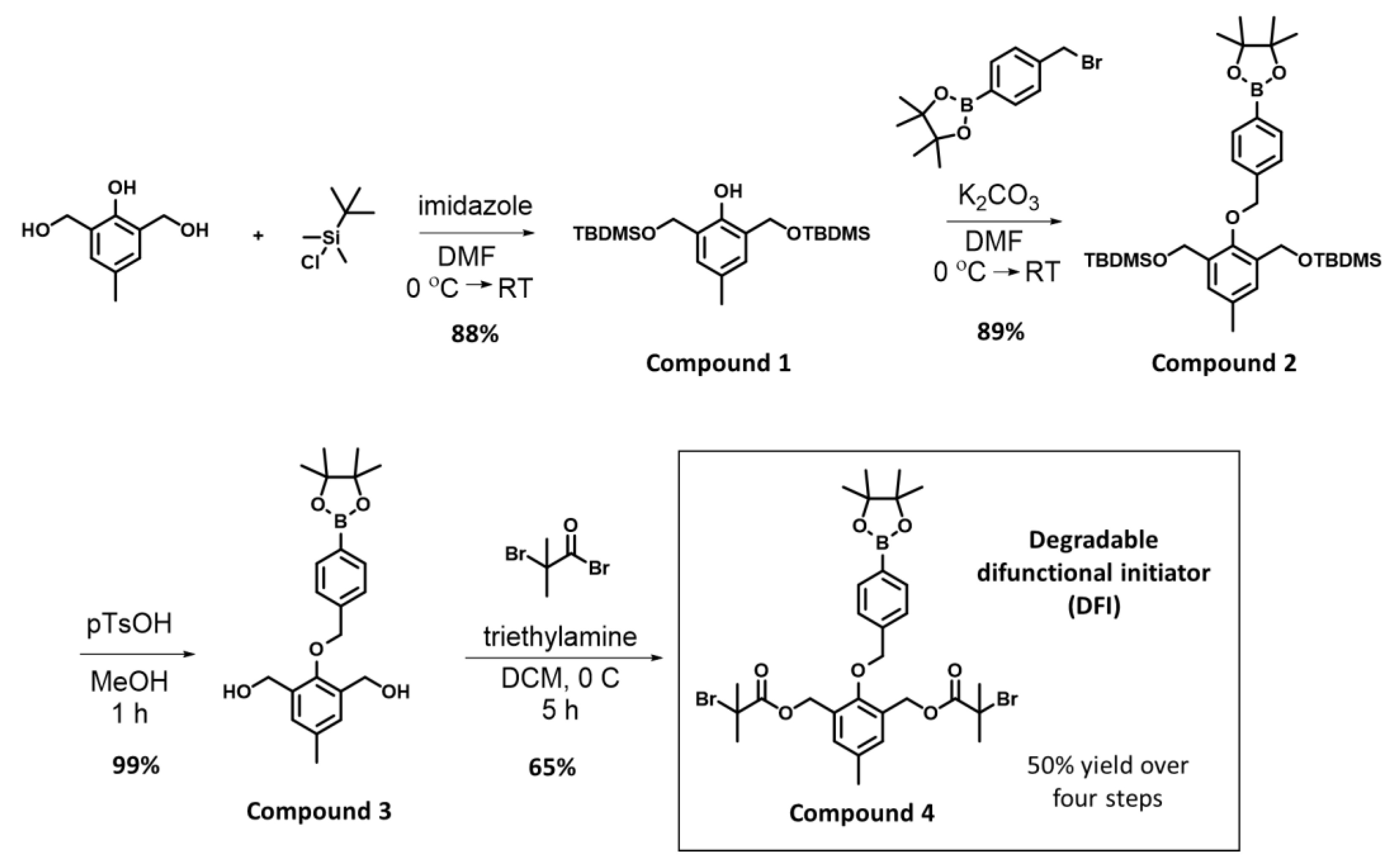
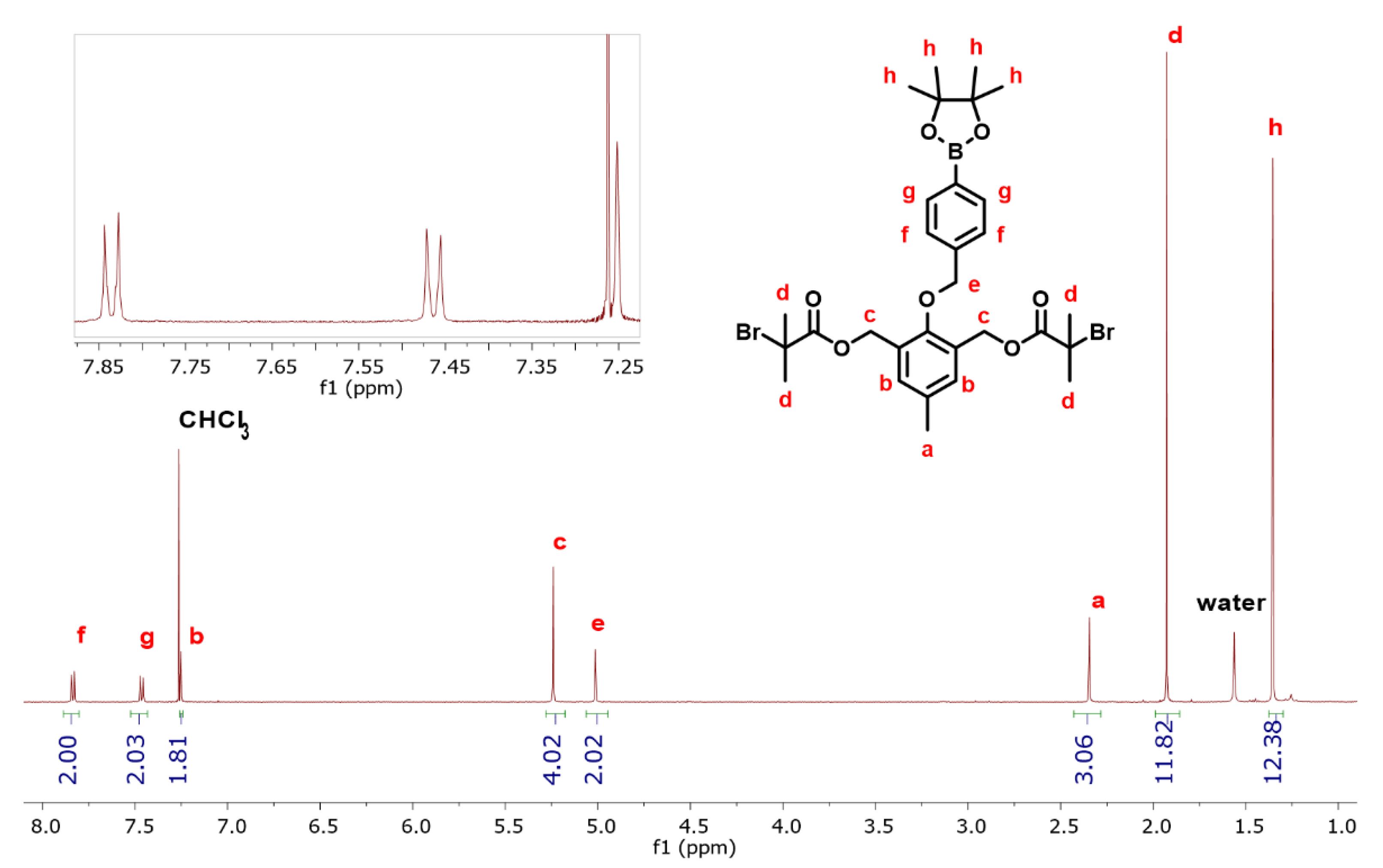
3.1. Synthesis of the DFI
3.2. Polymerization Using the DFI
3.3. Oxidative Degradation of pMMA with and without the DFI
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/“Living” Radical Polymerization. Halogen Atom Transfer Radical Polymerization Promoted by a Cu(I)/Cu(II) Redox Process. Macromolecules 1995, 28, 7901. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. “Living”/Controlled Radical Polymerization. Transition-Metal-Catalyzed Atom Transfer Radical Polymerization in the Presence of a Conventional Radical Initiator. Macromolecules 1995, 28, 7572. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3746. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization: Discovery and developments. Chem. Rec. 2004, 4, 159–175. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT ProcessA First Update. Aust. J. Chem. 2006, 59, 669–692. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” Atom Transfer Radical Polymerization: From Process Design to Preparation of Well-Defined Environmentally Friendly Polymeric Materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar] [CrossRef]
- Ouchi, M.; Terashima, T.; Sawamoto, M. Precision Control of Radical Polymerization via Transition Metal Catalysis: From Dormant Species to Designed Catalysts for Precision Functional Polymers. Acc. Chem. Res. 2008, 41, 1120–1132. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008, 37, 1087–1097. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process A Second Update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process—A Third Update. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Delplace, V.; Nicolas, J. Degradable vinyl polymers for biomedical applications. Nat. Chem. 2015, 7, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Rikkou, M.D.; Patrickios, C.S. Polymers prepared using cleavable initiators: Synthesis, characterization and degradation. Prog. Polym. Sci. 2011, 36, 1079–1097. [Google Scholar] [CrossRef]
