
Biomimetic Hydroxyapatite Crystals Growth on Phosphorylated Chitosan Films by In Vitro Mineralization Used as Dental Substitute Materials

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CS Films
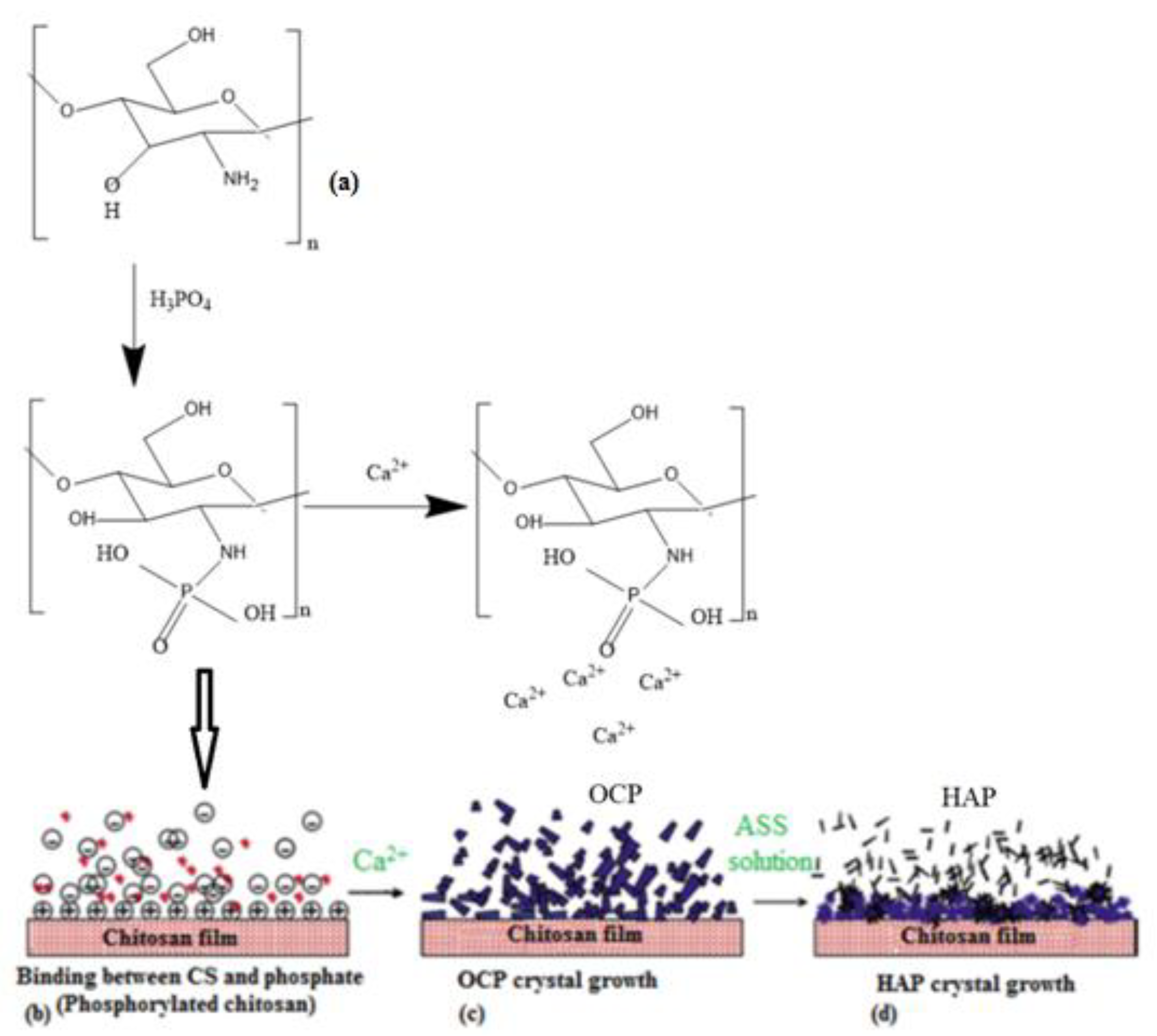
2.3. Biomimetic Mineralization of PCS Films
2.4. Characterization of Biomimetic Mineralized Films
2.5. Weight Test
2.6. Evaluation of Antimicrobial Activity of PCS In Vitro
3. Results and Discussion
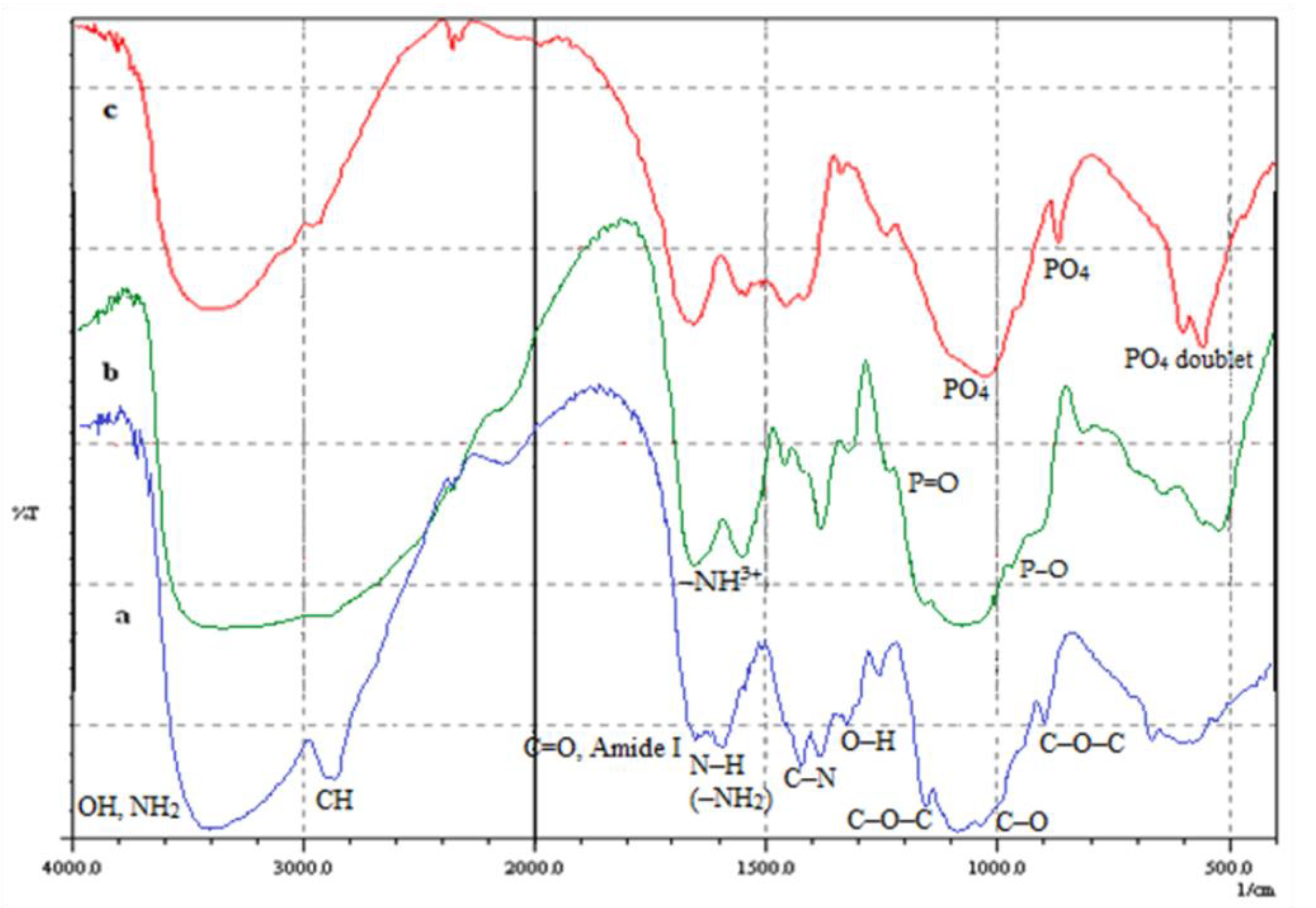
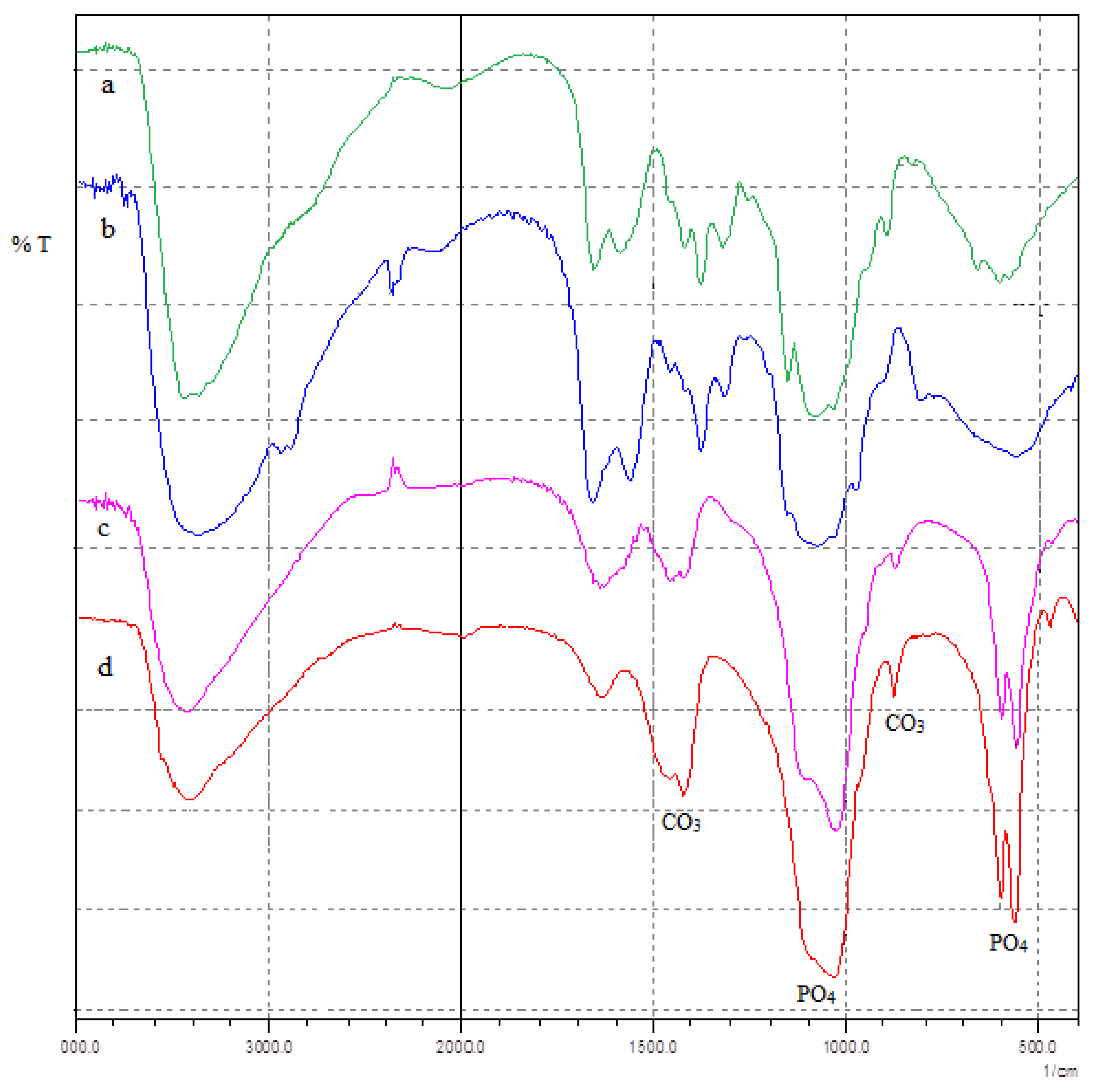
3.1. FTIR Results
3.2. XRD Results
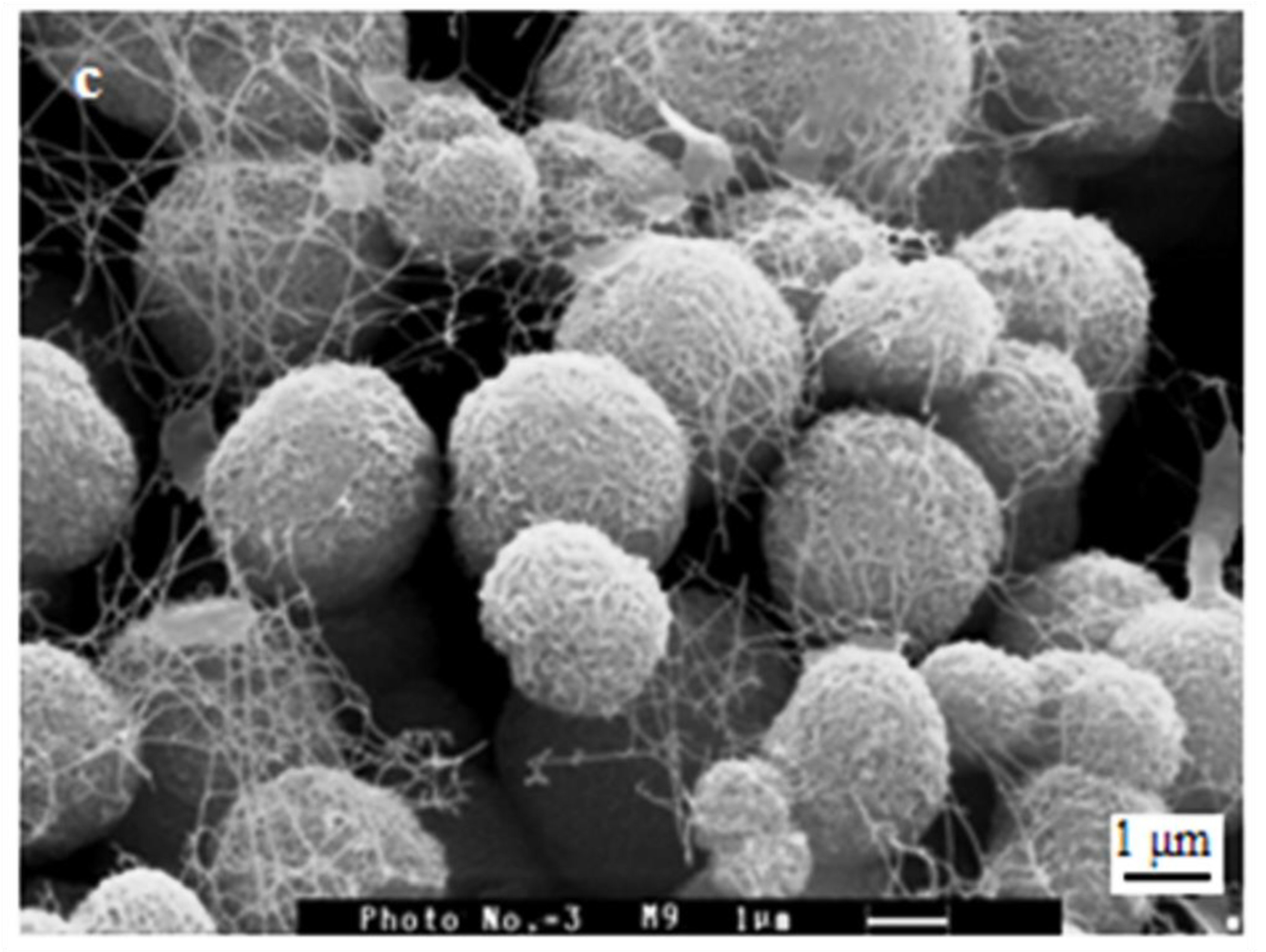
3.3. SEM and EDX Results
