Three grades of high density polyethylene selected for this study were linear grades varying in molecular mass and its distribution and in the level of branching. H3 shows a relatively low average molecular mass with a rather narrow distribution (M
w = 120,000 and M
w/M
n = 2.2, respectively), while exhibiting slight branching—number of short chain branches (SCB) is below 5 per 1000 C atoms in the main chain. As found in previous studies [
16], such a moderate level of branching suppresses crystallization, thus slightly reduces crystallinity compared to linear grades, but helps to maintain efficient stress transfer between lamellae, even thick extended-chain crystals, during mechanical tests. This facilitates ductile behavior of the material. While the previous experiments with more linear or lower molecular mass polyethylenes led also to extended-chain crystal morphology with the desired thickness range, the samples with thick crystals were reported to be brittle, fracturing at low strains [
8,
16]. The second grade—H4—is a highly linear HDPE, with molecular mass higher than H3, and polydispersity typical for HDPE’s (M
w/M
n = 5.9). This very linear PE was expected to crystallize easier than H3, which should result in higher crystallinity, and formation of thicker and less defected lamellar crystals under pressure, yet possibly more prone to premature fracture due to lower concentration of inter-crystalline links, similarly to samples of lower molecular mass tested earlier [
8,
16]. The H5 of relatively high average molecular mass (M
w = 478,000), a broad weight distribution (M
w/M
n = 12.2) and exhibiting rather low branching (below 3 branches per 1000 C) was also expected to develop crystallinity higher than H3 due to lower branching and very broad mass distribution, including the presence of low molecular fraction [
35,
36,
37,
38,
39], form thick crystals upon crystallization at elevated pressure and perhaps demonstrate a ductile behavior (a sufficient amount of inter-crystalline links was expected to form due to short branches [
36], as in H3).
3.1. Morphology of Non-Deformed Samples
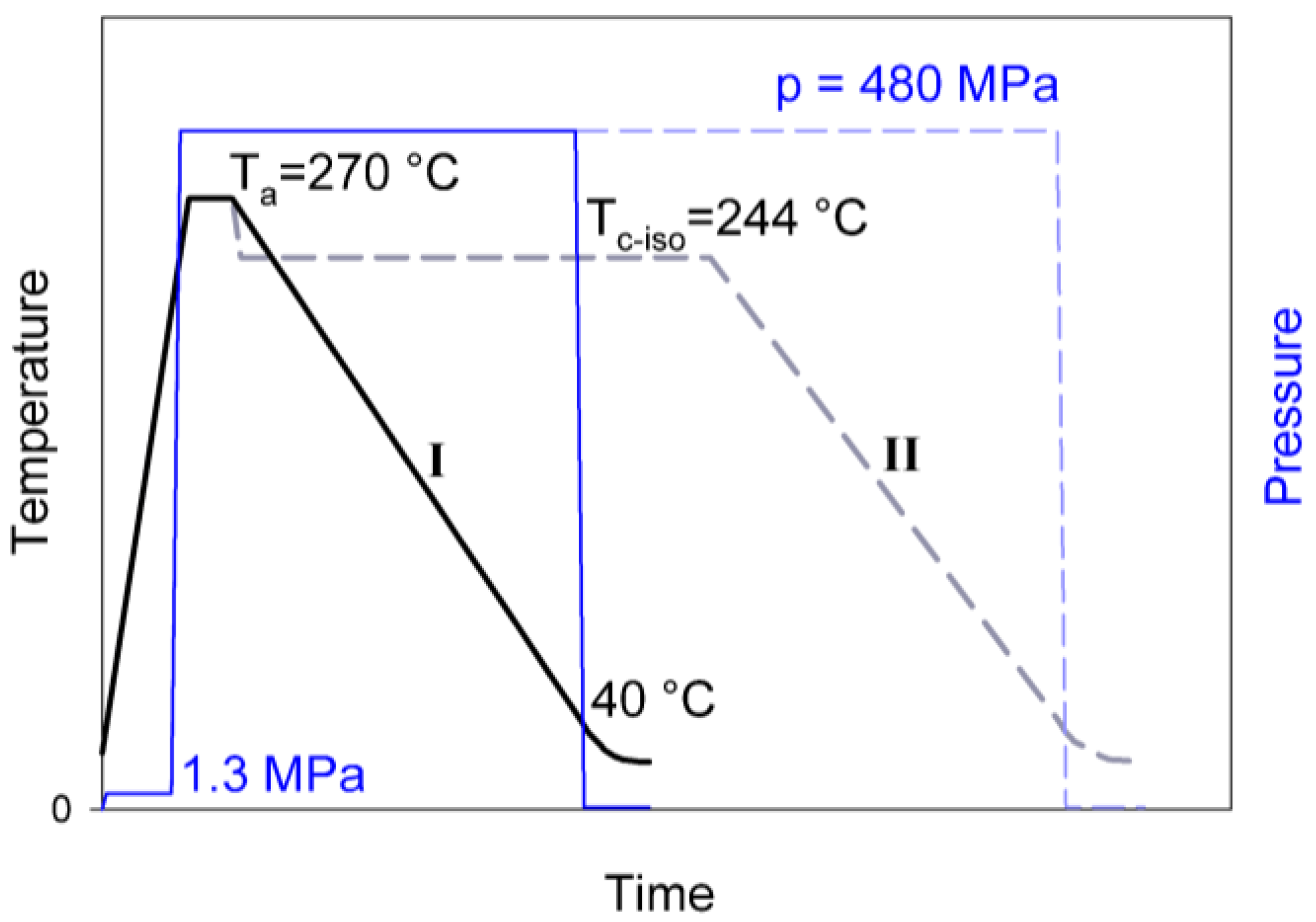
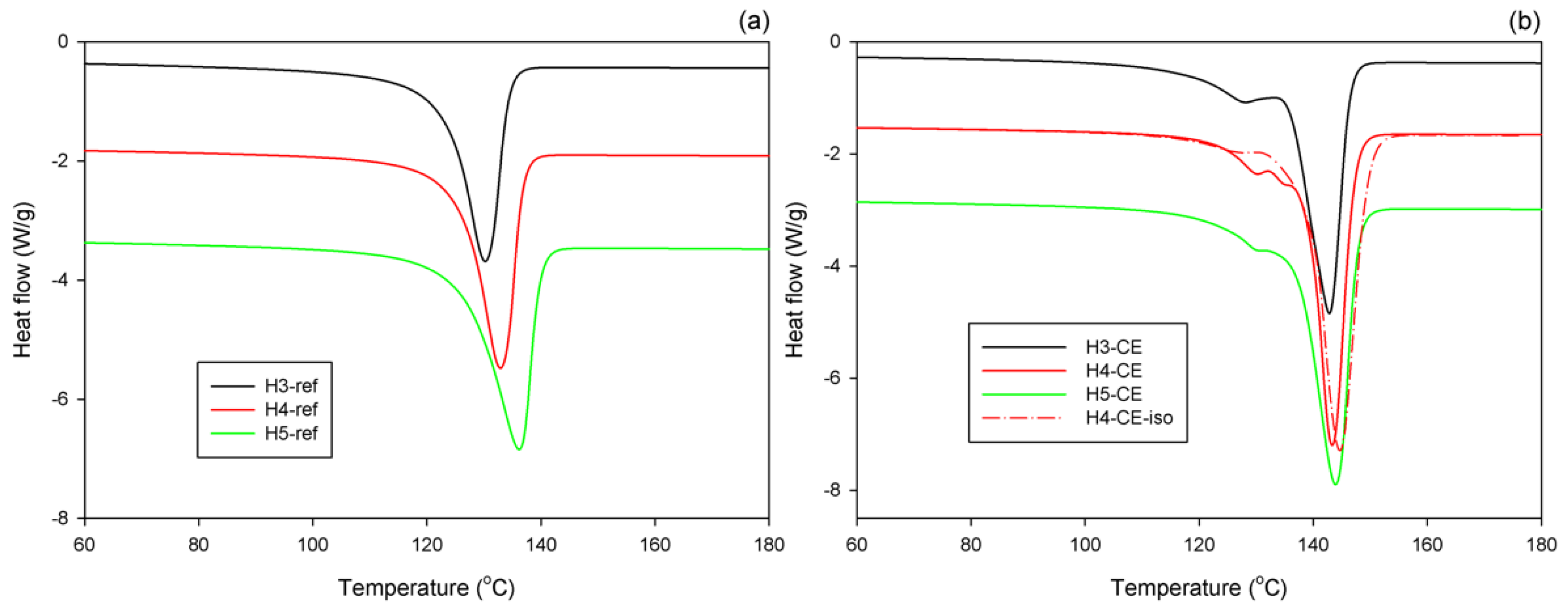
Figure 2 shows the DSC melting thermograms collected for the series of the reference samples, which were crystallized at conventional conditions (non-isothermal crystallization during fast cooling of the melt under near-atmospheric pressure), and CEPE-samples, crystallized non-isothermally under elevated pressure (cooling rate of 4 °C/min, p = 480 MPa). The results of evaluation of these curves are presented in
Table 2.
It can be seen in
Figure 2a that the reference samples of all three polymers exhibit a single melting peak, typical for high density polyethylene, with the peak temperature above 130 °C. The H3-ref sample shows the lowest melting peak temperature and crystallinity, probably due to the highest content of short branches in the chain (about five branches per 1000 C atoms in the chain, cf.
Table 1), hindering slightly crystallization process. More linear chains in H4-ref and H5-ref facilitate formation of thicker crystals (higher T
m) and higher overall crystallinity. The highest melting temperature (the thickest crystals) and the highest crystallinity were observed in H5-ref, which exhibits a high average molecular weight M
w, yet a very broad molecular weight distribution (M
w = 478,000, M
w/M