- Hsu, P.-H.; Almutairi, A. Recent progress of redox-responsive polymeric nanomaterials for controlled release. J. Mater. Chem. B 2021, 9, 2179–2188. [Google Scholar] [CrossRef]
- Stubelius, A.; Lee, S.; Almutairi, A. The Chemistry of Boronic Acids in Nanomaterials for Drug Delivery. Acc. Chem. Res. 2019, 52, 3108–3119. [Google Scholar] [CrossRef]
- Huber, S.; Mecking, S. Straightforward Synthesis of Conjugated Block Copolymers by Controlled Suzuki–Miyaura Cross-Coupling Polymerization Combined with ATRP. Macromolecules 2019, 52, 5917–5924. [Google Scholar] [CrossRef]
- D’Acunzo, F.; Masci, G. Playing construction with the monomer toy box for the synthesis of multi-stimuli responsive copolymers by reversible deactivation radical polymerization protocols. J. Polym. Sci. 2021, 59, 3059–3083. [Google Scholar] [CrossRef]
- Jazani, A.M.; Arezi, N.; Maruya-Li, K.; Jung, S.; Oh, J.K. Facile Strategies to Synthesize Dual Location Dual Acidic pH/Reduction-Responsive Degradable Block Copolymers Bearing Acetal/Disulfide Block Junctions and Disulfide Pendants. ACS Omega 2018, 3, 8980–8991. [Google Scholar] [CrossRef] [Green Version]
- de Gracia Lux, C.; Joshi-Barr, S.; Nguyen, T.; Mahmoud, E.; Schopf, E.; Fomina, N.; Almutairi, A. Biocompatible Polymeric Nanoparticles Degrade and Release Cargo in Response to Biologically Relevant Levels of Hydrogen Peroxide. J. Am. Chem. Soc. 2012, 134, 15758–15764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.-H.; Kong, J.; Seeliger, F.; Matyjaszewski, K. Mechanism of Halogen Exchange in ATRP. Macromolecules 2011, 44, 7546–7557. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Shipp, D.A.; Wang, J.