
Functionalization of Conductive Polymers through Covalent Postmodification




Abstract
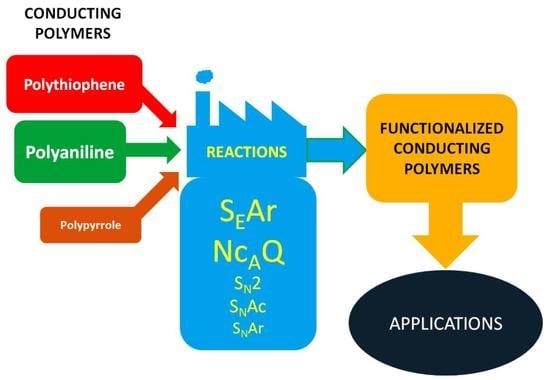
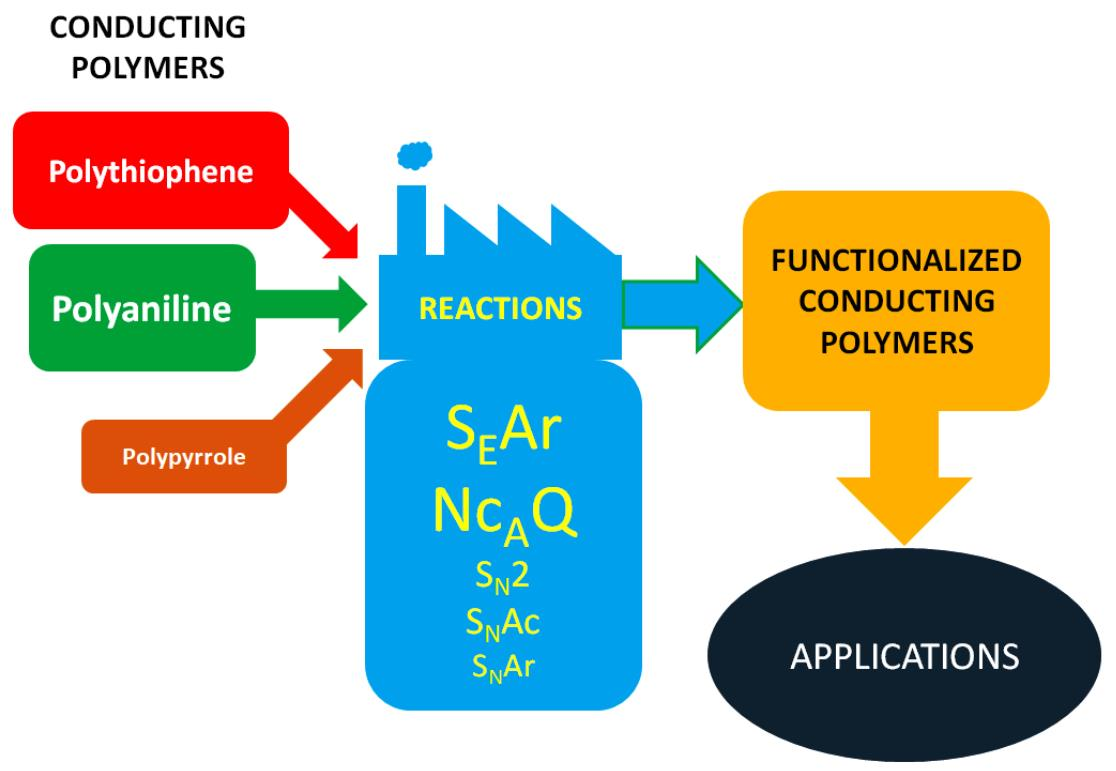
1. Introduction
2. Covalent Functionalization of Conductive Polymers
2.1. Polyaniline Functionalization
2.1.1. Electrophilic Substitution (SEAr) on Diphenylamine Rings
Electrophilic Aromatic Substitution in the Ring
- Sulfonation of Polyanilines
- Bromination (–Br) of Polyaniline
- Coupling with Diazonium Salts
Electrophilic Substitution in the N–H Group
- Amide Formation at the Nitrogen
- Tertiary Amine Formation at the Nitrogen
- Reversible Formation of Nitrosamine
2.1.2. Nucleophile Addition on Quinonimine Units
2.1.3. Reactions on Preattached Reactive Groups
2.2. Polythiophene Functionalization
2.2.1. Direct reactions on the Thiophene Ring
Nucleophilic Addition
Electrophilic Aromatic Substitution (SEAr) on Polythiophenes
Substitution of Lithiated Thiophene Rings
2.2.2. Reaction with Active Groups Present in Substituted Polythiophenes
Azide Moiety and Reaction with Alkynes
Amide Functionalization of Carboxy Substituted PT
Anionic and Cationic Moieties
Reaction with other Carboxylic Functionalities
Reactions with Attached Hydroxyl (–OH) Groups
Reaction with Miscellaneous Groups
2.2.3. Reactions with Substituted Poly(3,5-dioxythiophene) (PEDOT)
2.3. Polypyrrole Functionalization
2.4. Functionalization of Other Conducting Polymers
2.4.1. Poly(acetylene)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conducting Polymer | Added Group | Reaction Kind | Reaction Form | Reactants | Target Property | Ref. |
|---|---|---|---|---|---|---|
| PA (PPA) | –CONHAr | Amidation | Bulk | Aromatic amines | Optical absorption | [212] |
| PP (P(p-dMeoBz)) | –CN | Nu substitution | Film | CN- | Electrochemical | [213,215] |
| PPV | –HN–R–NH2 | Nu substitution | Bulk | H2N–R–NH2 | Cytocompatibility | [216] |
| PPV (MEH-PPV) | Multiple | (i) DCC catalyzed conjugation with –COOH (ii) “click” alkyne-azide | Bulk | various | Synthesis | [217] |
| PPV (MEH-PPV) | -Phtalocyanine | “click” alkyne-azide | Film | Functionalized phtalocyanine | Solar cells | [218] |
| PPV (DOH-PPV) | –X, Succinimide | Electrophilic addition | Bulk | NBS NCS | Fluorescence yield Solubility | [219] |
| PPV | –HN–R–NH2 | Silane chemistry | Bulk | diamines | Gene therapy | [220] |
| PPV | –Si–O–Si–NR–NH2 | Silane chemistry | Silane diamines | Cell adhesion | [221] | |
| PPE | OEG-oligopeptide | Reactive group (–COOH) | Film | OEG + oligoppetide | Gene therapy | [222] |
| PFO | PEG block | Terminal group | Bulk | PEG | Fluorescence Emission | [223] |
| PFO | Monosaccharides | Thioether | Bulk | Monosaccharides | Biocompatibility | [224] |
| PFO | PEG | “click” Diels-Alder | Bulk | Transcyclooctene | Fluorescence | [225] |
| PFO | PEG | “click” Diels-Alder | Bulk | Transcyclooctene term. PEG | Fluorescence 3D crosslink | [226] |
| P(FO-alt-T) | –X (halogen) | NCA | EchemFilm | X− (halide) | Optical | [226] |
2.4.2. Poly(phenylene)s
2.4.3. Poly(phenylenevinylene) (PPV)
2.4.4. Poly(phenylene ethynylene)
2.4.5. Poly(fluorene) (PFO)
3. Conclusions
4. Patents
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Aromatic amines |
| ACN | Acetonitrile |
| ATPhe | 4-aminotiophenol |
| CA | coupling agents |
| CE | Cellulose |
| CP | conducting polymer |
| CTA | cysteamine |
| DEHS | di-(2-ethylhexyl) sulfosuccinate |
| DMF | Dimethylformamide |
| DMFC | Direct methanol fuel cell |
| DMSO | Dimethylsulfoxide |
| Do | Diffusion coefficient |
| DOT | Dodecanotiol |
| DSC | Differential Scanning Calorimetry. |
| EChem | Electrochemical |
| FTIR | Fourier-Transform Infrared Spectroscopy |
| GO | Graphene oxide |
| HSPANI | highly sulfonated polyaniline (75–100%) |
| MALDI-TOF | Matrix assisted laser desorption/ionization-Time of Flight |
| MEH-PPV | poly(2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylene vinylene |
| MPS | mercaptopropansulfonate |
| NBS | N-bromosuccinimide |
| NCA | Nucleophilic conjugate addition |
| NCS | N-chlorosuccinimide |
| NFS | N-fluorosuccinimide |
| NMP | N-methylpyrrolydone |
| NMR | Nuclear Magnetic Resonance |
| Nu | NucleiphileNucleophile |
| NXSuc | N-halosuccucinimide |
| OEG | oligoethyleneglycol |
| OLED | Organic lifghtlight emitting diode |
| P3HT | poly(3-heylthiophene) |
| P3MeT | poly(3-methylthiophene) |
| P3OT | poly(3-octylthiophene) |
| PAMAM G4 | polyamidoamine dendrimer 4th generation |
| PANI (EBA) | polyaniline (emeraldine base form) |
| PANI (ES) | polyaniline (emeraldine salt form) |
| PANI (LEB) | polyaniline (leucoemeraldine base form) |
| PANI (PNB) | polyaniline (pernigraniline base form) |
| PANI | polyaniline |
| PBD | probe Beam Deflection |
| PEDOT | poly(ethylenedioxythiophene) |
| PEG | poly(ethyleneglycol) |
| PEI | poly(ethyleneimine) |
| PFO | poly(fluorene) |
| PNMANI | poly(N-methylaniline) |
| POH | poly[3-(6-hydroxyhexyl)thiophene] |
| PPDA | poly(p-phenylenediamine) |
| PPV | poly(phenylenevinylene) |
| PPy | polypyrrole |
| PSS | poly(styrene sulfonate) |
| PT | polythiophene |
| PVC | poly(vinylchloride) |
| RGO | Reduced graphene oxide |
| SEAr | Electrophilic aromatic substitution |
| SEN | Electrophilic substitution in the nitrogen |
| SNAc | Nucleophilic substitution in the acyl group |
| SNAl | Nucleophilic substitution in the alkyl group |
| SNAr | Nucleophilic aromatic substitution |
| SPAN | Sulfonated polyaniline (ca. 50%) |
| t-BOC | tert-butoxycarbonyl |
| Tg | Glass transition Temperature |
| THF | Tetrahydrofurane |
| TPP | tetraphenylporphyrin |
| VNHE | Volts vs. Normal Hydrogen Electrode |
| VRHE | Volts vs. Reversible Hydrogen Electrode |
| VSSCE | Volts vs. Saturated salt calomel electrode |
| XPS | X-ray Photoelectron Spectroscopy |
| XRD | X-ray diffraction |
References
- Reynolds, J.R.; Thompson, B.C.; Skotheim, T.A. Handbook of Conducting Polymers; 2 Volume Set; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Gall, D.; Jog, A.; Zhou, T. Narrow interconnects: The most conductive metals. In Proceedings of the 2020 IEEE International Electron Devices Meeting (IEDM), San Francisco, CA, USA, 12–18 December 2020; IEEE: Piscataway, NJ, USA, 2020; pp. 32.3.1–32.3.4. [Google Scholar]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- White, J.; Turnbull, A. Weathering of polymers: Mechanisms of degradation and stabilization, testing strategies and modelling. J. Mater. Sci. 1994, 29, 584–613. [Google Scholar] [CrossRef]
- Kim, J.; Zhang, G.; Shi, M.; Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 2021, 374, 212–216. [Google Scholar] [CrossRef]
- Dhandapani, E.; Thangarasu, S.; Ramesh, S.; Ramesh, K.; Vasudevan, R.; Duraisamy, N. Recent development and prospective of carbonaceous material, conducting polymer and their composite electrode materials for supercapacitor—A review. J. Energy Storage 2022, 52, 104937. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Stoeckel, M.-A.; Ruoko, T.-P.; Wu, H.-Y.; Liu, X.; Kolhe, N.B.; Wu, Z.; Puttisong, Y.; Musumeci, C.; Massetti, M. A high-conductivity n-type polymeric ink for printed electronics. Nat. Commun. 2021, 12, 2354. [Google Scholar] [CrossRef]
- Zahid, M.; Anum, R.; Siddique, S.; Shakir, H.F.; Rehan, Z. Polyaniline-based nanocomposites for electromagnetic interference shielding applications: A review. J. Thermoplast. Compos. Mater. 2021, 08927057211022408. [Google Scholar] [CrossRef]
- Fungo, F.; Jenekhe, S.A.; Bard, A.J. Plastic electrochromic devices: Electrochemical characterization and device properties of a phenothiazine-phenylquinoline donor− acceptor polymer. Chem. Mater. 2003, 15, 1264–1272. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Arias-Pardilla, J.; Pérez, J.; Vazquez, J.; Morallón, E.; Miras, M.C.; Barbero, C. Study of redox mechanism of poly(o-aminophenol) using in situ techniques: Evidence of two redox processes. J. Electroanal. Chem. 2005, 576, 139–145. [Google Scholar] [CrossRef]
- Molina, M.; Rivarola, C.; Miras, M.; Lescano, D.; Barbero, C. Nanocomposite synthesis by absorption of nanoparticles into macroporous hydrogels. Building a chemomechanical actuator driven by electromagnetic radiation. Nanotechnology 2011, 22, 245504. [Google Scholar] [CrossRef]