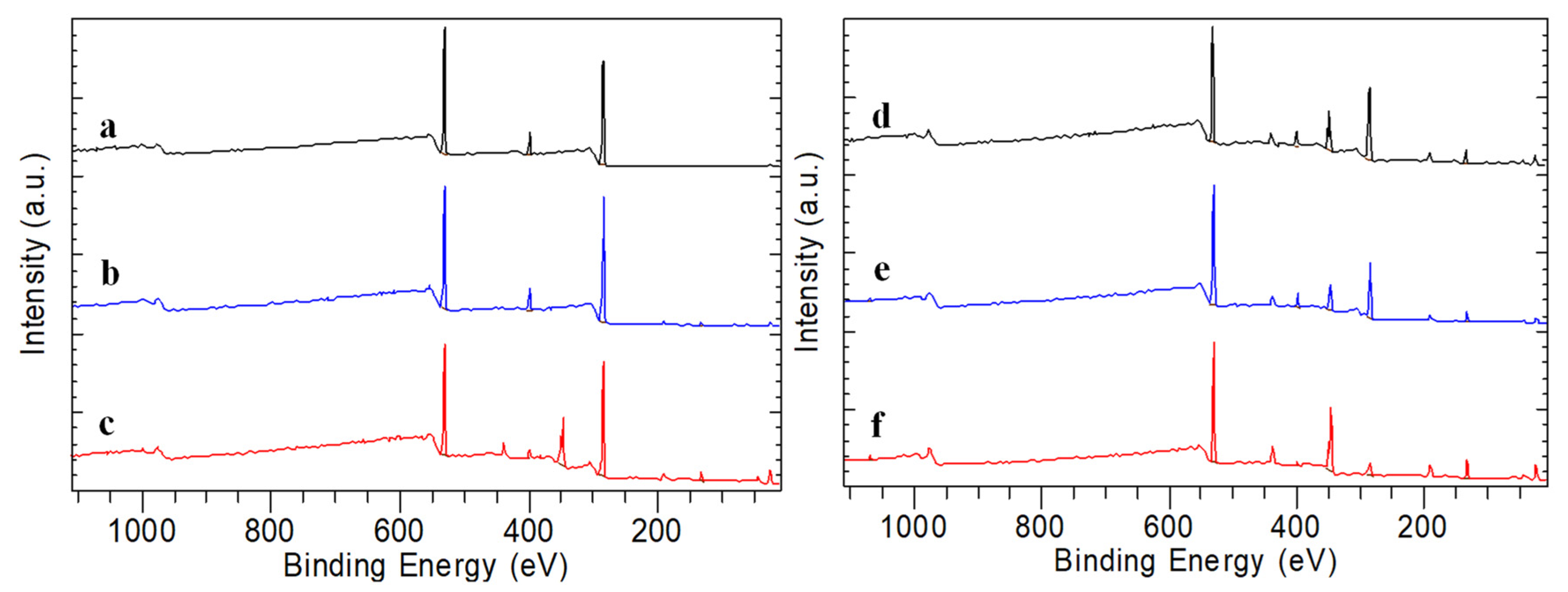
3.4. XPS Results
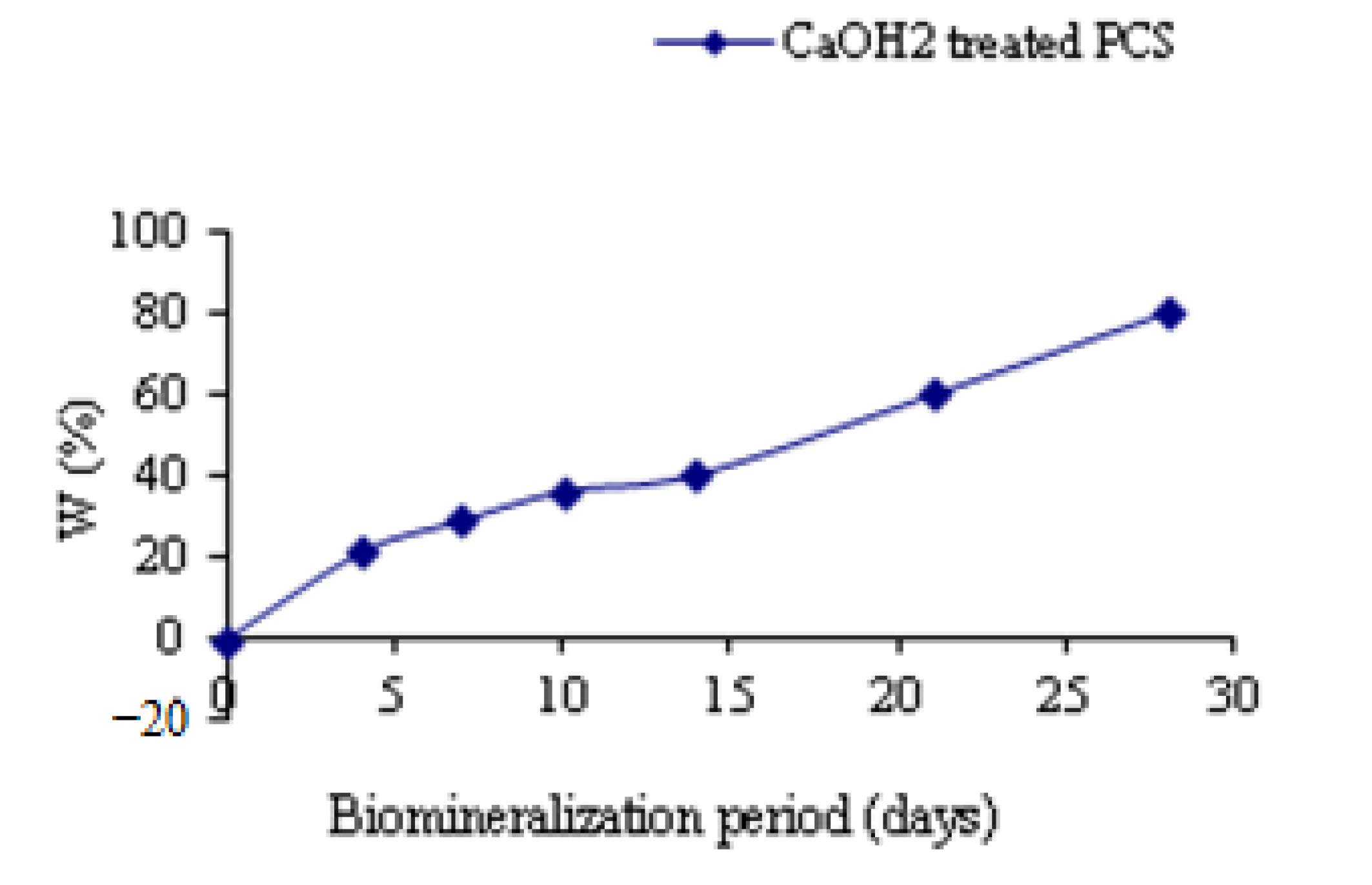
3.5. Weight Test Results
3.6. Antimicrobial Activity of PCS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, Q.X.; Zhong, D.Y.; Lü, R.; Qing Zhang, W.; Deng, J.; Guang Chen, X. In vitro evaluation of the biomedical properties of chitosan and quaternized chitosan for dental applications. Carbohydr. Res. 2009, 344, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Le Geros, R.Z. Apatite in biological systems. Prog Cryst. Growth Charact 1981, 4, 1–45. [Google Scholar] [CrossRef]