n = 12.2, respectively). This feature enables formation of relatively thick lamellae and reaching a high degree of crystallinity.
Figure 2b shows that the CEPE samples, crystallized non-isothermally at high pressure, demonstrate the main melting peak with the maximum at T
m2 = 142–144 °C, indicating the melting of very thick crystals, about 100–200 nm in thickness—cf.
Table 2. Since the crystalline stem length in these crystals is much bigger than in conventional folded-chain crystals and approaches the average chain contour length (260–350 nm, as estimated from M
n) such thick crystals are usually referred to as “extended-chain crystals” [
28]. The crystals of pseudo-hexagonal phase were formed under high pressure instead of ordinary orthorhombic phase [
27]. The high mobility of pseudo-hexagonal phase enabled a substantial crystal thickening and the formation of very thick, “extended-chain” lamellae. The crystals of pseudo-hexagonal phase were transformed to orthorhombic modification upon cooling, without reduction in size.
In addition to the main melting peak, a much smaller peak was observed on its low-temperature shoulder, at T
m1 = 129–130 °C. This peak indicates the melting of much thinner crystals, similar to the ordinary folded-chain crystals formed at conventional crystallization conditions, as e.g., in the reference samples. These folded-chain crystals formed probably in the later stage of the high-pressure process, when the temperature decreased, well after formation of extended-chain crystals [
26]. In fact, when the H4 was crystallized isothermally at T = 244 °C for a long time (sample H4-CE-iso) the size and the maximum temperature of the main melting peak of CE crystals (which formed first) increased, while the size of that secondary peak related to folded-chain crystals (grown later, upon cooling) decreased substantially, simply because much less material remained untransformed up to this stage; see
Figure 2b.
Due to high mobility in pseudo-hexagonal phase leading to crystal thickening, a considerable fraction of pre-existing chain entanglements can be resolved during crystallization and crystal thickening. This leads to a significant increase of crystallinity [
8,
29], while the remaining part of the material, incapable of crystallization and constituting an amorphous component, may demonstrate an entanglement density either lower (many entanglements resolved—as perhaps in the case of linear chains of relatively low molecular weight) or higher than in the initial melt, as some entanglements might be redistributed (swept) from the growing crystal into the amorphous phase instead of being fully resolved [
6]. This is confirmed by very high crystallinity of the CEPE samples—above 86 wt.%; in the case of slow, isothermal crystallization reaching even nearly 100 wt.% (H4-CE-iso). The small fraction of the folded-chain crystals grown in the final stage of the crystallization process (13–16 wt.%, as estimated for non-isothermally crystalized samples and only ca. 3 wt.% in H4-CE-iso; cf.
Table 2) is probably formed in these regions of the melt where entanglements were still preserved or even concentrated, so the crystallization in the extended-chain fashion there was difficult. Ultimately, only a relatively small fraction of the polymer was unable to convert to the crystalline phase (either thick extended-chain or thin folded-chain crystals) due to the high concentration of entanglements or due to other constraints, and then formed an amorphous component.