- Barbero, C.; Miras, M.; Kötz, R.; Haas, O. Sulphonated polyaniline (SPAN) films as cation insertion electrodes battery applications Part II: Exchange of mobile species in aqueous and non-aqueous solutions. J. Electroanal. Chem. 1997, 437, 191–198. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, A.; Riaz, U. Conducting Polymer-Based Micro-and Nano-batteries for Biomedical Applications: A Short Review. ChemistrySelect 2022, 7, e202201302. [Google Scholar] [CrossRef]
- Molina, M.; Rivarola, C.; Barbero, C. Effect of copolymerization and semi-interpenetration with conducting polyanilines on the physicochemical properties of poly(N-isopropylacrylamide) based thermosensitive hydrogels. Eur. Polym. J. 2011, 47, 1977–1984. [Google Scholar] [CrossRef]
- Schmidt, V.M.; Barbero, C.; Kötz, R. The ion exchange in polypyrrole in aqueous electrolytes. A probe beam deflection study of the effect of fixed negative charges. J. Electroanal. Chem. 1993, 352, 301–307. [Google Scholar] [CrossRef]
- Bongiovanni Abel, S.; Gallarato, L.A.; Dardanelli, M.S.; Barbero, C.A.; Rivarola, C.R.; Yslas, E.I. Photothermal lysis of Pseudomonas aeruginosa by polyaniline nanoparticles under near infrared irradiation. Biomed. Phys. Eng. Express 2018, 4, 045037. [Google Scholar] [CrossRef]
- Vines, J.B.; Lim, D.-J.; Park, H. Contemporary polymer-based nanoparticle systems for photothermal therapy. Polymers 2018, 10, 1357. [Google Scholar] [CrossRef]
- Yslas, E.I.; Ibarra, L.E.; Molina, M.A.; Rivarola, C.; Barbero, C.A.; Bertuzzi, M.L.; Rivarola, V.A. Polyaniline nanoparticles for near-infrared photothermal destruction of cancer cells. J. Nanoparticle Res. 2015, 17, 389. [Google Scholar] [CrossRef]
- Kushwaha, C.S.; Singh, P.; Shukla, S.K.; Chehimi, M.M. Advances in conducting polymer nanocomposite based chemical sensors: An overview. Mater. Sci. Eng. B 2022, 284, 115856. [Google Scholar] [CrossRef]
- Hackett, A.J.; Malmström, J.; Travas-Sejdic, J. Functionalization of conducting polymers for biointerface applications. Prog. Polym. Sci. 2017, 70, 18–33. [Google Scholar] [CrossRef]
- Wang, C.-C.; Wei, S.-C.; Luo, S.-C. Recent advances and biomedical applications of peptide-integrated conducting polymers. ACS Appl. Bio Mater. 2022, 5, 1916–1933. [Google Scholar] [CrossRef]
- Krinichnyi, V. Dynamics of spin charge carriers in polyaniline. Appl. Phys. Rev. 2014, 1, 021305. [Google Scholar] [CrossRef]
- Clark, R.L.; Yang, S.C. Poly-(2-methylaniline): The effect of chemical modification on the insulator/conductor transitions of polyaniline. Synth. Met. 1989, 29, 337–342. [Google Scholar] [CrossRef]
- Zaidi, N.A.; Foreman, J.P.; Tzamalis, G.; Monkman, S.C.; Monkman, A.P. Alkyl substituent effects on the conductivity of polyaniline. Adv. Funct. Mater. 2004, 14, 479–486. [Google Scholar] [CrossRef]
- Wei, W.; Hu, X.; Qi, X.; Yu, H.; Liu, Y.; Li, J.; Zhang, J.; Dong, W. A novel thermo-responsive hydrogel based on salecan and poly(N-isopropylacrylamide): Synthesis and characterization. Colloids Surf. B Biointerfaces 2015, 125, 1–11. [Google Scholar] [CrossRef]
- Bongiovanni Abel, S.; Yslas, E.I.; Rivarola, C.R.; Barbero, C.A. Synthesis of polyaniline (PANI) and functionalized polyaniline (F-PANI) nanoparticles with controlled size by solvent displacement method. Application in fluorescence detection and bacteria killing by photothermal effect. Nanotechnology 2018, 29, 125604. [Google Scholar] [CrossRef]
- Hiemenz, P.C.; Rajagopalan, R. Principles of Colloid and Surface Chemistry, Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Karger-Kocsis, J.; Kmetty, Á.; Lendvai, L.; Drakopoulos, S.X.; Bárány, T. Water-assisted production of thermoplastic nanocomposites: A review. Materials 2014, 8, 72–95. [Google Scholar] [CrossRef]
- Kolla, H.S.; Surwade, S.P.; Zhang, X.; MacDiarmid, A.G.; Manohar, S.K. Absolute molecular weight of polyaniline. J. Am. Chem. Soc. 2005, 127, 16770–16771. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Peacock, P.; Flippen, R.; Manohar, S.; MacDiarmid, A. The molecular weight of polyaniline by light scattering and gel permeation chromatography. Synth. Met. 1993, 60, 233–237. [Google Scholar] [CrossRef]
- Genies, E.; Syed, A.; Tsintavis, C. Electrochemical Study Of Polyaniline In Aqueous And Organic Medium. Redox And Kinetic Properties. Mol. Cryst. Liq. Cryst. 1985, 121, 181–186. [Google Scholar] [CrossRef]
- Watanabe, A.; Mori, K.; Iwasaki, Y.; Nakamura, Y. Molecular weight of electropolymerized polyaniline. J. Chem. Soc. Chem. Commun. 1987, 3–4. [Google Scholar] [CrossRef]
- Zheng, W.; Angelopoulos, M.; Epstein, A.J.; MacDiarmid, A. Experimental evidence for hydrogen bonding in polyaniline: Mechanism of aggregate formation and dependency on oxidation state. Macromolecules 1997, 30, 2953–2955. [Google Scholar] [CrossRef]
- Ponzio, E.A.; Echevarria, R.; Morales, G.M.; Barbero, C. Removal of N-methylpyrrolidone hydrogen-bonded to polyaniline free-standing films by protonation–deprotonation cycles or thermal heating. Polym. Int. 2001, 50, 1180–1185. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Cohen, I. Molecular orbital theory of the hydrogen bond. 20. Pyrrole and imidazole as proton donors and proton acceptors. J. Am. Chem. Soc. 1978, 100, 5285–5290. [Google Scholar] [CrossRef]
- Lii, J.H.; Chen, K.H.; Allinger, N.L. Alcohols, ethers, carbohydrates, and related compounds. IV. Carbohydrates. J. Comput. Chem. 2003, 24, 1504–1513. [Google Scholar] [CrossRef]
- Mazeau, K.; Heux, L. Molecular dynamics simulations of bulk native crystalline and amorphous structures of cellulose. J. Phys. Chem. B 2003, 107, 2394–2403. [Google Scholar] [CrossRef]
- Cao, Y.; Smith, P.; Heeger, A.J. Counter-ion induced processibility of conducting polyaniline. Synth. Met. 1993, 57, 3514–3519. [Google Scholar] [CrossRef]
- Harris, J.K.; Ratcliff, E.L. Ion diffusion coefficients in poly(3-alkylthiophenes) for energy conversion and biosensing: Role of side-chain length and microstructure. J. Mater. Chem. C 2020, 8, 13319–13327. [Google Scholar] [CrossRef]
- Konopka, S.; McDuffie, B. Diffusion coefficients of ferri-and ferrocyanide ions in aqueous media, using twin-electrode thin-layer electrochemistry. Anal. Chem. 1970, 42, 1741–1746. [Google Scholar] [CrossRef]
- Chambon, S.; Rivaton, A.; Gardette, J.-L.; Firon, M. Photo-and thermo-oxidation of poly(p-phenylene-vinylene) and phenylene-vinylene oligomer. Polym. Degrad. Stab. 2011, 96, 1149–1158. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Dao, L.; Leclerc, M.; Guay, J.; Chevalier, J. Synthesis and characterization of substituted poly(anilines). Synth. Met. 1989, 29, 377–382. [Google Scholar] [CrossRef]
- Barbero, C.; Kötz, R. Electrochemical formation of a self-doped conductive polymer in the absence of a supporting electrolyte. The copolymerization of o-aminobenzenesulfonic acid and aniline. Adv. Mater. 1994, 6, 577–580. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Acevedo, D.F.; Miras, M.C.; Motheo, A.J.; Barbero, C.A. Comparative study of 2-amino and 3-aminobenzoic acid copolymerization with aniline synthesis and copolymer properties. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5587–5599. [Google Scholar] [CrossRef]
- So, R.C.; Carreon-Asok, A.C. Molecular design, synthetic strategies, and applications of cationic polythiophenes. Chem. Rev. 2019, 119, 11442–11509. [Google Scholar] [CrossRef]
- Moodie, R.B.; Thomas, P.N.; Schofield, K. Electrophilic aromatic substitution. Part 18. Nitration of acetanilide and some analogues: A reconsideration. J. Chem. Soc. Perkin Trans. 1977, 2, 1693–1705. [Google Scholar] [CrossRef]
- Yue, J.; Epstein, A.J. Synthesis of self-doped conducting polyaniline. J. Am. Chem. Soc. 1990, 112, 2800–2801. [Google Scholar] [CrossRef]
- Yue, J.; Epstein, A.; MacDiarmid, A. Sulfonic acid ring-substituted polyaniline, a self-doped conducting polymer. Mol. Cryst. Liq. Cryst. 1990, 189, 255–261. [Google Scholar] [CrossRef]
- Ito, S.; Murata, K.; Teshima, S.; Aizawa, R.; Asako, Y.; Takahashi, K.; Hoffman, B.M. Simple synthesis of water-soluble conducting polyaniline. Synth. Met. 1998, 96, 161–163. [Google Scholar] [CrossRef]
- Yue, J.; Gordon, G.; Epstein, A.J. Comparison of different synthetic routes for sulphonation of polyaniline. Polymer 1992, 33, 4410–4418. [Google Scholar] [CrossRef]