- Holcomb, D.W.; Young, R.A. Thermal decomposition of human tooth enamel. Calcif. Tissue Int. 1980, 31, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Baranov, N.; Popa, M.; Atanase, L.I.; Ichim, D.L. Polysaccharide-Based Drug Delivery Systems for the Treatment of Periodontitis. Molecules 2021, 26, 2735. [Google Scholar] [CrossRef]
- Logeart-Avramoglou, D.; Anagnostou, F.; Bizios, R.; Petite, H. Engineering bone: Challenges and obstacles. J. Cell Mol. Med. 2005, 9, 72–84. [Google Scholar] [CrossRef]
- Murugan, R.; Ramakrishna, S. Bioresorbable composite bone paste using polysaccharide based nano hydroxyapatite. Biomaterials 2004, 25, 3829–3835. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics: From concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Fratzl, P. Characterizing natural fibre composites with hierarchical structure. Fibre Diffr. Rev. 2002, 210, 31–39. [Google Scholar]
- Dujardin, E.; Mann, S. Bio-inspired materials chemistry. Adv. Mater. 2002, 14, 775–788. [Google Scholar] [CrossRef]
- Ahmad, Z.; Mark, J.E. Biomimetic materials: Recent developments in organic–inorganic hybrids. Mater. Sci. Eng. C 1998, 6, 183–196. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Bracci, B.; Facchini, A.; Panzavolta, S.; Segatti, F.; Saturba, L. Nanocrystalline hydroxyapatite coatings on titanium: A new fast biomimetic method. Biomaterials 2005, 26, 4085–4089. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Himeno, T.; Kokubo, T.; Nakamura, T. Process and kinetics of bone like apatite formation on sintered hydroxyapatite in a simulated body fluid. Biomaterials 2005, 26, 4366–4373. [Google Scholar] [CrossRef]
- Oh, S.; Tobin, E.; Yang, Y.; Carnes, D.L.J.; Ong, J.L. In Vivo Evaluation of Hydroxyapatite Coatings of Different Crystallinities. Int. J. Oral Maxillofac. Implants 2005, 25, 726–731. Available online: https://www.researchgate.net/publication/7494377 (accessed on 24 May 2023).