-L.; Grimaud, T.; Patten, T.E. Utilizing Halide Exchange To Improve Control of Atom Transfer Radical Polymerization. Macromolecules 1998, 31, 6836–6840. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamago, S. Termination Mechanism in the Radical Polymerization of Methyl Methacrylate and Styrene Determined by the Reaction of Structurally Well-Defined Polymer End Radicals. Macromolecules 2015, 48, 6450–6456. [Google Scholar] [CrossRef]
- Kuivila, H.G. Electrophilic Displacement Reactions. III. Kinetics of the Reaction between Hydrogen Peroxide and Benzeneboronic Acid1. J. Am. Chem. Soc. 1954, 76, 870–874. [Google Scholar] [CrossRef]
- Kuivila, H.G.; Armour, A.G. Electrophilic Displacement Reactions. IX. Effects of Substituents on Rates of Reactions between Hydrogen Peroxide and Benzeneboronic Acid1-3. J. Am. Chem. Soc. 1957, 79, 5659–5662. [Google Scholar] [CrossRef]
- Brown, H.C. Hydroboration—A powerful synthetic tool. Tetrahedron 1961, 12, 117–138. [Google Scholar] [CrossRef]
- Huang, C.-F.; Kuo, S.-W.; Moravčíková, D.; Liao, J.-C.; Han, Y.-M.; Lee, T.-H.; Wang, P.-H.; Lee, R.-H.; Tsiang, R.C.-C.; Mosnáček, J. Effect of variations of CuIIX2/L, surface area of Cu0, solvent, and temperature on atom transfer radical polyaddition of 4-vinylbenzyl 2-bromo-2-isobutyrate inimers. RSC Adv. 2016, 6, 51816–51822. [Google Scholar] [CrossRef]
- Han, Y.-M.; Chen, H.-H.; Huang, C.-F. Polymerization and degradation of aliphatic polyesters synthesized by atom transfer radical polyaddition. Polym. Chem. 2015, 6, 4565–4574. [Google Scholar] [CrossRef]
- Wang, C.-H.; Song, Z.-Y.; Deng, X.-X.; Zhang, L.-J.; Du, F.-S.; Li, Z.-C. Combination of ATRA and ATRC for the Synthesis of Periodic Vinyl Copolymers. Macromol. Rapid Commun. 2014, 35, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Dong, B.-T.; Du, F.-S.; Li, Z.-C. Degradable Thermoresponsive Polyesters by Atom Transfer Radical Polyaddition and Click Chemistry. Macromolecules 2012, 45, 8580–8587. [Google Scholar] [CrossRef]