- Şahin, Y.; Pekmez, K.; Yıldız, A. Electrochemical synthesis of self-doped polyaniline in fluorosulfonic acid/acetonitrile solution. Synth. Met. 2002, 129, 107–115. [Google Scholar] [CrossRef]
- Wei, X.-L.; Wang, Y.; Long, S.; Bobeczko, C.; Epstein, A. Synthesis and physical properties of highly sulfonated polyaniline. J. Am. Chem. Soc. 1996, 118, 2545–2555. [Google Scholar] [CrossRef]
- Chen, S.-A.; Hwang, G.-W. Structure characterization of self-acid-doped sulfonic acid ring-substituted polyaniline in its aqueous solutions and as solid film. Macromolecules 1996, 29, 3950–3955. [Google Scholar] [CrossRef]
- Liu, G.; Freund, M.S. Nucleophilic substitution reactions of polyaniline with substituted benzenediazonium ions: A facile method for controlling the surface chemistry of conducting polymers. Chem. Mater. 1996, 8, 1164–1166. [Google Scholar] [CrossRef]
- Barbero, C.; Salavagione, H.J.; Acevedo, D.F.; Grumelli, D.E.; Garay, F.; Planes, G.A.; Morales, G.M.; Miras, M.C. Novel synthetic methods to produce functionalized conducting polymers I. Polyanilines. Electrochim. Acta 2004, 49, 3671–3686. [Google Scholar] [CrossRef]
- Stejskal, J.; Trchová, M.; Prokeš, J.; Sapurina, I. Brominated polyaniline. Chem. Mater. 2001, 13, 4083–4086. [Google Scholar] [CrossRef]
- McCoy, C.H.; Lorkovic, I.M.; Wrighton, M.S. Potential-dependent nucleophilicity of polyaniline. J. Am. Chem. Soc. 1995, 117, 6934–6943. [Google Scholar] [CrossRef]
- Lin, H.-K.; Chen, S.-A. Synthesis of new water-soluble self-doped polyaniline. Macromolecules 2000, 33, 8117–8118. [Google Scholar] [CrossRef]
- Hua, M.-Y.; Su, Y.-N.; Chen, S.-A. Water-soluble self-acid-doped conducting polyaniline: Poly (aniline-co-N-propylbenzenesulfonic acid-aniline). Polymer 2000, 41, 813–815. [Google Scholar] [CrossRef]
- Han, C.-C.; Jeng, R.-C. Concurrent reduction and modification of polyaniline emeraldinebase with pyrrolidine and other nucleophiles. Chem. Commun. 1997, 553–554. [Google Scholar] [CrossRef][Green Version]
- Han, C.-C.; Lu, C.-H.; Hong, S.-P.; Yang, K.-F. Highly conductive and thermally stable self-doping propylthiosulfonated polyanilines. Macromolecules 2003, 36, 7908–7915. [Google Scholar] [CrossRef]
- Han, C.-C.; Hong, S.-P.; Yang, K.-F.; Bai, M.-Y.; Lu, C.-H.; Huang, C.-S. Highly conductive new aniline copolymers containing butylthio substituent. Macromolecules 2001, 34, 587–591. [Google Scholar] [CrossRef]
- Salavagione, H.; Morales, G.; Miras, M.; Barbero, C. Synthesis of a self-doped polyaniline by nucleophilic addition. Acta Polym. 1999, 50, 40–44. [Google Scholar] [CrossRef]
- Yue, J.; Epstein, A.J. Electronic control of pH at sulfonated polyaniline electrodes. J. Chem. Soc. Chem. Commun. 1992, 1540–1542. [Google Scholar] [CrossRef]
- Morales, G.M.; Llusa, M.; Miras, M.C.; Barbero, C. Effects of high hydrochloric acid concentration on aniline chemical polymerization. Polymer 1997, 38, 5247–5250. [Google Scholar] [CrossRef]
- Barbero, C.; Morales, G.; Grumelli, D.; Planes, G.; Salavagione, H.; Marengo, C.; Miras, M. New methods of polyaniline functionalization. Synth. Met. 1999, 101, 694–695. [Google Scholar] [CrossRef]
- Grumelli, D.E.; Forzani, E.S.; Morales, G.M.; Miras, M.C.; Barbero, C.A.; Calvo, E.J. Microgravimetric study of electrochemically controlled nucleophilic addition of sulfite to polyaniline. Langmuir 2004, 20, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Blomquist, M.; Bobacka, J.; Ivaska, A.; Levon, K. Electrochemical and spectroscopic study on thiolation of polyaniline. Electrochim. Acta 2013, 90, 604–614. [Google Scholar] [CrossRef]
- Pyshkina, O.A.; Kim, B.; Korovin, A.N.; Zezin, A.; Sergeyev, V.G.; Levon, K. Interpolymer complexation of water-soluble self-doped polyaniline. Synth. Met. 2008, 158, 999–1003. [Google Scholar] [CrossRef]
- Amaya, T.; Hatai, T.; Kurata, I.; Hirao, T. Synthesis of self-doped polyaniline bearing phosphonic acid moiety via Pd-catalyzed phosphonation of poly(2-bromoaniline). Tetrahedron Lett. 2018, 59, 1715–1718. [Google Scholar] [CrossRef]
- Kumar, D.R.; Dhakal, G.; Shafi, P.M.; Sayed, M.S.; Lee, J.; Lee, Y.R.; Shim, J.-J. Sulfite food additive electrochemical determination by nucleophilic addition on poly(4-aminodiphenylamine)-4-aminothiophenol-Au composite electrode. Microchem. J. 2022, 181, 107635. [Google Scholar] [CrossRef]
- Shoji, E.; Freund, M.S. Poly (aniline boronic acid): A new precursor to substituted poly(aniline)s. Langmuir 2001, 17, 7183–7185. [Google Scholar] [CrossRef]
- Durgaryan, A.H.; Miraqyan, N.A.; Arakelyan, R.H.; Durgaryan, N.A. Investigation of addition reaction of sodium thiosulfate pentahydrate to quinonediimine groups. Polym. Bull. 2019, 76, 3929–3940. [Google Scholar] [CrossRef]
- Miraqyan, N.A.; Arakelyan, R.H.; Durgaryan, N.A.; Durgaryan, A.H. Synthesis and investigation of poly(p-phenylenediamine)–poly(1,4-benzoquinonediimine-N,N-diyl-1,4-phenylene). Chem. Pap. 2018, 72, 1517–1524. [Google Scholar] [CrossRef]
- Bhadra, S.; Kim, N.H.; Lee, J.H. Synthesis of water soluble sulfonated polyaniline and determination of crystal structure. J. Appl. Polym. Sci. 2010, 117, 2025–2035. [Google Scholar] [CrossRef]
- Mendes, L.C.; Falco, A.P.S.; Pinho, M.S.; Marques, P.O. Sulfonated polyaniline: Influence of sulfonation routes on its thermal and structural characteristics. Mater. Res. 2011, 14, 466–471. [Google Scholar] [CrossRef]
- Raffa, D.; Leung, K.; Battaglini, F. A microelectrochemical enzyme transistor based on an N-alkylated poly(aniline) and its application to determine hydrogen peroxide at neutral pH. Anal. Chem. 2003, 75, 4983–4987. [Google Scholar] [CrossRef]
- Salavagione, H.J. Preparation and characterization of “clickable” polyaniline derivatives on graphene modified electrodes. J. Electroanal. Chem. 2016, 765, 118–125. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Miras, M.C.; Barbero, C. Chemical lithography of a conductive polymer using a traceless removable group. J. Am. Chem. Soc. 2003, 125, 5290–5291. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Miras, M.C.; Barbero, C. Photolithographic patterning of a conductive polymer using a polymeric photoacid generator and a traceless removable group. Macromol. Rapid Commun. 2006, 27, 26–30. [Google Scholar] [CrossRef]
- Lee, C.-W.; Seo, Y.-H.; Lee, S.-H. A soluble polyaniline substituted with t-BOC: Conducting patterns and doping. Macromolecules 2004, 37, 4070–4074. [Google Scholar] [CrossRef]
- Yue, J.; Wang, Z.H.; Cromack, K.R.; Epstein, A.J.; MacDiarmid, A.G. Effect of sulfonic acid group on polyaniline backbone. J. Am. Chem. Soc. 1991, 113, 2665–2671. [Google Scholar] [CrossRef]
- Barbero, C.; Miras, M.C.; Schnyder, B.; Hass, O.; Kötz, R. Sulfonated polyaniline films as cation insertion electrodes for battery applications. Part 1.—Structural and electrochemical characterization. J. Mater. Chem. B 1994, 4, 1775–1783. [Google Scholar] [CrossRef]
- Barbero, C.; Miras, M.; Kötz, R.; Haas, O. Comparative study of the ion exchange and electrochemical properties of sulfonated polyaniline (SPAN) and polyaniline (PANI). Synth. Met. 1993, 55, 1539–1544. [Google Scholar] [CrossRef]
- Epstein, A.; Smallfield, J.; Guan, H.; Fahlman, M. Corrosion protection of aluminum and aluminum alloys by polyanilines: A potentiodynamic and photoelectron spectroscopy study. Synth. Met. 1999, 102, 1374–1376. [Google Scholar] [CrossRef]
- Min, Y.; Yang, Y.; Poojari, Y.; Liu, Y.; Wu, J.-C.; Hansford, D.J.; Epstein, A.J. Sulfonated polyaniline-based organic electrodes for controlled electrical stimulation of human osteosarcoma cells. Biomacromolecules 2013, 14, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mu, S. Electrochromic properties of sulfonic acid ring-substituted polyaniline in aqueous and non-aqueous media. Synth. Met. 2004, 144, 143–149. [Google Scholar] [CrossRef]
- Epstein, A.J.; Wang, Y.; Gebler, D.; Fu, D.; Swager, T. Control of light-emitting polymer devices using polymer/polymer interfaces. MRS Online Proc. Libr. 1997, 488, 75–85. [Google Scholar] [CrossRef]
- Bai, H.; Xu, Y.; Zhao, L.; Li, C.; Shi, G. Non-covalent functionalization of graphene sheets by sulfonated polyaniline. Chem. Commun. 2009, 1667–1669. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.X.; Cheng, H.M. Water-soluble multiwalled carbon nanotubes functionalized with sulfonated polyaniline. J. Phys. Chem. B 2006, 110, 9095–9099. [Google Scholar] [CrossRef]