- Stigler, M.; Bezemer, J.; de Groot, K.; Layrolle, P. Incorporation of different antibiotics into carbonated hydroxyapatite coatings on titanium implants, release and antibiotics efficacy. J. Control Release 2004, 99, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Mihalache, A.; Rata, D.M.; Cadinoiu, A.N.; Patras, X.; Sindilar, E.V.; Bacaita, S.E.; Popa, M.; Atanase, L.I.; Daraba, O.M. Bupivacaine-loaded chitosan hydrogels for topical anesthesia in dentistry. Polym. Int. 2020, 69, 1152–1160. [Google Scholar] [CrossRef]
- Di Martino, A.; Sittinger, M.; Risbud, M.V. Chitosan: A versatile biopolymer for orthopaedic tissue-engineering. Biomaterials 2005, 26, 5983–5990. [Google Scholar] [CrossRef]
- Bumgardner, J.D.; Wiser, R.; Gerard, P.D.; Bergin, P.; Chestnutt, B.; Marin, M.; Ramsey, V.; Elder, S.H.; Gilbert, J.A. Chitosan: Potential use as a bioactive coating for orthopedic and craniofacial/dental implants. J. Biomater. Sci. Polym. 2003, 14, 423–438. [Google Scholar] [CrossRef]
- Qi, L.; Xu, Z.; Jiang, X.; Hu, C.; Zou, X. Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr. Res. 2004, 339, 2693–2700. [Google Scholar] [CrossRef]
- Elder, S.H.; Nettles, D.L.; Bumgardner, J.D. Synthesis and characterization of chitosan scaffolds for cartilage-tissue engineering. Methods Mol. Biol. 2004, 238, 41–48. [Google Scholar] [CrossRef]
- Atanase, L.I. Micellar Drug Delivery Systems Based on Natural Biopolymers. Polymers 2021, 13, 477. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Q. Chemical structure analyses of phosphorylated chitosan. J. Carbohydr. Res. 2014, 386, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Prabaharan, M.; Reis, R.L.; Mano, J.F. Graft copolymerized chitosan—Present status and applications. Carbohydr. Polym. 2005, 62, 142–158. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhu, Y.; Feng, Q.L.; Cui, F.Z.; Ma, J.B. Responses of Osteo-and Fibroblast Cells to Phosphorylated Chitin. J. Bioact. Compat. Polym. Biomed. Appl. 2003, 18, 135–146. [Google Scholar] [CrossRef]
- Varma, H.K.; Yokogawa, Y.; Espinosa, F.F.; Kawamoto, Y.; Nishizawa, K.; Nagata, F.; Kameyama, T. Porous calcium phosphate coating over phosphorylated chitosan film by a biomimetic method. Biomaterials 1999, 20, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, J.; Wang, Y.; He, B. Structural characterization of phosphorylated chitosan and their applications as effective additives of calcium phosphate cements. Biomaterials 2001, 222, 247–2255. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, W.; Nishizawa, K.; Nagata, F.; Kameyama, T. Bioactive Properties of Chitin/Chitosan—Calcium Phosphate Composite Materials. J. Sol-Gel Sci. Technol. 2001, 21, 105–113. [Google Scholar] [CrossRef]
- Amaral, I.F.; Granja, P.L.; Barbosa, M.A. In vitro Mineralisation of Chitosan Membranes Carrying Phosphate Functionalities. Key Eng. Mater. 2004, 254, 577–580. [Google Scholar] [CrossRef]
- Chen, W.; Li, Q.L.; Zen, Q.; Li, G.; Jiang, H.; Liu, L.; Darvell, B.V. Biomimetic Mineralization and Bioactivity of Phosphorylated Chitosan. Key Eng. Mater. 2005, 288–289, 429–432. [Google Scholar] [CrossRef]
- Amaral, I.F.; Granja, P.L.; Barbosa, M.A. Chemical modification of chitosan by phosphorylation: An XPS, FT-IR and SEM study. J. Biomater. Sci. Polym. Ed. 2005, 16, 1575–1593. [Google Scholar] [CrossRef]
- Chesnutt, B.M.; Yuan, Y.; Brahmandam, N.; Yang, Y.; Ong, J.L.; Haggard, W.O.; Bumgardner, J.D. Characterization of biomimetic calcium phosphate on phosphorylated chitosan films. J. Biomed. Mater. Res. A. 2007, 82, 343–353. [Google Scholar] [CrossRef]
- Merlusca, I.P.; Ibanescu, C.; Tuchilus, C.; Danu, M.; Atanase, L.I.; Popa, I.M. Characterization of neomycin-loaded xanthan-chitosan hydrogels for topical applications. Cellulose Chem. Technol. 2018, 53, 709–719. [Google Scholar] [CrossRef]
- Rata, D.M.; Cadinoiu, A.N.; Popa, M.; Atanase, L.I.; Daraba, O.M.; Popescu, I.; Romila, L.E.; Ichim, D.L. Biocomposite Hydrogels for the Treatment of Bacterial Infections: Physicochemical Characterization and In Vitro Assessment. Pharmaceutics 2021, 13, 2079. [Google Scholar] [CrossRef]