The highest melting point (presumably the thickest crystals) and the highest crystallinity were found in the samples of H4 (H4-CE and H4-CE-iso), which is essentially a highly linear PE. Samples H3-CE and H5-CE demonstrated a slightly lower melting temperature as well as crystallinity, probably due to higher branching, which made the crystallization process more difficult.
The morphology of CEPE-samples was examined with scanning electron microscopy (SEM).
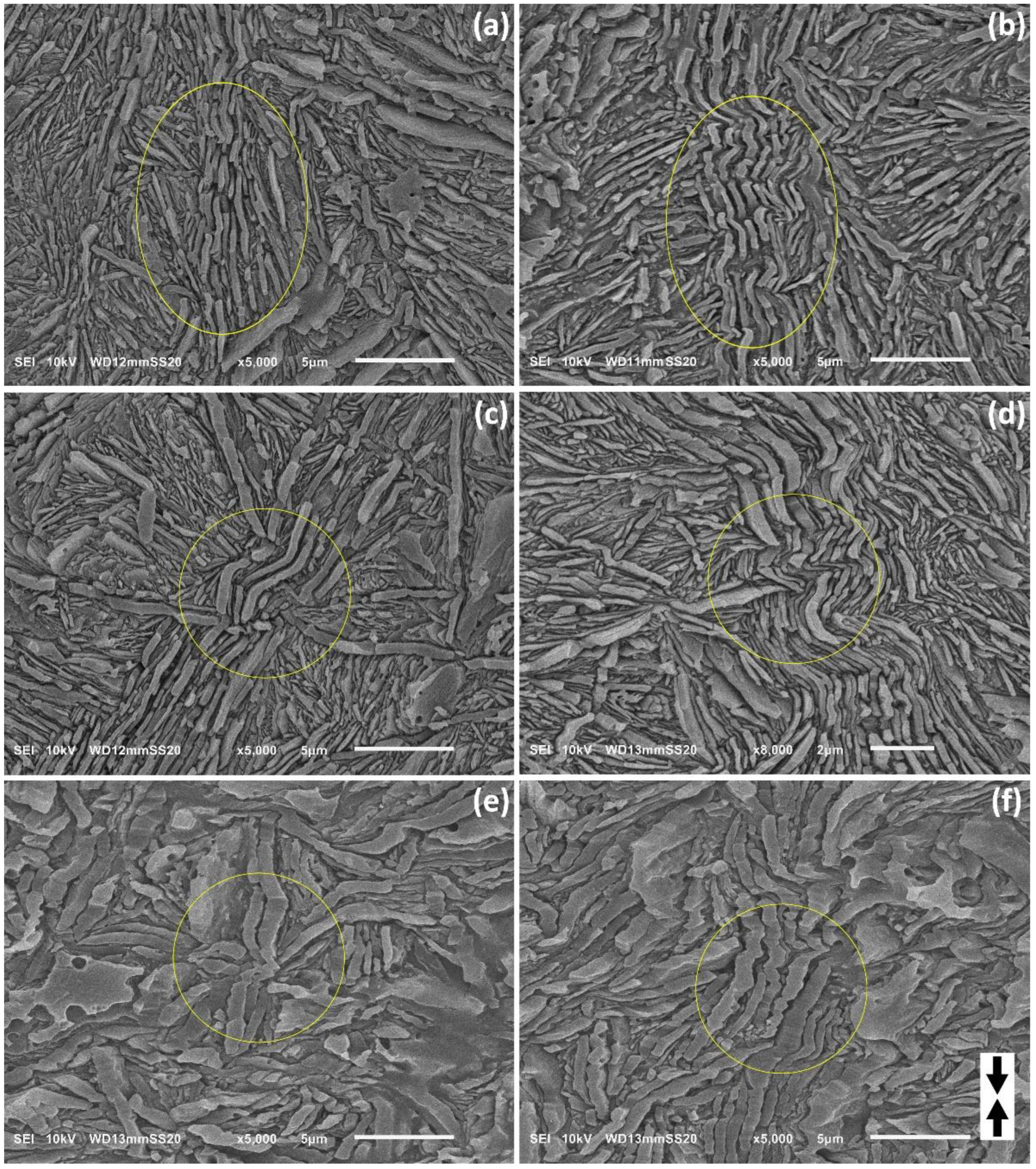
Figure 3 presents the SEM micrographs of the non-deformed samples, etched with permanganic etchant in order to expose their lamellar morphology [
34]. This treatment etches preferentially the amorphous material between lamellae, while the crystalline lamellae remain nearly undamaged, being etched a little deeper only locally, in points of high concentration of defects, e.g., dislocations [
40]. The plane of observation was exposed for etching and subsequent observation, using the microtoming method, therefore there were no features in the micrographs associated with deformation, fracture or related damage to the material that might occur in samples prepared by other methods, e.g., freeze-fracturing. Therefore, the micrographs show probably the actual morphology of non-deformed samples.
SEM observations demonstrated that the lamellae in the examined samples generally did not show any preferred orientation. Nevertheless, in order to better visualize the structure as well as for easier determination of lamella thickness, the micrographs chosen for presentation in
Figure 3 were taken in selected areas where majority of the lamellae were oriented edge-on. Note an important feature: some parallel striations can be recognized on the exposed faces of the edge-on oriented lamellae, roughly perpendicular to the lamella plane; see e.g.,
Figure 3d,f. These striations, produced by slightly uneven etching of the crystalline phase (etching rate depends on the direction in the crystal and the perfection of the crystal), visualize the direction of the chain in the lamellae [
40].
It can be seen from the presented micrographs that the lamellae in H3-CE and H4-CE are very thick, long—often much longer than 10 μm—and flat, with no twists. Usually, several adjacent lamellae are arranged parallel to each other and form stacks. These stacks are organized in higher-level radial structures resembling spherulites, radiating from the center of the spherulite and often extending up to the spherulite boundary. Frequently, especially in H3-CE, between two very thick lamellae one or more parallel but much thinner lamellae can be observed. The long, edge-on oriented lamellae in many instances give the impression to be shattered into shorter blocks. However, observations of tilted and flat-on oriented lamellae demonstrated that the apparent discontinuities seen at the lamellae edges are actually the result of locally deeper etching in places with high concentration of defects, such as e.g., dislocations [
40]. This can create quite deep notches on the edge of an otherwise long and continuous lamella, which may then look likely to be broken into shorter pieces when observed in an edge-on orientation.