- Dutta, K.; Das, S.; Kumar, P.; Kundu, P.P. Polymer electrolyte membrane with high selectivity ratio for direct methanol fuel cells: A preliminary study based on blends of partially sulfonated polymers polyaniline and PVdF-co-HFP. Appl. Energy 2014, 118, 183–191. [Google Scholar] [CrossRef]
- Sun, P.; Li, Z.; Wang, S.; Yin, X. Performance enhancement of polybenzimidazole based high temperature proton exchange membranes with multifunctional crosslinker and highly sulfonated polyaniline. J. Membr. Sci. 2018, 549, 660–669. [Google Scholar] [CrossRef]
- Zhou, M.; Li, W.; Gu, T.; Wang, K.; Cheng, S.; Jiang, K. A sulfonated polyaniline with high density and high rate Na-storage performances as a flexible organic cathode for sodium ion batteries. Chem. Commun. 2015, 51, 14354–14356. [Google Scholar] [CrossRef]
- Wei, Y.; Yan, Y.; Zou, Y.; Shi, M.; Deng, Q.; Zhao, N.; Wang, J.; You, C.; Yang, R.; Xu, Y. Sulfonated polyaniline coated bamboo-derived biochar/sulfur cathode for Li-S batteries with excellent dual conductivity and polysulfides affinity. Electrochim. Acta 2019, 310, 45–57. [Google Scholar] [CrossRef]
- Tatsuma, T.; Ogawa, T.; Sato, R.; Oyama, N. Peroxidase-incorporated sulfonated polyaniline–polycation complexes for electrochemical sensing of H2O2. J. Electroanal. Chem. 2001, 501, 180–185. [Google Scholar] [CrossRef]
- Lee, W.-J.; Kim, Y.-J.; Kaang, S. Electrical properties of polyaniline/sulfonated polycarbonate blends. Synth. Met. 2000, 113, 237–243. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, L.; Tang, D.; Fu, X.; Tang, J.; Guo, X.; Hu, C. Sulfonated polyaniline as a solid organocatalyst for dehydration of fructose into 5-hydroxymethylfurfural. Green Chem. 2017, 19, 1932–1939. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, G.; Ding, H.; Shan, Y. Fabrication and photovoltaic properties of self-assembled sulfonated polyaniline/TiO2 nanocomposite ultrathin films. Mater. Chem. Phys. 2007, 102, 249–254. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, J.; Wu, C. Polyacrylamide hydrogels with trapped sulfonated polyaniline. Eur. Polym. J. 2005, 41, 1342–1349. [Google Scholar] [CrossRef]
- Guo, Y.; Ji, C.; Ye, Y.; Chen, Y.; Yang, Z.; Xue, S.; Niu, Q.J. High performance nanofiltration membrane using self-doping sulfonated polyaniline. J. Membr. Sci. 2022, 652, 120441. [Google Scholar] [CrossRef]
- Krutyakov, Y.A.; Kudrinsky, A.; Olenin, A.Y.; Lisichkin, G. Synthesis of highly stable silver colloids stabilized with water soluble sulfonated polyaniline. Appl. Surf. Sci. 2010, 256, 7037–7042. [Google Scholar] [CrossRef]
- Wei, X.; Epstein, A.J. Synthesis of highly sulfonated polyaniline. Synth. Met. 1995, 74, 123–125. [Google Scholar] [CrossRef]
- Wei, X.-L.; Fahlman, M.; Epstein, A. XPS study of highly sulfonated polyaniline. Macromolecules 1999, 32, 3114–3117. [Google Scholar] [CrossRef]
- Ioffe, D. Synthesis of monobrominated anilines via bromination-isomerization. Mendeleev Commun. 1993, 31, 16. [Google Scholar]
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, L.; Jiang, Z.; Wu, D.; Li, Z.; Zhou, X. Sulfonato–Cu (salen) Complex Catalyzed N-Arylation of Aliphatic Amines with Aryl Halides in Water. Eur. J. Org. Chem. 2010, 26, 4971–4975. [Google Scholar] [CrossRef]
- Aksungur, T.; Erol, F.; Seferoğlu, N.; Seferoğlu, Z. Experimental and theoretical studies on monoazo dye including diphenylamine and N-methyldiphenylamine derivatives. J. Mol. Struct. 2020, 1217, 128449. [Google Scholar] [CrossRef]
- DeArmitt, C.; Armes, S.P.; Winter, J.; Uribe, F.; Gottesfeld, S.; Mombourquette, C. A novel N-substituted polyaniline derivative. Polymer 1993, 34, 158–162. [Google Scholar] [CrossRef]
- Planes, G.A.; Morales, G.M.; Miras, M.C.; Barbero, C. A soluble and electroactive polyaniline obtained by coupling of 4-sulfobenzenediazonium ion and poly(N-methylaniline). Synth. Met. 1998, 97, 223–227. [Google Scholar] [CrossRef]
- Acevedo, D.F.; Rivarola, C.R.; Miras, M.C.; Barbero, C.A. Effect of chemical functionalization on the electrochemical properties of conducting polymers. Modification of polyaniline by diazonium ion coupling and subsequent reductive degradation. Electrochim. Acta 2011, 56, 3468–3473. [Google Scholar] [CrossRef]
- Arias-Pardilla, J.; Salavagione, H.J.; Barbero, C.; Morallón, E.; Vázquez, J. Study of the chemical copolymerization of 2-aminoterephthalic acid and aniline.: Synthesis and copolymer properties. Eur. Polym. J. 2006, 42, 1521–1532. [Google Scholar] [CrossRef]
- Acevedo, D. Estudio de la Sintesis y Propiedades de Nuevos Materiales Aplicando Tecnicas de Quimica Combinatoria. Ph.D. Thesis, UNRC, Río Cuarto, Argentina, 2005. [Google Scholar]
- Monge, N.E.; Miras, M.C.; Barbero, C.A. High-throughput screening method to detect amphiphilic counterions able to solubilize conducting polymers. J. Comb. Chem. 2010, 6, 814–817. [Google Scholar] [CrossRef]
- Acevedo, D.; Miras, M.; Barbero, C.; Potyrailo, R.A.; Maier, W. Combinatorial Synthesis and Screening of Photochromic Dyes and Modified Conducting Polymers. In Combinatorial High-Throughput Discovery Optimization of Catalysts Materials; CRC Press: Boca Raton, FL, USA, 2006; p. 239. [Google Scholar]
- Rodriguez, R.C.; Moncada, A.B.; Acevedo, D.F.; Planes, G.A.; Miras, M.C.; Barbero, C.A. Electroanalysis using modified hierarchical nanoporous carbon materials. Faraday Discuss. 2013, 164, 147–173. [Google Scholar] [CrossRef]
- Yang, M.; Ma, J.; Zhang, C.; Yang, Z.; Lu, Y. General synthetic route toward functional hollow spheres with double-shelled structures. Angew. Chem. Int. Ed. 2005, 44, 6727–6730. [Google Scholar] [CrossRef]
- Maleki, A.; Nematollahi, D. Electrochemical synthesis and mechanestic study of quinone imines exploiting the dual character of N, N-dialkyl-p-phenylenediamines. Org. Lett. 2011, 13, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- Han, C.-C.; Chen, H.-Y. Highly conductive and electroactive fluorine-functionalized polyanilines. Macromolecules 2007, 40, 8969–8973. [Google Scholar] [CrossRef]
- Han, C.C.; Yang, K.F.; Hong, S.P.; Balasubramanian, A.; Lee, Y.T. Syntheses and characterizations of aniline/butylthioaniline copolymers: Comparisons of copolymers prepared by the new concurrent reduction and substitution route and the conventional oxidative copolymerization method. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1767–1777. [Google Scholar] [CrossRef]
- Han, C.-C.; Bai, M.-Y.; Yang, K.-F.; Lee, Y.-S.; Lin, C.-W. A novel method for making highly dispersible conducting polymer and concentric graphitic carbon nano-spheres based on an undoped and functionalized polyaniline. J. Mater. Chem. B 2008, 18, 3918–3925. [Google Scholar] [CrossRef]
- Lahiff, E.; Bell, S.; Diamond, D. Functionalised Nanostructured Polyaniline—A New Substrate for Building Adaptive Sensing Surfaces. MRS Online Proc. Libr. 2007, 1054, FF05-05. [Google Scholar] [CrossRef]
- Lahiff, E.; Lynam, C.; Gilmartin, N.; Wallace, G.G.; O’Kennedy, R.; Diamond, D. Controllable chemical modification of Polyaniline Nanofibres. MRS Online Proc. Libr. 2009, 1240, Art nr 602. [Google Scholar] [CrossRef]
- Lahiff, E.; Woods, T.; Blau, W.; Wallace, G.G.; Diamond, D. Synthesis and characterisation of controllably functionalised polyaniline nanofibres. Synth. Met. 2009, 159, 741–748. [Google Scholar] [CrossRef]
- Crean, C.; Lahiff, E.; Gilmartin, N.; Diamond, D.; O’Kennedy, R. Polyaniline nanofibres as templates for the covalent immobilisation of biomolecules. Synth. Met. 2011, 161, 285–292. [Google Scholar] [CrossRef]
- Florea, L.; Lahiff, E.; Diamond, D. Modified polyaniline nanofibres for ascorbic acid detection. MRS Online Proc. Libr. 2011, 1312, 903. [Google Scholar] [CrossRef]
- Yánez-Heras, J.; Planes, G.A.; Williams, F.; Barbero, C.A.; Battaglini, F. Sequential electrochemical polymerization of aniline and their derivatives showing electrochemical activity at neutral pH. Electroanalysis 2010, 22, 2801–2808. [Google Scholar] [CrossRef]