- Brugnerotto, H.; Lizardi, J.; Goycoolea, F.M. An infrared investiguation in relation with chitin and chitosan characterization. J. Polymer. 2001, 24, 2569–2580. [Google Scholar]
- Knaul, J.Z.; Kasaai, M.R.; Bui, V.T.; Creber, C.A.M. Characterization of deacetylated chitosan and chitosan molecular weight. Can. J. Chem. 1988, 76, 1699–1706. [Google Scholar] [CrossRef]
- Arvidson, H.; Johansson, E.G. Galvanic currents between dental alloys in vitro. Scand J. Dent. Res. 1985, 93, 467–473. [Google Scholar] [CrossRef]
- Suge, S.; Ishikawa, K.; Kawasaki, A. Duration of Dentinal Tubule Occlusion Formed by Calcium Phosphate Precipitation Method: In vitro Evaluation Using Synthetic Saliva. J. Dent. Res. 1995, 74, 1709–1714. [Google Scholar] [CrossRef]
- Shellis, R.P. A synthetic saliva for cultural studies of dental plaque. Arch. Oral Biol. 1978, 23, 485–489. [Google Scholar] [CrossRef]
- Soderholm, J.M.B.; Mukherjeel, R.; Longmatel, J. Filler Leachability of Composites Stored in Distilled Water or Artificial Saliva. J. Dent. Res. 1996, 75, 1692–1699. [Google Scholar] [CrossRef]
- Speciale, A.; Musumeci, R.; Biandino, G.; Millazo, I.; Caccamo, F.; Nicoletti, G. Minimal inhibitory concentration and time-kill determination of morifloxacin against aerobic and anaerobic isolates. Int. J. Antimicrob. Agents 2002, 19, 111–118. [Google Scholar] [CrossRef]
- Roeges, N.P.G. A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures; John Wiley: Chichester, UK, 1994. [Google Scholar]
- Kuptsov, A.H.; Zhizhin, G.N. Handbook of Fourrier Transform Raman and Infrared Spectra of Polymers, 1st ed.; Elsevier: Amsterdam, The Netherlands, 29 October 1998. [Google Scholar]
- Dudek, G.; Turczyn, R.; Konieczny, K. Robust poly(vinyl alcohol) membranes containing chitosan/chitosan derivatives microparticles for pervaporative dehydration of ethanol. Sep. Purif. Technol. 2020, 234, 116094. [Google Scholar] [CrossRef]
- Suzuki, O. Biological Role of Synthetic Octacalcium Phosphate in Bone Formation and Mineralization. J. Oral Biosci. 2010, 52, 6–14. [Google Scholar] [CrossRef]
- Horváthová, R.; Müller, L.; Helebrant, A.; Greil, P.; Müller, F.A. In vitro transformation of OCP into carbonated HA under physiological conditions. Mater. Sci. Eng. C 2008, 28, 1414–1419. [Google Scholar] [CrossRef]
- Suzuki, M. Octacalcium phosphate (OCP)-based bone substitute materials. Jpn. Dent. Sci. Rev. 2013, 49, 58–71. [Google Scholar] [CrossRef]
- Granja, F.L.; Barbosa, M.A.; Pouysegu, L. Cellulose phosphates as biomaterials. Mineralization of chemically modified regenerated cellulose hydrogels. J. Mater. Sci. 2001, 36, 2163–2172. [Google Scholar] [CrossRef]
- Drouet, C. Apatite Formation: Why It May Not Work as Planned, and How to Conclusively Identify Apatite Compounds. BioMed Res. Int. 2013, 2013, 490946. [Google Scholar] [CrossRef]
- Komlev, V.S.; Fedotov, A.Y. Mineralization of the Chitosan–Octacalcium Phosphate Composite Material in a Simulated Human Body Fluid. Dokl. Chem. 2014, 459, 215–218. [Google Scholar] [CrossRef]
- Manafi, S.A.; Yazdani, B.; Rahimiopour, M.R.; Sadrnezhaad, S.K.; Amin, M.H.; Razavi, M. Synthesis of nano-hydroxyapatite under a sonochemical/hydrothermal condition. Biomed. Mater. 2008, 3, 025002. [Google Scholar] [CrossRef]
- Li, B.; Huang, L.; Wang, X.; Ma, J.; Xie, F. Biodegradation and compressive strength of phosphorylated chitosan/chitosan/hydroxyapatite bio-composites. Mater. Des. 2011, 32, 4543–4547. [Google Scholar] [CrossRef]
- Cui, H.; Wu, N.; Ma, X.; Niu, F. Superior intrinsic flame-retardant phosphorylated chitosan aerogel as fully sustainable thermal insulation bio-based material. Polym. Degrad. Stab. 2023, 207, 110213. [Google Scholar] [CrossRef]