In contrast, lamellae seen in H5-CE, are much shorter than in H3-CE or H4-CE—only about 2–5 μm in length—and while still arranged in parallel stacks, do not show clear higher level organization. Such crystallization habit is probably related to a significantly higher molecular weight of this PE grade comparing to H3 and H4.
In all samples studied the lamellae were found extremely thick—the average lamella thickness, determined from micrographs (at least 400 lamellae measured in each image) ranged from about 120 nm in H3-CE to approx. 230 nm in H5-CE and above 260 nm in H4-CE—cf.
Figure 3 and
Table 2. Comparing these thicknesses to the contour length of the average chain, which is approx. 320, 260 and 350 nm for H3, H4 and H5, respectively (as estimated on the basis of M
n), one can conclude that in the studied samples crystallized under high pressure, the chains were actually built into the crystals in a well extended fashion, with probably only occasional folding. Moreover, the lamellae observed in the sample H4-CE-iso, crystallized, and annealed isothermally for 1 h at high pressure, were found even thicker—up to 2 μm—with the average thickness of about 420 nm, which exceeds well the average chain length ≈ 260 nm. This indicates that the individual crystalline stems had to be frequently formed from more than one chain, and consequently some chain ends had to be incorporated inside the crystal.
The average lamella thickness determined from SEM micrographs is noticeably larger than the crystalline stem length
l* that determines the crystal thickness, estimated from the melting temperature with the Gibbs-Thomson equation (Equation (4))—see
Table 2. This discrepancy demonstrates that a great caution is needed when using this equation for estimation of the thickness of lamellar crystals. It seems that the melting point data can only be used for a rough approximation of the crystal thickness, especially in the range of high melting temperatures, close to the equilibrium melting temperature, where the dependence of
l* on T
m becomes very steep, and any single, even small, experimental error in T
m may result in a large inaccuracy in the estimation of lamella thickness.
3.2. Deformation Behavior
The reference samples, crystallized under conventional conditions, as well as samples crystallized under high pressure, composed of extended-chain lamellae, were deformed at room temperature in the uniaxial compression mode with the constant true strain rate. All reference samples were found ductile as they deformed easily to high strains, well above e = 1.6. The H3-CE and H5-CE samples also appeared ductile in this range. In contrast, samples of H4-CE exhibited different behavior—while a few samples deformed in a ductile manner even to high strains, many others, especially those of slightly higher crystallinity, demonstrated brittle behavior, fracturing at low strains, usually prior to reaching the yield point. Moreover, practically all of the H4-CE-iso samples of extremely high crystallinity, appeared brittle. This brittleness was probably due to a strong deficit of amorphous chains in these samples, which could provide a sufficient connectivity between crystals through amorphous layers and transmit stress between these crystals, like tie-molecules or similar.
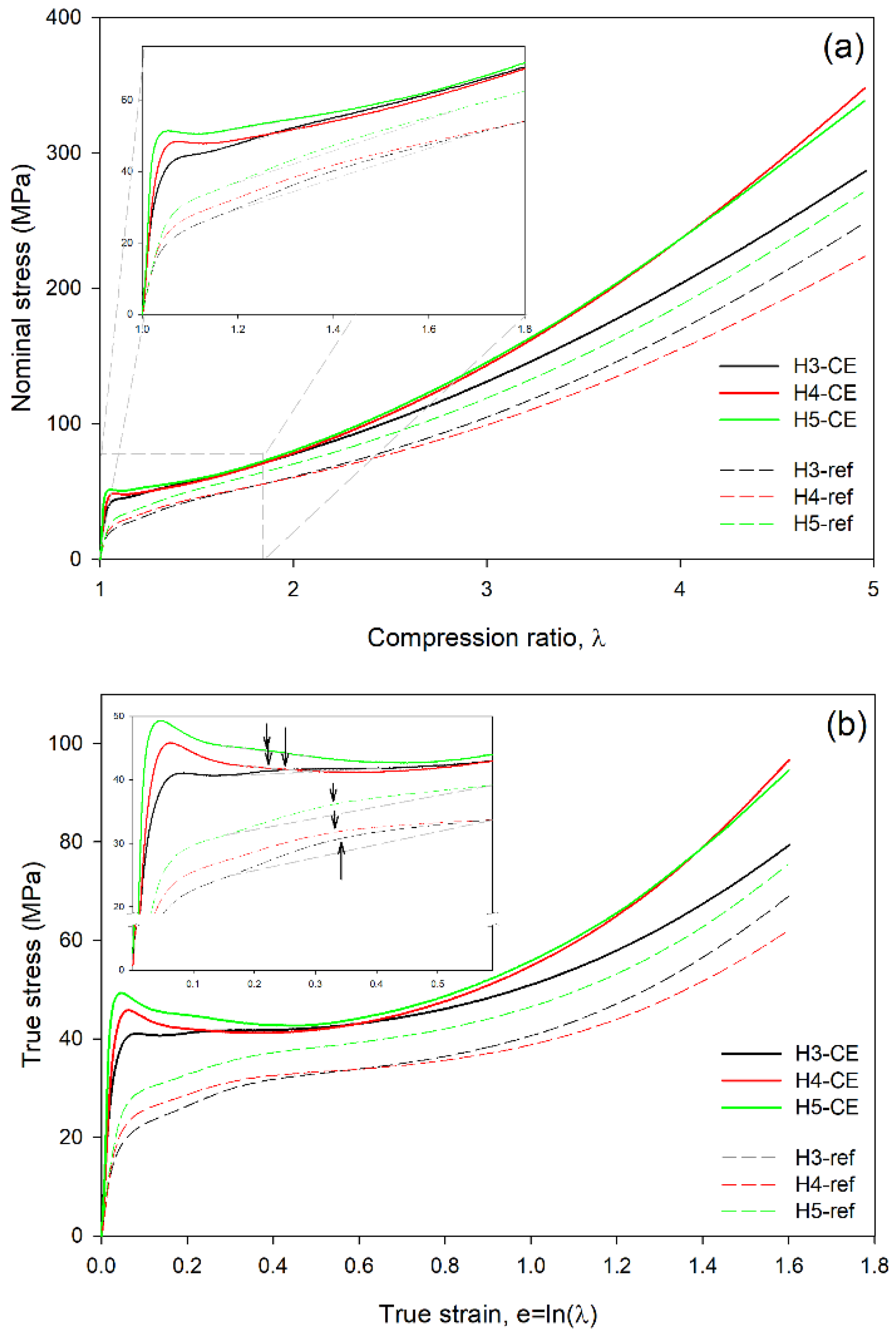
Figure 4a presents the dependencies of the nominal stress vs. compression ratio determined experimentally in the reference and the CEPE samples. From these data the true stress-true strain curves were calculated using Equations (1) and (2) and plotted in
Figure 4b. It can be seen that in the compression curves of the reference samples, crystallized at conventional conditions and containing thin folded-chain lamellae, both the nominal and true stress increase continuously with increasing deformation, without any maximum that could define the yield point (in such a case the yield can be alternatively estimated with the offset construction [
41]). This is a typical response of polyethylene to compression [
9]. In contrast, all CEPE samples show a maximum in stress defining the yield point, seen clearly especially in the true stress-true strain curves. The elastic modulus (determined from initial slope) and the stress at yield are notably higher in CEPE samples than in the respective reference samples, which reflects their higher crystallinity and thickness of crystal. The stress maximum is followed by a short strain softening region extending up to the true strain of about e = 0.5, where stress-hardening stage begins in all samples. Comparing the mechanical response of the reference samples, it can be seen that the stress in the yielding and plastic flow regions increase from H3-ref to H4-ref and then to H5-ref, which reflects the increasing thickness of the lamellar crystals in these samples (cf.