- Yslas, E.I.; Cavallo, P.; Acevedo, D.F.; Barbero, C.A.; Rivarola, V.A. Cysteine modified polyaniline films improve biocompatibility for two cell lines. Mater. Sci. Eng. C 2015, 51, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Neira-Carrillo, A.; Yslas, E.; Marini, Y.A.; Vásquez-Quitral, P.; Sánchez, M.; Riveros, A.; Yáñez, D.; Cavallo, P.; Kogan, M.J.; Acevedo, D. Hybrid biomaterials based on calcium carbonate and polyaniline nanoparticles for application in photothermal therapy. Colloids Surf. B Biointerfaces 2016, 145, 634–642. [Google Scholar] [PubMed]
- Amaya, T.; Kurata, I.; Inada, Y.; Hatai, T.; Hirao, T. Synthesis of phosphonic acid ring-substituted polyanilines via direct phosphonation to polymer main chains. RSC Adv. 2017, 7, 39306–39313. [Google Scholar] [CrossRef]
- Salavagione, H.J.; Arias-Pardilla, J.; Vázquez, J.; Miras, M.C.; Morallón, E.; Barbero, C. Spectroelectrochemical study of the oxidation of diaminophenols on platinum electrodes in acidic medium. Electrochim. Acta 2005, 50, 5414–5422. [Google Scholar] [CrossRef]
- Hoogenboom, R. Thiol-Alkin-Chemie: Ein leistungsfähiges Syntheseverfahren für hochfunktionalisierte Verbindungen. Angew. Chem. Int. Ed. 2010, 122, 3489–3491. [Google Scholar]
- McCullough, R.D.; Tristram-Nagle, S.; Williams, S.P.; Lowe, R.D.; Jayaraman, M. Self-orienting head-to-tail poly(3-alkylthiophenes): New insights on structure-property relationships in conducting polymers. J. Am. Chem. Soc. 1993, 115, 4910–4911. [Google Scholar] [CrossRef]
- Kaloni, T.P.; Giesbrecht, P.K.; Schreckenbach, G.; Freund, M.S. Polythiophene: From fundamental perspectives to applications. Chem. Mater. 2017, 29, 10248–10283. [Google Scholar] [CrossRef]
- Huynh, T.-P.; Sharma, P.S.; Sosnowska, M.; D’Souza, F.; Kutner, W. Functionalized polythiophenes: Recognition materials for chemosensors and biosensors of superior sensitivity, selectivity, and detectability. Prog. Polym. Sci. 2015, 47, 1–25. [Google Scholar] [CrossRef]
- Qi, Z.; Rees, N.G.; Pickup, P.G. Electrochemically induced substitution of polythiophenes and polypyrrole. Chem. Mater. 1996, 8, 701–707. [Google Scholar] [CrossRef]
- Kurioka, T.; Inagi, S. Electricity-Driven Post-Functionalization of Conducting Polymers. Chem. Rec. 2021, 21, 2107–2119. [Google Scholar] [CrossRef]
- Inagi, S.; Hosaka, K.; Hayashi, S.; Fuchigami, T. Anodic Halogenations of Poly (Thiophene) Derivatives; ECS Meeting Abstracts; IOP Publishing: Bristol, UK, 2009; p. 1655. [Google Scholar]
- Hayashi, S.; Inagi, S.; Hosaka, K.; Fuchigami, T. Post-functionalization of poly(3-hexylthiophene) via anodic chlorination. Synth. Met. 2009, 159, 1792–1795. [Google Scholar] [CrossRef]
- Shida, N.; Okazaki, D.; Kurioka, T.; Nishiyama, H.; Seferos, D.S.; Tomita, I.; Inagi, S. Anodic Chlorination of Selenophene-Containing Polymers: Reaction Efficiency and Selective Reaction of Single Segment in Rod-Rod Diblockcopolymer. ChemElectroChem 2017, 4, 1824–1827. [Google Scholar] [CrossRef]
- Kurioka, T.; Shida, N.; Tomita, I.; Inagi, S. Post-Functionalization of Aromatic C–H Bonds at the Main Chains of π-Conjugated Polymers via Anodic Chlorination Facilitated by Lewis Acids. Macromolecules 2021, 54, 1539–1547. [Google Scholar] [CrossRef]
- Hayashi, S.; Inagi, S.; Fuchigami, T. Efficient electrochemical polymer halogenation using a thin-layered cell. Polym. Chem. 2011, 2, 1632–1637. [Google Scholar] [CrossRef]
- Li, Y.; Vamvounis, G.; Holdcroft, S. Facile functionalization of poly(3-alkylthiophene)s via electrophilic substitution. Macromolecules 2001, 34, 141–143. [Google Scholar] [CrossRef]
- Yadav, P.K.; Prakash, O.; Ray, B.; Maiti, P. Functionalized polythiophene for corrosion inhibition and photovoltaic application. J. Appl. Polym. Sci. 2021, 138, 51306. [Google Scholar] [CrossRef]
- Koo, B.; Sletten, E.M.; Swager, T.M. Functionalized poly(3-hexylthiophene)s via lithium–bromine exchange. Macromolecules 2015, 48, 229–235. [Google Scholar] [CrossRef]
- Tanaka, S.; Rosli, S.K.B.; Takada, K.; Taniai, N.; Yoshitomi, T.; Ando, H.; Matsumoto, K. Effects of bromination of poly(3-hexylthiophene) on the performance of bulk heterojunction solar cells. RSC Adv. 2017, 7, 46874–46880. [Google Scholar] [CrossRef]
- Boufflet, P.; Casey, A.; Xia, Y.; Stavrinou, P.N.; Heeney, M. Pentafluorobenzene end-group as a versatile handle for para fluoro “click” functionalization of polythiophenes. Chem. Sci. 2017, 8, 2215–2225. [Google Scholar] [CrossRef]
- Nam, C.-Y.; Qin, Y.; Park, Y.S.; Hlaing, H.; Lu, X.; Ocko, B.M.; Black, C.T.; Grubbs, R.B. Photo-cross-linkable azide-functionalized polythiophene for thermally stable bulk heterojunction solar cells. Macromolecules 2012, 45, 2338–2347. [Google Scholar] [CrossRef]
- Bu, H.-B.; Götz, G.; Reinold, E.; Vogt, A.; Schmid, S.; Segura, J.L.; Blanco, R.; Gómez, R.; Bäuerle, P. Efficient post-polymerization functionalization of conducting poly(3, 4-ethylenedioxythiophene)(PEDOT) via ‘click’-reaction. Tetrahedron 2011, 67, 1114–1125. [Google Scholar] [CrossRef]
- Buga, K.; Kepczynska, K.; Kulszewicz-Bajer, I.; Zagórska, M.; Demadrille, R.; Pron, A.; Quillard, S.; Lefrant, S. Poly (alkylthiophene) with pendant dianiline groups via postpolymerization functionalization: Preparation, spectroscopic, and spectroelectrochemical characterization. Macromolecules 2004, 37, 769–777. [Google Scholar] [CrossRef]
- Richter, T.V.; Bühler, C.; Ludwigs, S. Water-and ionic-liquid-soluble branched polythiophenes bearing anionic and cationic moieties. J. Am. Chem. Soc. 2012, 134, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, C.; Zhang, Y.; Sun, J.; Cao, L.; Ji, J.; Feng, F. Facile crosslinking of polythiophenes by polyethylenimine via ester aminolysis for selective Cu (II) detection in water. Biosens. Bioelectron. 2018, 109, 255–262. [Google Scholar] [PubMed]
- Lanzi, M.; Costa-Bizzarri, P.; Paganin, L.; Cesari, G. Synthesis by post-polymerization functionalization of sensitive polythiophenes for selective chemo-recognition purposes. React. Funct. Polym. 2007, 67, 329–340. [Google Scholar] [CrossRef]
- Liu, J.; McCullough, R.D. End group modification of regioregular polythiophene through postpolymerization functionalization. Macromolecules 2002, 35, 9882–9889. [Google Scholar] [CrossRef]
- Barsch, U.; Beck, F. Anodic overoxidation of polythiophenes in wet acetonitrile electrolytes. Electrochim. Acta 1996, 41, 1761–1771. [Google Scholar] [CrossRef]
- Krische, B.; Zagorska, M. The polythiophene paradox. Synth. Met. 1989, 28, 263–268. [Google Scholar] [CrossRef]
- Pud, A. Stability and degradation of conducting polymers in electrochemical systems. Synth. Met. 1994, 66, 1–18. [Google Scholar] [CrossRef]
- Inagi, S.; Hosaka, K.; Hayashi, S.; Fuchigami, T. Solid-phase halogenation of a conducting polymer film via electrochemical polymer reaction. J. Electrochem. Soc. 2010, 157, E88. [Google Scholar] [CrossRef]
- Streitwieser, A., Jr. Solvolytic displacement reactions at saturated carbon atoms. Chem. Rev. 1956, 56, 571–752. [Google Scholar]
- Isse, A.A.; Lin, C.Y.; Coote, M.L.; Gennaro, A. Estimation of standard reduction potentials of halogen atoms and alkyl halides. J. Phys. Chem. B 2011, 115, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Kurioka, T.; Nishiyama, H.; Tomita, I.; Inagi, S. Improvement of Current Efficiency in Anodic Chlorination of Poly (3-hexylthiophene) by using a Boron Trifluoride-Diethyl Ether Complex. ChemElectroChem 2018, 5, 753–755. [Google Scholar] [CrossRef]
- Patil, Y.; Sassona, Y. Chemoselective reduction of nitroarenes to aromatic amines with commercial metallic iron powder in water under mild reaction conditions. Org. Chem. Curr. Res 2015, 4, 1–4. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Wang, J.; Li, H.; Zhou, K.; Gui, R.; Xian, K.; Qi, Q.; Yang, X.; Chen, Y. Brominated Polythiophene Reduces the Efficiency-Stability-Cost Gap of Organic and Quantum Dot Hybrid Solar Cells. Adv. Energy Mater. 2022, 12, 2201975. [Google Scholar] [CrossRef]
- Florio, S.; Salomone, A. Heterocycle-mediated ortho-functionalization of aromatic compounds: The DoM methodology and synthetic utility. Synthesis 2016, 48, 1993–2008. [Google Scholar] [CrossRef]
- Yin, J.; Zhou, W.; Zhang, L.; Xie, Y.; Yu, Z.; Shao, J.; Ma, W.; Zeng, J.; Chen, Y. Improved glass transition temperature towards thermal stability via thiols solvent additive versus DIO in polymer solar cells. Macromol. Rapid Commun. 2017, 38, 1700428. [Google Scholar] [CrossRef]