- Mizutani, M.; Palermo, E.F.; Thoma, L.M.; Satoh, K.; Kamigaito, M.; Kuroda, K. Design and Synthesis of Self-Degradable Antibacterial Polymers by Simultaneous Chain- and Step-Growth Radical Copolymerization. Biomacromolecules 2012, 13, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.-T.; Li, Z.-L.; Zhang, L.-J.; Du, F.-S.; Li, Z.-C. Synthesis of linear functionalized polyesters by controlled atom transfer radical polyaddition reactions. Polym. Chem. 2012, 3, 2523–2530. [Google Scholar] [CrossRef]
- Mizutani, M.; Satoh, K.; Kamigaito, M. Metal-Catalyzed Radical Polyaddition for Aliphatic Polyesters via Evolution of Atom Transfer Radical Addition into Step-Growth Polymerization. Macromolecules 2009, 42, 472–480. [Google Scholar] [CrossRef]
- Satoh, K.; Mizutani, M.; Kamigaito, M. Metal-catalyzed radical polyaddition as a novel polymer synthetic route. Chem. Commun. 2007, 1260–1262. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, L.; Sims, H.; Nguyen, N.; Collins, C.; Palmer, J.; Wasson, F. A Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide. Polymers 2022, 14, 1733. https://doi.org/10.3390/polym14091733
Hill L, Sims H, Nguyen N, Collins C, Palmer J, Wasson F. A Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide. Polymers. 2022; 14(9):1733. https://doi.org/10.3390/polym14091733
Chicago/Turabian StyleHill, Lawrence, Hunter Sims, Ngoc Nguyen, Christopher Collins, Jeffery Palmer, and Fiona Wasson. 2022. "A Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide" Polymers 14, no. 9: 1733. https://doi.org/10.3390/polym14091733
APA StyleHill, L., Sims, H., Nguyen, N., Collins, C., Palmer, J., & Wasson, F. (2022). A Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide. Polymers, 14(9), 1733. https://doi.org/10.3390/polym14091733