Table 2). The crystal thickness is known to control the yielding and plastic flow behavior of semicrystalline polymers with relatively thin folded-chain crystals [
16,
42]. In the strain-hardening stage, e > 0.6, the stress-strain curves of all three control samples show similar slope, although H3-ref demonstrates slightly stronger hardening effect than H4-ref or H5-ref. Since the strain-hardening is controlled primarily by the density of the molecular network in the amorphous phase [
6,
11], the observed response of the reference samples can indicate similar properties of the molecular network, perhaps with a slightly higher network density in the H3-ref sample. On the other hand, the H3-CE sample with extended-chain crystals shows lower slope of the curve in this strain range, i.e., weaker strain-hardening, while H4-CE and H5-CE demonstrate similar strain-hardening, which appears stronger than in H3-CE or the reference samples. This may indicate that the density of the network of entangled chains in H3-CE is slightly lower than in the reference samples, whereas the concentration of entanglements in amorphous phase of H4-CE and H5-CE is a little higher, probably as a consequence of some redistribution of pre-existing entanglements into amorphous phase during formation and thickening of pseudo-hexagonal crystals under high pressure. High-pressure crystallization in H3, of molecular weight lower than H4 or H5, probably resulted in resolving of more chain entanglements, which led to a less dense molecular network in H3-CE sample than in the reference material or species of higher molecular weight, H4 and H5.
Another feature observed in the true stress—true strain curves of the reference as well as CEPE samples is the so-called ‘second yield,’ which can be distinguished as a hump or bulge (low and very broad local maximum) extending from about e ≈ 0.2 to e ≈ 0.5 (centered around e ≈ 0.35) in the reference samples and located at slightly lower strains, from e ≈ 0.15 to e ≈ 0.35 (centered about e ≈ 0.22–0.25) in the CEPE samples; see the inset of
Figure 4b displaying enlarged initial part of the stress-strain curves.
The second yield, similarly to the primary yield, is generally associated with the deformation of the crystalline phase. Initially, the second yield was attributed to the activation of the block slip along the direction of the chain (so-called ‘coarse’ chain slip), which can ultimately lead to lamellar fragmentation [
43,
44,
45,
46,
47]. Later, Sedighiamiri et al. [
48] proposed to associate the second yield with activation of the transverse slip systems rather than with the coarse chain slip. In the recent studies [
20,
21,
22] we demonstrated that the second yield is actually related to the microbuckling instability occurring in the stacks of lamellae that were initially oriented along the direction of compression. The lamellae of such specific, longnitudinal orientation are not able to deform according to the usual crystallographic mechanisms because the corresponding resolved shear stresses are much lower than critical. Consequently, the deformation is locally locked and the stresses increase with increasing strain. Then, the stacked lamella can respond to increasing compressive in-plane stress with bending or undulation, a phenomenon referred to as microbuckling instability [
18,
20]. Due to phase connectivity the neighboring lamellae are forced to bend jointly, which leads soon to development of bigger joint folds or angular kinks from a small initial ripple. Kinking, triggered by microbuckling, was observed in many semicrystalline polymers, e.g., polyethylene (PE), isotactic polypropylene (iPP), polyoxymethylene (POM) or isotactic polystyrene (iPS) [
17,
49,
50,
51,
52,
53].
The microbuckling in semicrystalline polymers is generally analogous to buckling in other layered systems. The layer buckling phenomenon was first investigated in structural geology. Several theories of rock folding were developed, mainly using the elastic approach (see, e.g., [
54]). Later, similar considerations were made for liquid crystals and highly oriented block copolymers with lamellar morphology [
55,
56,
57,
58]. It was concluded that a necessary condition for buckling to occur in a layered structure consisting of alternating hard and soft layers is a large difference in the layer stiffness. In addition, a strong coupling between layers is also required if buckling is to occur in tension, as in the case of some semicrystalline polymeric materials. Buckling is driven by the simple, natural tendency of a material to deform in a way that minimizes energy—buckling of hard and soft layers proves less expensive in terms of free energy than the alternative mechanism by dilatation of soft layers or voiding at interface [
59]. Buckling has been found to be controlled by strain. Read et al. [
55] demonstrated that buckling starts at a critical strain due to a geometrical instability that can even happen in the elastic range. Such a small sinusoidal instability (undulation) continues to evolve into a fold, angular kink or a chevron shape as the strain increases above the instability point. Non-linear phenomena, such as the plastic deformation in hard layers, can be also involved in buckling and subsequent formation of the kink, thus enhancing its angular profile [
55]. The development of sharp angular kinks or chevrons turned out to be energetically advantageous compared to round folds, especially if the fraction of the hard phase was high [
59]. The elastic theories predict the dependence of the critical strain for buckling initiation on the ratio of the layer stiffness (determined by the product of layer thickness and its elastic modulus) of the soft and hard layers [
18,
55]. FEM model calculations performed for the oriented block copolymer structure [
55] demonstrated that the first sinusoidal undulations were generated when the ratio of the elastic moduli exceeded 500 (at constant layer thickness). In semicrystalline polymers deformed above T
g, this ratio is expected to be much higher (e.g., in polyethylene, PE, an overall modulus of elasticity of crystalline phase is about 50 GPa—elastic constants calculated for anisotropic PE crystal range from c
11 ≈ c
22 ≈ 10 GPa to c
33 ≈ 300 GPa [
60], while that of amorphous layer is below 40 MPa [
61], i.e., smaller by 3 order of magnitude), which can indicate their high susceptibility to microbuckling upon deformation.