- Zotti, G.; Zecchin, S.; Schiavon, G.; Vercelli, B.; Berlin, A. Novel polythiophene regular copolymers from 3′,4′-diamino-and 3′,4′-dinitro-terthiophenes: Modulation of electronic properties via post-polymerization functionalization. Electrochim. Acta 2005, 50, 1469–1474. [Google Scholar] [CrossRef]
- Hayashi, S.; Inagi, S.; Fuchigami, T. Synthesis of 9-substituted fluorene copolymers via chemical and electrochemical polymer reaction and their optoelectronic properties. Macromolecules 2009, 42, 3755–3760. [Google Scholar] [CrossRef]
- Xiao, L.; Sun, J.; Liu, L.; Hu, R.; Lu, H.; Cheng, C.; Huang, Y.; Wang, S.; Geng, J. Enhanced photothermal bactericidal activity of the reduced graphene oxide modified by cationic water-soluble conjugated polymer. ACS Appl. Mater. Interfaces 2017, 9, 5382–5391. [Google Scholar]
- Bao, B.; Yuwen, L.; Zhan, X.; Wang, L. Water-soluble hyperbranched polyelectrolytes with high fluorescence quantum yield: Facile synthesis and selective chemosensor for Hg2+ and Cu2+ ions. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 3431–3439. [Google Scholar] [CrossRef]
- Salatelli, E.; Angiolini, L.; Brazzi, A.; Lanzi, M.; Scavetta, E.; Tonelli, D. Synthesis, characterization and electrochemical properties of new functional polythiophenes. Synth. Met. 2010, 160, 2681–2686. [Google Scholar] [CrossRef]
- Guselnikova, O.A.; Postnikov, P.S.; Fitl, P.; Tomecek, D.; Sajdl, P.; Elashnikov, R.; Kolska, Z.; Chehimi, M.M.; Švorčík, V.; Lyutakov, O. Tuning of PEDOT: PSS properties through covalent surface modification. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 378–387. [Google Scholar] [CrossRef]
- Istif, E.; Mantione, D.; Vallan, L.; Hadziioannou, G.; Brochon, C.; Cloutet, E.; Pavlopoulou, E. Thiophene-Based Aldehyde Derivatives for Functionalizable and Adhesive Semiconducting Polymers. ACS Appl. Mater. Interfaces 2020, 12, 8695–8703. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Hazra, S.; Shit, A.; Chatterjee, D.P.; Banerjee, A.; Nandi, A.K. Development of Polythiophene–Tripeptide Covalent Conjugates Showing Excellent Structure-Dependent Photophysical and Photocurrent Properties. J. Phys. Chem. C 2021, 125, 17518–17529. [Google Scholar] [CrossRef]
- DEMADRILLE, R.; BUGA, K.; ZAGORSKA, M.; QUILLARD, S.; RANNOU, P.; KULSZEWICZ-BAJER, I.; PRON, A. Spectroscopic and Spectroelectrochemical Studies of New Poly (thiophene)–Based Conjugated Polymers Containing Oligoaniline Side Chains. Nonlinear Opt. 2005, 32, 39–57. [Google Scholar]
- Buga, K.; Pokrop, R.; Zagórska, M.; Demadrille, R.; Genoud, F. Random and regioregular thiophene-based copolymers containing oligoaniline side chains: Synthesis, spectroscopic and spectroelectrochemical investigations. Synth. Met. 2005, 153, 137–140. [Google Scholar] [CrossRef]
- Baüerle, P.; Hiller, M.; Scheib, S.; Sokolowski, M.; Umbach, E. Post-polymerization functionalization of conducting polymers: Novel poly(alkylthiophene)s substituted with easily. Replaceable activated ester groups. Adv. Mater. 1996, 8, 214–218. [Google Scholar] [CrossRef]
- Paganin, L.; Lanzi, M.; Costa-Bizzarri, P.; Bertinelli, F.; Masi, C. Synthesis and Characterization of a Highly Soluble Poly [3-(10-hydroxydecyl) thiophene]. In Macromolecular Symposia; WILEY-VCH Verlag: Weinheim, Germany, 2004; pp. 11–20. [Google Scholar]
- Iraqi, A.; Pickup, D.F. Preparation and characterisation of tractable regioregular head-to-tail poly(3-alkylthiophene)s with ferrocene substituents. Polym. Int. 2006, 55, 780–783. [Google Scholar] [CrossRef]
- Salatelli, E.; Benelli, T.; Caretti, D.; Cocchi, V.; Giorgini, L.; Lanzi, M.; Mazzocchetti, L. Novel porphyrin-containing regioregular poly(alkylthiophene) copolymers tested as polymeric solar cells. Polymer 2016, 97, 314–322. [Google Scholar] [CrossRef]
- Jayaraman, S.; Rajarathnam, D.; Srinivasan, M. Formation of polythiophene multilayers on solid surfaces by covalent molecular assembly. Mater. Sci. Eng. B 2010, 168, 45–54. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, M.; Wang, Y.; Pan, Q.; Gong, Q.; Xia, Z.; Li, Y. Remarkably enhanced performances of novel polythiophene-grafting-graphene oxide composite via long alkoxy linkage for supercapacitor application. Carbon 2019, 147, 519–531. [Google Scholar] [CrossRef]
- Lan, X.; Liu, C.; Wang, T.; Hou, J.; Xu, J.; Tan, R.; Nie, G.; Jiang, F. Effect of functional groups on the thermoelectric performance of carbon nanotubes. J. Electron. Mater. 2019, 48, 6978–6984. [Google Scholar] [CrossRef]
- Ranu, B.C.; Bhar, S. Dealkylation of ethers. A review. Org. Prep. Proced. Int. 1996, 28, 371–409. [Google Scholar] [CrossRef]
- Pei, Q.; Zuccarello, G.; Ahlskog, M.; Inganäs, O. Electrochromic and highly stable poly(3,4-ethylenedioxythiophene) switches between opaque blue-black and transparent sky blue. Polymer 1994, 35, 1347–1351. [Google Scholar] [CrossRef]
- Deepa, M.; Bhandari, S.; Kant, R. A comparison of charge transport behavior in functionalized and non-functionalized poly 3, 4-(ethylenedioxythiophene) films. Electrochim. Acta 2009, 54, 1292–1303. [Google Scholar] [CrossRef]
- Balog, M.; Rayah, H.; Le Derf, F.; Sallé, M. A versatile building block for EDOT or PEDOT functionalization. New J. Chem. 2008, 32, 1183–1188. [Google Scholar] [CrossRef]
- Daugaard, A.E.; Hvilsted, S.; Hansen, T.S.; Larsen, N.B. Conductive polymer functionalization by click chemistry. Macromolecules 2008, 41, 4321–4327. [Google Scholar] [CrossRef]
- Bu, H.-B.; Götz, G.; Reinold, E.; Vogt, A.; Schmid, S.; Blanco, R.; Segura, J.L.; Bäuerle, P. “Click”-functionalization of conducting poly(3,4-ethylenedioxythiophene)(PEDOT). Chem. Commun. 2008, 1320–1322. [Google Scholar] [CrossRef]
- Wei, B.; Ouyang, L.; Liu, J.; Martin, D.C. Post-polymerization functionalization of poly(3,4-propylenedioxythiophene)(PProDOT) via thiol–ene “click” chemistry. J. Mater. Chem. B 2015, 3, 5028–5034. [Google Scholar] [CrossRef]
- Daugaard, A.E.; Hansen, T.S.; Larsen, N.B.; Hvilsted, S. Microwave assisted click chemistry on a conductive polymer film. Synth. Met. 2011, 161, 812–816. [Google Scholar] [CrossRef]
- Goll, M.; Ruff, A.; Muks, E.; Goerigk, F.; Omiecienski, B.; Ruff, I.; González-Cano, R.C.; Navarrete, J.T.L.; Delgado, M.C.R.; Ludwigs, S. Functionalized branched EDOT-terthiophene copolymer films by electropolymerization and post-polymerization “click”-reactions. Beilstein J. Org. Chem. 2015, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Besbes, M.; Trippé, G.; Leviallain, E.; Mazari, M.; Le Derf, F.; Perepichka, I.F.; Derdour, A.; Gorgues, A.; Sallé, M.; Roncali, J. Rapid and Efficient Post-Polymerization Functionalization of Poly (3,4-ethylenedioxythiophene)(PEDOT) Derivatives on an Electrode Surface. Adv. Mater. 2001, 13, 1249–1252. [Google Scholar] [CrossRef]
- Bhandari, S.; Deepa, M.; Srivastava, A.K.; Kant, R. Post-polymerization functionalization of poly(3,4-ethylenedioxythiophene) films by 1-fluoro-2-nitro-4-azidobenzene: Electrochromism and redox behavior. J. Mater. Chem. B 2009, 19, 2336–2348. [Google Scholar] [CrossRef]
- Weng, M.; Yu, X. Electrochemical oxidation of para-aminophenol with rare earth doped lead dioxide electrodes: Kinetics modeling and mechanism. Front. Chem. 2019, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Gulprasertrat, N.; Chapromma, J.; Aree, T.; Sritana-anant, Y. Synthesis of functionalizable derivatives of 3,4-ethylenedioxythiophene and their solid-state polymerizations. J. Appl. Polym. Sci. 2015, 132, 42233. [Google Scholar] [CrossRef]
- Huang, X.; Yang, L.; Emanuelsson, R.; Bergquist, J.; Strømme, M.; Sjödin, M.; Gogoll, A. A versatile route to polythiophenes with functional pendant groups using alkyne chemistry. Beilstein J. Org. Chem. 2016, 12, 2682–2688. [Google Scholar] [CrossRef]
- Qu, K.; Kondengaden, S.M.; Li, J.; Wang, X.; Sevilla, M.D.; Li, L.; Zeng, X. Carbohydrate-functionalized polythiophene biointerface: Design, fabrication, characterization and application for protein analysis. Appl. Surf. Sci. 2019, 486, 561–570. [Google Scholar] [CrossRef]
- Camurlu, P.; Guven, N.; Bicil, Z. Ferrocene clicked polypyrrole derivatives: Effect of spacer group on electrochemical properties and post-polymerization functionalization. Des. Monomers Polym. 2016, 19, 212–221. [Google Scholar] [CrossRef]