- Ohtsu, N.; Hiromoto, S.; Yamane, M.; Satoh, K.; Tomozawa, M. Chemical and crystallographic characterizations of hydroxyapatite- and octacalcium phosphate-coatings on magnesium synthesized by chemical solution deposition using XPS and XRD. Surf. Coat. Technol. 2013, 218, 114–118. [Google Scholar] [CrossRef]
- Szekeres, M.; Fodor, G.; Fazekas, A.; Radnai, M.; Turz, K.; Dékany, I. Formation of octacalcium phosphate by heterogeneous nucleation on titania surface. Colloid Polym. Sci. 2005, 283, 587–592. [Google Scholar] [CrossRef]
- Nehrke, G.; Cappellen, P.V. Framboidal vaterite aggregates and their transformation into calcite: A morphological study. J. Cryst. Growth 2006, 287, 528–530. [Google Scholar] [CrossRef]
- Petrakova, N.V.; Teterina, A.Y.; Mikheeva, P.V.; Akhmedova, S.A.; Kuvshinova, E.A.; Sviridova, I.K.; Sergeeva, N.S.; Smirnov, I.V.; Fedotov, A.Y.; Kargin, Y.F.; et al. In Vitro Study of Octacalcium Phosphate Behavior in Different Model Solutions. ACS Omega 2021, 6, 7487–7498. [Google Scholar] [CrossRef]
- McLeod, K.; Kumar, S.; Dutta, N.K.; Smart, R.S.C.; Voelcker, N.H.; Anderson, G.I. X-ray photoelectron spectroscopy study of the growth kinetics of biomimetically grown hydroxyapatite thin-film coatings. Appl. Surf. Sci. 2010, 256, 7178–7185. [Google Scholar] [CrossRef]
- Rouxhet, P.G.; Genet, M.J. XPS analysis of bio-organic systems. Surf. Interface Anal. 2011, 43, 1453–1470. [Google Scholar] [CrossRef]
- Genet, M.J.; Dupont-Gillain, C.C.; Rouxhet, P.G. Medical Applications of Colloids; Matijevic, E., Ed.; Springer: New York, NY, USA, 2008; p. 177. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ions | Natural Saliva (mM/L) | Artificial Saliva (mM/L) |
|---|---|---|
| Ca2+ | 1.43 | 1.43 |
| Mg2+ | 0.21 | 0.21 |
| K+ | 22.7 | 20.50 |
| Na+ | 10.13 | 11.55 |
| CO32− | 6.45 | 6.45 |
| CL− | 23.22 | 23.22 |
| PO43− | 5.24 | 5.10 |
| Samples | N (%) | C (%) | O (%) | Ca (%) | P (%) | Ca/P |
|---|---|---|---|---|---|---|
| PCS | 5.82 | 67.89 | 24.64 | 1.52 | ||
| PCS-Ca(OH)2-treated | 2.44 | 58.34 | 28.14 | 7.75 | 3.32 | 2.33 |
| PCS-Ca(OH)2-treated four days biomineralized | 4.07 | 51.95 | 31.88 | 6.65 | 5.45 | 1.22 |
| PCS-Ca(OH)2-treated 15 days biomineralized | 5.69 | 44.16 | 39.4 | 5.41 | 4.8 | 1.13 |
| PCS-Ca(OH)2-treated 28 days biomineralized | 1.28 | 18.85 | 47.84 | 16.33 | 11.93 | 1.37 |
| Stoichiometric HAP | 59.1 | 22.7 | 13.63 | 1.67 |
| Elements | CS | PCS | ||
|---|---|---|---|---|
| Expected | XPS | Expected | XPS | |
| N/C286.2 | 0.2 | 0.195 | 0.2 | 0.280 |
| (O+N)/C286.2 | 1.2 | 1.103 | 1.2 | 0.720 |
| C287.7/C286.2 | 0.3 | 0.282 | 0.3 | 0.260 |
| (O+N)/(C286.3+C287.7) | 0.923 | 0.860 | 0.923 | 0.571 |
| O/C286.2 | 1 | 0.908 | 1 | 0.624 |
| O532.6/C286.2 | 0.9 | 0.792 | 0.9 | 0.881 |
| N/C287.7 | 0.667 | 0.693 | 0.667 | 1.071 |
| C284.8/C | 0.071 | 0.420 | 0.071 | 0.553 |
| O532.6/O531.2 | 9 | 6.851 | 2.25 | 2.92 |
| O/N | 5 | 4.643 | 6.5 | 4.234 |
| O/P | 13 | 16.21 | ||
| O531.2/P | 4 | 4.13 | ||
| Microorganisms | MIC of PCS (%) |
|---|---|
| Candida albicans | 0.10 |
| Staphylocoques aureus | 0.05 |
| Escherichia coli | 0.025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmani, F.; Larbi Bouamrane, O.; Ben Bouabdallah, A.; Atanase, L.I.; Hellal, A.; Apintiliesei, A.N. Biomimetic Hydroxyapatite Crystals Growth on Phosphorylated Chitosan Films by In Vitro Mineralization Used as Dental Substitute Materials. Polymers 2023, 15, 2470. https://doi.org/10.3390/polym15112470
Rahmani F, Larbi Bouamrane O, Ben Bouabdallah A, Atanase LI, Hellal A, Apintiliesei AN. Biomimetic Hydroxyapatite Crystals Growth on Phosphorylated Chitosan Films by In Vitro Mineralization Used as Dental Substitute Materials. Polymers. 2023; 15(11):2470. https://doi.org/10.3390/polym15112470
Chicago/Turabian StyleRahmani, Fathia, Omar Larbi Bouamrane, Amina Ben Bouabdallah, Leonard I. Atanase, Abdelkader Hellal, and Aurelian Nichita Apintiliesei. 2023. "Biomimetic Hydroxyapatite Crystals Growth on Phosphorylated Chitosan Films by In Vitro Mineralization Used as Dental Substitute Materials" Polymers 15, no. 11: 2470. https://doi.org/10.3390/polym15112470