Microbuckling is limited to the specifically oriented lamellae and can accommodate only a minor strain. Nevertheless, it appears to be a significant transformation in the deformation sequence, since kinking triggered by microbuckling results in a rapid and fairly large irreversible rotation of the lamellae that were involved in the kink. Such rotation leads to an increase of the resolved shear stress in the potential slip planes of the just reoriented lamellae, enabling activation of new deformation mechanisms of lower plastic resistance, such as crystallographic slip. It opens a new path of relatively easy deformation that allows a significant part of the material volume, previously locked, to join the deformation by relatively easy crystallographic mechanisms, similar to those previously available only for other lamellae already preferably oriented to slip, as e.g., those in diagonal parts of the spherulites. It was reported that microbuckling and kinking can also contribute to some fragmentation of lamellae, although limited only to kink tips [
22].
Since microbuckling instability can effectively modify the deformation path, it often manifests macroscopically in the stress–strain curve as a characteristic low and broad hump, commonly referred to as the second yield. We reported in our previous papers [
20,
21,
22,
25] a hump related to the second yield in the stress-strain curves of several grades of polyethylene, observed around e ≈ 0.3–0.4 in the plane-strain compression. Concurrently, characteristic changes were observed in the 2D-SAXS and 2D-WAXS images, including the formation of a distinctive four-point SAXS pattern, which clearly indicated lamella kinking and development of a chevron-like morphology.
Experimental evidence has shown that microbuckling is driven by significantly different levels of stiffness of the hard (crystalline) and soft (amorphous) layers and their strong interfacial connectivity [
12,
17,
18,
55]. It was found that the critical strain for microbuckling (i.e., the strain at which microbuckling is initiated) depends primarily on the ratio of stiffness of the layers, as well as on the concentration of the stress transmitters at the crystal–amorphous interface (like tie-molecules, entangled loops, etc.) [
20,
21]. The dependence of the critical strain of microbuckling on layer stiffness was confirmed experimentally for semicrystalline polymers—in the recent studies of deformation instabilities in samples of non-oriented and oriented PE [
20,
21,
22,
25], we found that microbuckling instability occurred in PE samples of various structure at the true strain ranging approximately from 0.3 to 0.4. The critical strain was found proportional to the ratio of the stiffness of the amorphous and crystalline layers, which is consistent with the general prediction. The stiffness of the crystalline and amorphous layers is determined by the thickness and the modulus of elasticity of the respective phase. In particular, when the elastic properties of crystalline and amorphous components have not been altered, the critical strain of microbuckling depends on the ratio of the thickness of the amorphous and crystalline layers, although the actual thickness of the layers can vary [
20].
The hump assigned to the second yield in the reference samples, uniaxially compressed in this study, was observed at the position similar to that found previously in various PE samples deformed in the plane-strain compression [
20,
21,
22,
25]—centered around e ≈ 0.35. In contrast, a less distinct bulge, but centered at lower strains (e = 0.22–0.25), was discerned in CEPE-samples. The shift of the second yield to lower strains in CEPE seems reasonable, if we recall that the microbuckling is controlled by the ratio of stiffness of the amorphous and crystalline layers. The elastic moduli of the crystalline phase in the reference and CEPE-samples are identical, while the moduli of the amorphous phase are most probably very similar (as suggested by similar strain hardening behavior). Therefore, the key parameter for microbuckling should be the ratio of the layer thickness. As the crystallinity of the CEPE-samples is significantly higher than the reference samples, the thickness ratio
la/lc is clearly lower in the CEPE- than in the reference samples. This implies an earlier microbuckling in CEPE-samples that should occur at a lower critical strain, and perhaps the size of the hump associated with the second yield should be smaller than in the reference samples of conventional morphology.
3.3. Microscopic Observations
Microscopic observation of the deformed samples was needed to characterize the structural changes accompanying deformation and obtain direct evidence of active deformation mechanisms, including microbuckling instability in CEPE samples during their deformation. For this purpose, microscopic observations of samples deformed to various strains were carried out using a scanning electron microscope.
Figure 5 presents a set of SEM micrographs of the sample H3-CE, both non-deformed and deformed to strains ranging from e = 0.2 to e = 1.4.
Figure 6 and
Figure 7 present similar sets of SEM micrographs of deformed samples H4-CE and H5-CE. In all micrographs the loading (compression) direction is roughly vertical. The presented micrographs document well the evolution of lamellar morphology with increasing strain, including the formation of angular kinks in the lamellae that were initially oriented along the LD and the development of these kinks with increasing strain, which occurred in all PE grades studied here. Kinking appeared collective, involving several neighboring lamellae. The most clear picture of microbuckling, followed by the formation of collective lamellar kinks, can be seen on the micrographs of the H3-CE sample; cf.