- Camurlu, P.; Karagoren, N. Clickable, versatile poly(2,5-dithienylpyrrole) derivatives. React. Funct. Polym. 2013, 73, 847–853. [Google Scholar] [CrossRef]
- Raicopol, M.; Andronescu, C.; Atasiei, R.; Hanganu, A.; Pilan, L. Post-polymerization electrochemical functionalization of a conducting polymer: Diazonium salt electroreduction at polypyrrole electrodes. J. Electrochem. Soc. 2014, 161, G103. [Google Scholar] [CrossRef]
- Jang, K.S.; Lee, H.; Moon, B. Synthesis and characterization of water soluble polypyrrole doped with functional dopants. Synth. Met. 2004, 143, 289–294. [Google Scholar] [CrossRef]
- Das, S.; Dutta, K.; Kundu, P.P. Sulfonated polypyrrole matrix induced enhanced efficiency of Ni nanocatalyst for application as an anode material for DMFCs. Mater. Chem. Phys. 2016, 176, 143–151. [Google Scholar] [CrossRef]
- Bieniarz, C.; Husain, M.; Tarcha, P.J. Evidence for the Addition of Nucleophiles to the Surface of Polypyrrole Latex. Macromolecules 1999, 32, 792–795. [Google Scholar] [CrossRef]
- Frontera, E. Química Combinatoria de Nanomateriales. Ph.D. Thesis, UNRC, Río Cuarto, Argentina, 2012. [Google Scholar]
- Miodek, A.; Mejri-Omrani, N.; Khoder, R.; Korri-Youssoufi, H. Electrochemical functionalization of polypyrrole through amine oxidation of poly(amidoamine) dendrimers: Application to DNA biosensor. Talanta 2016, 154, 446–454. [Google Scholar] [CrossRef]
- Raicopol, M.; Andronescu, C.; Atasiei, R.; Hanganu, A.; Manea, A.M.; Rau, I.; Kajzar, F.; Pilan, L. Synthesis of conducting azopolymers by electrochemical grafting of a diazonium salt at polypyrrole electrodes. Synth. Met. 2015, 206, 84–91. [Google Scholar] [CrossRef]
- Huisgen, R. Centenary lecture-1,3-dipolar cycloadditions. In Proceedings of the Chemical Society of London; Royal Soc Chemistry Thomas Graham House, Science Park, Milton Rd: Cambridge, UK, 1961; p. 357. [Google Scholar]
- Assresahegn, B.D.; Brousse, T.; Bélanger, D. Advances on the use of diazonium chemistry for functionalization of materials used in energy storage systems. Carbon 2015, 92, 362–381. [Google Scholar] [CrossRef]
- Gomberg, M.; Bachmann, W.E. The synthesis of biaryl compounds by means of the diazo reaction. J. Am. Chem. Soc. 1924, 46, 2339–2343. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.; Kim, C.; Kang, E.; Tan, K. Difference in doping behavior between polypyrrole films and powders. Synth. Met. 1995, 72, 243–248. [Google Scholar] [CrossRef]
- Gustafsson, G.; Lundström, I.; Liedberg, B.; Wu, C.; Inganäs, O.; Wennerström, O. The interaction between ammonia and poly(pyrrole). Synth. Met. 1989, 31, 163–179. [Google Scholar] [CrossRef]
- Guimard, N.K.; Gomez, N.; Schmidt, C.E. Conducting polymers in biomedical engineering. Prog. Polym. Sci. 2007, 32, 876–921. [Google Scholar] [CrossRef]
- Hu, J.; Wang, J.; Nguyen, T.H.; Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, H.; Louis, E.J.; MacDiarmid, A.G.; Chiang, C.K.; Heeger, A.J. Synthesis of electrically conducting organic polymers: Halogen derivatives of polyacetylene,(CH) x. J. Chem. Soc. Chem. Commun. 1977, 578–580. [Google Scholar] [CrossRef]
- Pauly, A.C.; Theato, P. Synthesis and characterization of poly(phenylacetylenes) featuring activated ester side groups. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 211–224. [Google Scholar] [CrossRef]
- Fabre, B.; Simonet, J. Post-polymerization electrochemical functionalization of a conducting polymer: Anodic cyanation of poly(p-dimethoxybenzene). J. Electroanal. Chem. 1996, 416, 187–189. [Google Scholar] [CrossRef]
- Duchateau, J.; Lutsen, L.; Guedens, W.; Cleij, T.J.; Vanderzande, D. Versatile post-polymerization functionalization of poly(p-phenylene vinylene) copolymers containing carboxylic acid substituents: Development of a universal method towards functional conjugated copolymers. Polym. Chem. 2010, 1, 1313–1322. [Google Scholar] [CrossRef]
- Fabre, B.; Kanoufi, F.; Simonet, J. Electrochemical and XPS investigations of the anodic substitution of an electronic conducting polymer. Cyanation of poly[(1,4-dimethoxybenzene)-co-(3-methylthiophene)]. J. Electroanal. Chem. 1997, 434, 225–234. [Google Scholar] [CrossRef]
- Andreades, S.; Zahnow, E. Anodic cyanations of aromatic compounds. J. Am. Chem. Soc. 1969, 91, 4181–4190. [Google Scholar] [CrossRef]
- Bradley, D. Precursor-route poly(p-phenylenevinylene): Polymer characterisation and control of electronic properties. J. Phys. D Appl. Phys. 1987, 20, 1389. [Google Scholar] [CrossRef]
- Cid, J.-J.; Duchateau, J.; Van Severen, I.; Ganivet, C.R.; de la Torre, G.; Vazquez, P.; Cleij, T.; Lutsen, L.; Vanderzande, D.; Torres, T. Synthesis and characterization of high molecular weight phthalocyanine-PPV copolymers through post-polymerization functionalization. J. Porphyr. Phthalocyanines 2011, 15, 659–666. [Google Scholar] [CrossRef]
- Campo, B.J.; Duchateau, J.; Ganivet, C.R.; Ballesteros, B.; Gilot, J.; Wienk, M.M.; Oosterbaan, W.D.; Lutsen, L.; Cleij, T.J.; de la Torre, G. Broadening the absorption of conjugated polymers by “click” functionalization with phthalocyanines. Dalton Trans. 2011, 40, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, Z.; Huang, X.; Lu, G.; Manners, I.; Winnik, M.A.; Feng, C. Water-Dispersible, Colloidally Stable, Surface-Functionalizable Uniform Fiberlike Micelles Containing a π-Conjugated Oligo (p-phenylenevinylene) Core of Controlled Length. Macromolecules 2020, 53, 8009–8019. [Google Scholar] [CrossRef]
- Borah, R.; Ingavle, G.C.; Kumar, A.; Sandeman, S.R.; Mikhalovsky, S.V. Surface-functionalized conducting nanofibers for electrically stimulated neural cell function. Biomacromolecules 2021, 22, 594–611. [Google Scholar] [CrossRef] [PubMed]
- Wosnick, J.H.; Mello, C.M.; Swager, T.M. Synthesis and application of poly(phenylene ethynylene)s for bioconjugation: A conjugated polymer-based fluorogenic probe for proteases. J. Am. Chem. Soc. 2005, 127, 3400–3405. [Google Scholar] [CrossRef]
- Neher, D. Polyfluorene homopolymers: Conjugated liquid-crystalline polymers for bright blue emission and polarized electroluminescence. Macromol. Rapid Commun. 2001, 22, 1365–1385. [Google Scholar] [CrossRef]
- Li, Y.; Vamvounis, G.; Holdcroft, S. Control of conjugation length and enhancement of fluorescence efficiency of poly(p-phenylenevinylene)s via post-halogenation. Chem. Mater. 2002, 14, 1424–1429. [Google Scholar] [CrossRef]
- Marsitzky, D.; Klapper, M.; Müllen, K. End-functionalization of poly(2,7-fluorene): A key step toward novel luminescent rod-coil block copolymers. Macromolecules 1999, 32, 8685–8688. [Google Scholar] [CrossRef]
- Xue, C.; Donuru, V.R.; Liu, H. Facile, versatile prepolymerization and postpolymerization functionalization approaches for well-defined fluorescent conjugated fluorene-based glycopolymers. Macromolecules 2006, 39, 5747–5752. [Google Scholar] [CrossRef]
- Kardelis, V.; Denk, M.M.; Adronov, A. Click-Functionalization of a Poly (Tetrazine-co-Fluorene)-Conjugated Polymer with a Series of trans-Cyclooctene Derivatives. Angew. Chem. Int. Ed. 2021, 133, 3017–3023. [Google Scholar] [CrossRef]
- Malik, A.H.; Hussain, S.; Kalita, A.; Iyer, P.K. Conjugated polymer nanoparticles for the amplified detection of nitro-explosive picric acid on multiple platforms. ACS Appl. Mater. Interfaces 2015, 7, 26968–26976. [Google Scholar] [CrossRef]










| Polymer (Form) | % Funct | Added Group | Reaction | Polymer Form | Reactants | Target Property | Reaction Time (Temperature) | Ref. |
|---|---|---|---|---|---|---|---|---|
| PANI (EB) | 50 | –SO3− | SEAr | Bulk | SO3−/H2SO4 | Water solubility | 2 h (r.t) | [48] |
| PANI (EB) | 50 | –SO3− | SEAr | Bulk | SO3−/H2SO4 | Water solubility | 2 h (r.t.) | [49] |
| PANI PN/s | 50 | –SO3− | SEAr | Bulk | HSO3Cl H2O | Water solubility | 5 h (80 °C) 4 h (100 °C) | [50] |
| PANI(EB) | 25 | –SO3 | SEAr | Bulk | Emeraldine hydrogen sulfate | 130 °C | [51] | |
| Aniline (Echem polym.) | 24–89 | –SO3H | SEAr | Film | FSO3H/can | Solubility in wide range of solvents | r.t. | [52] |
| PANI LE | 75 | –SO3− | SEAr | Bulk | SO3−/H2SO4 | solubility | 1 h (5 °C) | [53] |
| PANI EB | 50 | –SO3− | SEAr | Film | SO3−/H2SO4 | Self-doping, thermostability | 3 h (25 °C) | [54] |
| PANI–LE (Reduced state) | -- | NO2C6H4– | CANQ | Film | NO2C6H4–N2+BF4− | Spatial functionalization control | Var. | [55] |
| PNMANI | 50–100 | –SO3− | SEAr | Bulk | SO3−/H2SO4 | Ion exchange | 1 h (25 °C) | [56] |
| PANI EB | 50 | –Br | SEAr (Halogenation) | Bulk | Br2/HBr | Conductivity increase | [57] | |
| PANI LE/EB | Var. | Amide | NuSAc | Echem film | Anhydrides | Film modification | Var. | [58] |
| PANI EB | 30 | Aromatic amide (with SO3H) | SNAc | Bulk | o-sulfobenzoic anhydride | Water solubility | 24 h (25 °C) | [59] |
| PANI EB | 47 | –C3H6– C6H4–SO3− | SN2 | Bulk | (1) H-Na (2) p-(3-BrC3H6)–C6H4–SO3−Na+ | Water solubility Self-doping | (1) 2 h (45 °C) (2) 24 h (r.t.) | [60] |
| PANI EB | -- | pyrrolidinium | CANQ | Bulk | pyrrolidine | Synthesis | 96 h (25 °C) | [61] |
| PANI EB | 20.7 | C12H25SH | CANQ | Bulk | Dodecanotiol | Synthesis | 6 min (25 °C) | [62] |
| PANI EB | 15–26 | sulfonic acid moiety | CANQ | Film | 3-mercapto-1-propanesulfonic acid sodium salt Acetic acid (cat) | Conductivity Thermally Stability Self-doping | 14 h (r.t.) | [62] |
| PANI EB | 38–121 | butylthio group | CANQ | Film | butane-1-thiol | Solubility in wide range of solvents | 14 h (r.t.) | [63] |
| PANI EB | 50 | SO32−/SO3H− | CANQ | Bulk | SO3H− | Self-doping | 2 h (80 °C) | [64] |
| PANI PN | Sulfinic Anions | CANQ | Bulk | C6H5SO2− | Modification | 2 h (70 °C) | [65] | |
| PANI ES | Var. | Chloride Ions | CANQ | Bulk | HCl | Synthesis | <1 h (5 °C) | [66] |
| PANI (LE) | Low | SNAr activated aromatic halides | SNAr | Bulk | 4-NO2–C6H4–Cl 2-NO2–C6H4–Cl 2,4-diNO2–C6H3−Cl | Modification | 2 h (70 °C) | [67] |
| PANI (LE) | Low | –CO–CH3 | SNAc | Bulk | Ethanoic anhydride (neat) | Solubility | 2 h (25 °C) | [67] |
| PANI Echem | 25–100 | SO32−/SO3H− | CANQ | Film | –SO3H | Controlled modification | Var. | [68] |
| PANI Echem | -- | –S–R–Ferrocene | CANQ | Film | HOCH2CH2SH | Linking of redox groups | 60–72 h (r.t.) | [69] |
| PANI ES | 100 | –SO3H | SEAr | Bulk | ClSO3H | Interpolymer complex | 5 h (80 °C) | [70] |
| P(2BrANI) | 100 | –PO3H2 | SNAr | film | HPo(OEt)2 Pd0 (cat) | Self-doping | 2 days (120 °C) | [71] |
| PANI brushes linked by S-Me | 50 | –SO3H | CANQ | Polymer brushes | SO32− | Sulfite determination | – | [72] |
| P(ANI boronic acid) | 100 | OH− X–(halide) | ipso-reaction | Echem film | –OH –X | Synthesis | 10 min (25 °C) | [73] |
| PPDA | <100 | –S–SO3− | CANQ | Bulk | S2O32− | Solubility | 6 h (0–5 °C) | [74] |
| PPDA | 25–50 | N2H4 | CANQ | Bulk | –NH–NH2 | Reduction | 12 h (0–5 °C) | [75] |
| PANI (ES) | 93–94 | –SO3- | SEAr | Bulk | SO3/H2SO4 | Crystalline structure | 6 h (25 °C) | [76] |
| PANI (ES) | 50 | –SO3− | SEAr | Bulk | SO3/H2SO4 | Synthesis | 0.5–2 h (25 °C) | [77] |
| PANI (ES) | 0 | none | SEAr | Bulk | H2SO4 | Synthesis | 24 h (100–150 °C) | [77] |
| PANI (LE) | -- | >N–(CH2)3–SO3− | SN2 | Echem film | propanesultone | Self-doping sensor at pH 7 | 12 h (r.t.) | [78] |
| Poly(3-ethynylaniline)-co-ANI) | 100 | –C=C–SR | Thiol-yne click | Bulk | PEG-dithiol | Linking w/graphene | 5 min (r.t.) | [79] |
| PANI(ES) | -- | –N–NO | SEN | Film | NO+ | Lithography | 15 min (r.t.) | [80] |
| PANI(ES) | -- | –N–NO | SEN | Film | NO+ | Photolithography | 15 min (r.t.) | [81] |
| PANI(EB) | 25 | –N–tBOC | SN | Bulk | t-BOC | Photolithography | 3 h (80 °C) | [82] |
| Polymer (Form) | % Funct | Added group | Reaction | Polymer Form | Reactants | Target Property | Refs |
|---|---|---|---|---|---|---|---|
| PT, P3MeT, poly(2,2′-bithiophene) | var. (25 to 80) | Halide (Cl, Br) | Nucleophilic addition | Film | Cl−, Br− | Solubility | [136] |
| PT, P3MeT, poly(2,2′-bithiophene) | var. (25 to 80) | CH3O– | Nucleophilic addition | Film | CH3O– | Solubility | [136] |
| P3MeT | 50 | –Br | Nucleophilic addition | Film | Et4N+Br−/ACN | Solubility | [137] |
| P3MeT | 70 | –Cl | Nucleophilic addition | Film | Et4N+Cl−/ACN | Solubility | [137] |
| poly(thiophene-alt-fluorene) | - | Halide (Cl, Br) | Nucleophilic additI | Film | Et4N+Br−/CAN Et4N+Cl−/ACN | Modification | [138] |
| P3HT | 72 | –Cl | Nucleophilic addIon | Bulk | Et4N+Cl−/ACN | Modification | [139] |
| P3HS and P3HT-b-P3HS | 49/65 | –Cl | Nucleophilic addition | Film | boron trifluoride-diethyl ether (BFEE) | Modification | [140] |
| P3HT | 96 | –Cl | Nucleophilic addition | Film | AlCl3/MeCN | Changes in optoelectronic properties | [141] |
| PTF and PBT | >70 | –Cl | Nucleophilic addition | Film | Et4NX in thin layered cell | Improving in optical properties | [142] |
| P3HT | 93 | –Br | SEAr | Bulk | NBS | Electrooptical properties | [143] |
| P3HT | 98 | –Cl | SEAr | Bulk | NCS | Electrooptical properties | [143] |
| P3HT | 93 | –NO2 | SEAr | Bulk | Fuming sulfuric acid | Electrooptical properties | [143] |
| Diazoted-P3HT | 58 | –CN | SEAr | Film | copper (I) cyanide | Thermal stability/Corrosion inhibition | [144] |
| P3HT | - | –Br | SEAr | Film | NBS | Modification for other functionalizations | [145] |
| P3HT | var. (2 to 84) | –Br | SEAr | Film | NBS | Changes in photovoltaic properties | [146] |
| P3OT | 64 | pentafluorobenzene (PFB) | Lithiation (SNAr) | Film | Click reaction | Energy transfer | [147] |
| P3HT-Si5 | - | Azide moiety | Nucleophilic susbstitution | Film | P3HT-Si5 → P3HTOH5 Bu4NN3 | Thermal stability | [148] |
| poly(azidomethyl-EDOT) | 90 | alkyl | Nucleophilic susbstitution | Film | Cu(CH3CI) PF6, using ACN, TFH, or benzonitrile | Modification | [149] |
| PT | - | Dianiilines groups | Amidation | Film | N-phenyl-1,4-phenylenediamine | Solution processability | [150] |
| P3HT | - | –COOH | Deprotonation | Film | deprotonation of P3T and subsequent reaction with CO2 | Solubility | [151] |
| P3HT | 50–60 | –MIM | Nucleophilic substitution (carbanion formation + hydrosilation) | Film | 3-bromopropene, methylimidazolium, N-methylimidazole | Solubility | [151] |
| PT-E | - | –Amide | Ester aminolysis | Bulk | PEI in N-methyl-2-pyrrolidone | Water solubility | [152] |
| PT-Br | 79 | –ester group | Nucleophilic substitution | Bulk/film | Sodium hexanoate in DMF | Ponderal and microstructural features | [153] |
| HT-PHT | - | –OH | Protected cross-coupling | Bulk | thienylzinc compounds/THF | Modification | [154] |
| PT-ESTER | >90 | –OH | Hydrolysis | Bulk/film | KOH/Methanol | Ponderal and microstructural features | [153] |
| Polymer (Form) | Added Group | Reaction Kind | Reaction Form | Reactants | Target Property | Ref |
|---|---|---|---|---|---|---|
| P(Py-N3) | Ferrocene | Click Azide-yne | Bulk | Ethynylferrocene Cu2+ (cat) | Electrochromic properties | [198] |
| Poly(T-Py(R-N3)-T) | Ferrocene | Click Azide-yne | Echem film | Ethynylferrocene | Electrochromic | [199] |
| PPy | Aromatic compounds | reduction of aryl diazonium salt | Echem Film | Diazonium salt + nBu4N+BF4− | Sensing and biological devices | [200] |
| PPy | –SO3− | SEAr sulfonation | Bulk | HSO3Cl | Water solubility | [201] |
| PPy | –SO3− (*) | SEAr sulfonation | Granules | HSO3Cl | Catalyst support | [202] |
| PPy nanoparticles | 2-mercaptoethanol ethanolamine | NuA | Latex particles | Mercaptoacetic Ethanollamine | Potential in immobilization of proteins | [203] |
| PPy | Multiple | NuA | Film | DOT CTA MPS ATPhe | Combinatorial synthesis | [204] |
| PPy | PAMAM(G4)-Ferrocene | Cation radical addition (†) | Film | PAMAM G4 | Biosensing | [205] |
| PPy | p-nitroazobenzene | Film | MeCN solutions containing 1 mM of diazonium salt and 0.1 M TBABF4− | Photoactivity | [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abel, S.B.; Frontera, E.; Acevedo, D.; Barbero, C.A. Functionalization of Conductive Polymers through Covalent Postmodification. Polymers 2023, 15, 205. https://doi.org/10.3390/polym15010205
Abel SB, Frontera E, Acevedo D, Barbero CA. Functionalization of Conductive Polymers through Covalent Postmodification. Polymers. 2023; 15(1):205. https://doi.org/10.3390/polym15010205
Chicago/Turabian StyleAbel, Silvestre Bongiovanni, Evelina Frontera, Diego Acevedo, and Cesar A. Barbero. 2023. "Functionalization of Conductive Polymers through Covalent Postmodification" Polymers 15, no. 1: 205. https://doi.org/10.3390/polym15010205
APA StyleAbel, S. B., Frontera, E., Acevedo, D., & Barbero, C. A. (2023). Functionalization of Conductive Polymers through Covalent Postmodification. Polymers, 15(1), 205. https://doi.org/10.3390/polym15010205