Figure 5. The lamellae in this sample prior to deformation were quite uniform in thickness, long compared to their thickness, straight and usually organized in long stacks of nearly parallel lamellae, which in turn, formed spherulites at a higher morphological level. The first signs of activity of the microbuckling can be recognized already at the strain as low as e = 0.2, where local undulations can be observed to arise, initially along individual lamella. Apparently, such instability spreads soon over the adjacent lamellae, which leads to a collective kinking of several neighboring lamellae (e > 0.3). It can be seen in the micrographs that the formation of angular kinks is preferred over round folds. This behavior is in line with the theoretical prediction that angular kinks, involving severe plastic deformation, should be energetically advantageous compared to round folds, especially when the fraction of the hard (crystalline) phase is high [
59]. The development of such angular kinks, engaging several neighboring lamellae, can be observed at e = 0.3–0.4. A further increase in strain leads to a further development of kinks, tightening the apex angle at the kink tip and progressive rotation of lamellae in the kink limbs out of the loading direction. Frequently, several kinks develop along the length of the stacked lamellae, which give rise to a chevron-like morphology that gradually replaces the initial spherulitic morphology, at e ≥ 0.7. On the other hand, the other lamellae, oriented diagonally in spherulites, supposed to deform preferably by slip mechanism from the beginning (after passing the yield point), do not undergo any kinking and therefore remain straight up to high strains, only decreasing its thickness. This reduction in thickness and concurrent rotation of the chain direction inside lamella can confirm deformation by crystallographic slip along the direction of the chain as an active mechanism operating in these lamellae [
1,
2,
3]. The morphology of the deformed material at higher strains consists then of relatively long straight lamellae and much shorter, but also straight, parts of the lamellae in the kink limbs, separated by kink tips. No damage to the lamellae in the tip regions was found, and the lamellae, although heavily deformed, mostly retained their integrity. Importantly, all of the straight sections of lamellae in kink limbs are oriented at an angle to the loading direction, which orientation favors chain slip, which becomes the primary deformation mechanism of nearly all lamellae beyond the microbuckling instability. The deformation instability, developing into kinks, was observed by SEM to occur in H3-CE at the true strain of e ≈ 0.2–0.3. This range of strains coincides well with the local maximum recognized in the stress-strain curve, centered around e ≈ 0.25 and assigned to the second macroscopic yield. It confirms again the hypothesis that the second yield is directly related to the microbuckling instability [
20,
22]. This instability occurs at the strain lower than that found previously in conventionally crystallized PE (e ≈ 0.35, as observed in the reference sample H3-ref or in the PE samples of various structure examined in the previous studies [
20,
21,
22,
25]). Such a reduction in the critical strain is associated with a substantial increase in the thickness of the crystalline layer relative to the amorphous layer and an increase in the crystallinity of the CEPE material, which results in a significant decrease in the ratio of the amorphous to crystalline layer stiffness, which has been shown to control primarily the microbuckling behavior. As a result, the critical strain of microbuckling instability decreases significantly, as expected [
20,
22].
As can be seen in
Figure 6, microbuckling and kinking in the H4-CE sample is slightly less pronounced than in H3-CE (
Figure 5). We did not find any indication of microbuckling at e = 0.2. However, at e = 0.3, some small lamellar kinks can be recognized, similarly to H3-CE. These kinks involve adjacent lamellae and develop, sharpening the apex angle, with increasing strain. The intensity of kinking and the average tilt of lamellae in kink limbs with respect to LD tend to be generally lower than in H3-CE at similar strain. These differences can be attributed to different initial morphology—lamellae in H4-CE are similar in length to these seen in H3-CE, but are much thicker, thus have a lower aspect ratio. This can appear to be another factor modifying microbuckling behavior—apparently, microbuckling is more intense when the aspect ratio of lamellae is higher. Less intense microbuckling and kinking resulted in a noticeably smaller hump in the stress-strain curve, assigned to the second yield in H4-CE (see
Figure 4). On the other hand, this hump in H4-CE is located at strains slightly lower than that in H3-CE (e ≈ 0.22 vs. e ≈ 0.25, respectively). As the crystallinity in the H4-CE is significantly higher than in the H3-CE, the thickness ratio
la/lc, so the ratio of respective compliances, must be lower in H4-CE sample than in H3-CE. This implies a possible earlier initiation of microbuckling in the H4-CE. Unfortunately, this effect was not confirmed by direct SEM observations. However, in the case of less intense microbuckling, as in H4-CE, the events of its initiation could be not recognized or simply overlooked in sample deformed to only e = 0.2.
Figure 7 presents the set of SEM micrographs obtained for H5-CE sample at various deformation stages. The initial morphology of this material is different from H3 or H4 in that the lamellae are much shorter than in H3-CE or H4-CE, thus exhibit the lower aspect ratio,
L/lc < 10. As a result, microbuckling and subsequent kinking, although present, is less intense than in the previously reported samples with much longer lamellae. The first kinks can be recognized in the micrographs not earlier than at the true strain of e = 0.5, while the mature kinks, resulting from cooperative deformation of adjacent lamellae, can be observed at e ≥ 0.7. Moreover, it seems that fewer neighboring lamellae are engaged in joint kinks. We attribute such a less intense microbuckling to the reduced aspect ratio of the lamellae in the structure, which perhaps results in weaker constraints imposed on the short lamellae in bulk. Consequently, a limited activity of other deformation mechanisms (as e.g., rotation of the lamellar stacks) may be allowed, especially for the shortest lamellae. Nevertheless, the long lamellae, oriented parallel to LD, still underwent microbuckling and then kinking similar to that in previously discussed samples. As in the case of H4-CE, a less intense kinking led to a less pronounced hump attributed to the second macroscopic yield in the stress-strain curve (see
Figure 4).
The SEM micrographs of H3-CE and H4-CE samples deformed to relatively low strains were reviewed in order to find the mechanism of initiation of lamellar kinks. The selected micrographs are presented in
Figure 8.
Figure 8a—H3-CE at e = 0.2—evidences that the kinks are initiated and start to develop in long lamellae oriented along the direction of compression due to the instability leading to their undulation—small quasi-periodic ripples can be recognized along the individual lamellae already at e = 0.2; see
Figure 8a. As the strain increases, the amplitude of the undulation increases and the instability is gradually transmitted to adjacent lamellae, so that several lamellae in a stack begin to deform cooperatively, creating a larger lamellar kink—see
Figure 8b; e = 0.4. This mechanism is similar to that observed in buckling of several other materials [
55,
56].
Microbuckling instability can be additionally triggered in the lamellae under longitudinal loading by some fluctuations of their thickness — deformation resistance generally decreases with decreasing thickness of the lamella [
42], therefore their locally thinner parts are more susceptible to any deformation instability and apparently microbuckling may be easier to initiate there. This effect can be observed in the sample H4-CE, demonstrating significant roughness of the lamella surface and varying thickness—see
Figure 8e,f. It can be clearly seen in these micrographs that multiple small, but already sharp kinks were preferentially formed in thinner parts of the lamellae. Very sharp tips of these kinks indicate that localized plastic deformation by crystallographic slip mechanism had a large share in their formation.
Another factor contributing to microbuckling instability and the formation of kinks is perhaps the non-uniformity of the stress field around the longitudinally compressed lamella. This stress field can be locally disturbed, for example, by the other lamella with very different orientation, which is in contact with the side of the lamella preferably oriented for buckling. The presence of such side lamella and perhaps its deformation or translation can then contribute to generation of an additional shear stress component normal to the plane of the considered lamella under compression. Such a local perturbation of the stress field may be large enough to trigger microbuckling instability in this part of the lamella. In fact, kinks were often observed to develop near the point where the stack of lamellae oriented initially along the loading direction was in lateral contact with another lamella of nearly perpendicular orientation, as can be seen in the micrographs of
Figure 8c,d. In addition, it was also observed that stacks of inclined lamellae in the immediate vicinity of a longitudinally compressed lamellae, and undergoing extensive shear deformation, possibly locally exerted additional shear stress on these compressed lamella, which encouraged their microbuckling, and subsequent kinking.
As mentioned above, the effect of plastic deformation due to crystallographic slip was noticed in the tips of the lamellar kinks. Further SEM observations of the deformed samples of H4-CE revealed that the same crystallographic slip system along the chain direction was involved throughout the entire kink, including both the tip region as well as the limbs.
Figure 9 presents the SEM micrographs of H4-CE sample deformed to e = 0.5. In all thick lamellae oriented edge-on, parallel striations can be easily recognized—roughly perpendicular to the lamella plane in the initial non-deformed material (cf.
Figure 3d–f and evidently tilted with respect to the lamella plane in the deformed samples (
Figure 9). As already discussed, these striations reflect the actual direction of the chain in the lamella. The striations visible in practically all lamellae of the deformed sample—including these inclined that remain straight during the deformation as well as those which were initially oriented parallel to the loading direction and later underwent kinking—are all oriented preferentially in nearly the same direction, tilted towards the transverse direction. Such preferred tilted orientation of the chain direction (rotated inside individual lamellae out of the initial orientation) within the straight, diagonal lamellae and thinning of these lamellae with increasing tilt evidences the activity of the crystallographic chain slip as the primary deformation mechanism in these lamellae [
1,
3]. The same chain slip mechanism had to be, however active, also in the lamellae that developed kinks. The same uniform orientation of the chain direction throughout the entire kinked lamellae clearly indicates a well advanced single chain slip system. Its operation as a primary active deformation mechanism facilitated the development of sharp angular kink with straight limbs from an initial small undulation, which possibly may have started with some elastic bending. This crystallographic slip mechanism was not allowed at the very beginning of the deformation process because the resolved shear stress along the chain direction in the longitudinally loaded lamella was too low to activate this slip mechanism. However, once microbuckling produced the first undulations, the corresponding resolved shear along the chain direction started to increase locally due to modification of the lamella shape, resulting in some rotation of the chain direction. This, at some point, enabled the chain slip mechanism to be activated. Subsequently, this relatively easy deformation mechanism became to dominate further plastic deformation, resulting in development of mature angular kinks with straight legs.
Figure 10 illustrates the further development of the lamellar kink with increasing strain (here the applied true strain e = 1.5). It can be observed that in this well advanced angular kink seen in the micrograph, the lamellae are continuous, not disrupted or broken even at the sharp tip of the kink, where they change their orientation abruptly. The lamellae are significantly thinned in the straight limbs of the kink, which again confirms their intense deformation by chain slip mechanism. Due to compressive load and shearing of the amorphous layers that accompanied the slip within crystalline lamellae (referred to as interlamellar slip [
1,
3]), adjacent lamellae are very close each to the other in the straight limbs. The intense intra- and interlamellar shear within both limbs of the kink quite often forced the local delamination and the formation of very small cavities between lamellae in the apical region, where the shear strain was smaller than in the limbs. However, such a local separation of the lamellae at the kink tip allowed close packing of the entire lamellar structure to be maintained without fracturing the lamellae. In addition, it can be observed that the apex angle of successive lamellae changes gradually across the kink and the kink becomes sharper with distance from the initiation point due to the strong deformation of adjacent lamellae by intense chain slip. This results in bigger cavities created between lamellae in the apex region at the far edge of the kink than in its center. The cavitation of the material was limited to the apical line of kinks described here and was not observed elsewhere in the deformed material, so globally it can be considered a marginal effect. This behavior observed in compression is opposite to tensile deformation, where extensive cavitation often plays much more important role.
Figure 11 presents another micrograph of a H3-CE sample deformed to high strains, e = 1.4–1.6. These micrographs show a very interesting feature of the deformation of samples with crystals of extended-chain morphology: the lamella preserved their continuity up to very high strains, e > 1.6 (compression ratio λ > 5). All lamellae—including these initially diagonal, which deformed from the beginning by chain slip mechanism supported by interlamellar slip, as well as those initially parallel to the LD, which first kinked due to microbuckling instability, and then continued to deform by chain slip—do not show any fragmentation and retain their integrity up to high strains; cf.
Figure 11a. As can be seen in the inset of
Figure 11b, the deformed lamellae were exceptionally thinned due to advancing slip, yet did not fragment even at very high strain. This behavior is very different from that of the conventionally crystallized samples consisting of relatively thin folded-chain lamellae, where a severe fragmentation of lamellae is usually observed during deformation in both tension and compression [
6,
20,
62]. It was found that lamella fragmentation occurs in polyethylene in the true strain range of e = 0.6–1.0 (relatively weak at e = 0.6, but extensive at e = 1) as a result of slip instability, which results in its strong localization and finally leads to the lamella rupture [
20]. The lamellae were already well thinned at this stage of the deformation process due to progressing chain slip. Any fluctuation in the lamella thickness or stress concentration at the crystal-amorphous interface caused by the highly stretched ‘stress transmitter’ chains ST in the adjacent amorphous layer, quickly led to the localization of the crystallographic slip in a narrow zone. Further intense slip, however limited to such narrow zones, led very soon to tearing of the already very thin lamella, as the total translation of the crystalline stem relative to the adjacent stem due to slip mechanism approached stem length. Consequently, an extensive fragmentation of lamellae was observed. In the case discussed here, the extended-chain lamellae are one order of magnitude thicker than a folded-chain crystals, therefore a much higher amount of shear is required do tear of such a very thick lamella, even if the slip localizes (actually, the lamella deformation in samples of extended-chain crystal morphology was found relatively homogeneous and only a very few traces of severe slip localization were noticed). As a result, the thick, extended-chain lamellae can be heavily deformed by intense slip and still maintain their integrity. Especially if the concentration of the ST chains, which ensure connectivity of crystalline and amorphous phases, but impose some constraints on extensive crystal deformation, is increased, as probably in the case of H4-CE and H5-CE. Greater concentration of ST chains results in a smoother distribution of the stress at interface, and thus reduced susceptibility to slip instability that would result in slip localization and eventual crystal fragmentation [
20,
21].

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}