
Unusual Aspects of Charge Regulation in Flexible Weak Polyelectrolytes
 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Fundamental Concepts on Weak Polyelectrolytes
2.1. Ionization Properties
2.1.1. Protonation Equilibria
2.1.2. Electrostatic Interactions
2.1.3. An Illustrative Example
2.2. Conformational Properties
2.2.1. Basic Models
2.2.2. The Rotational Isomeric State (RIS) Model
2.2.3. Elasticity
3. Theoretical Models for Weak Polyelectrolytes
3.1. The Site Binding (SB) Model
3.1.1. Short-Range (SR) Interactions
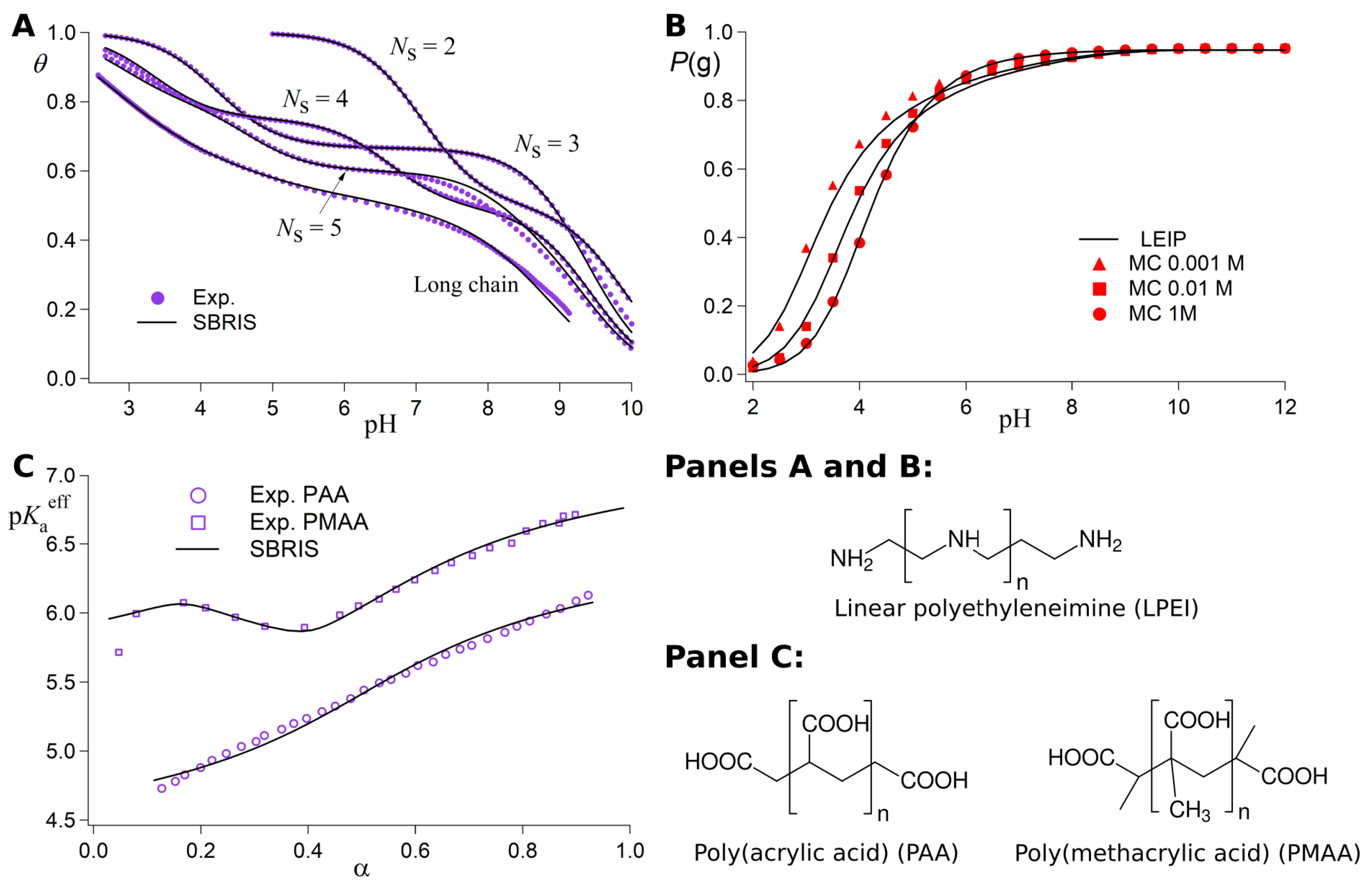
3.1.2. Long-Range (LR) Interactions: Local Effective Interaction Parameters (LEIP)
3.2. The Site Binding Rotational Isomeric State (SBRIS) Model
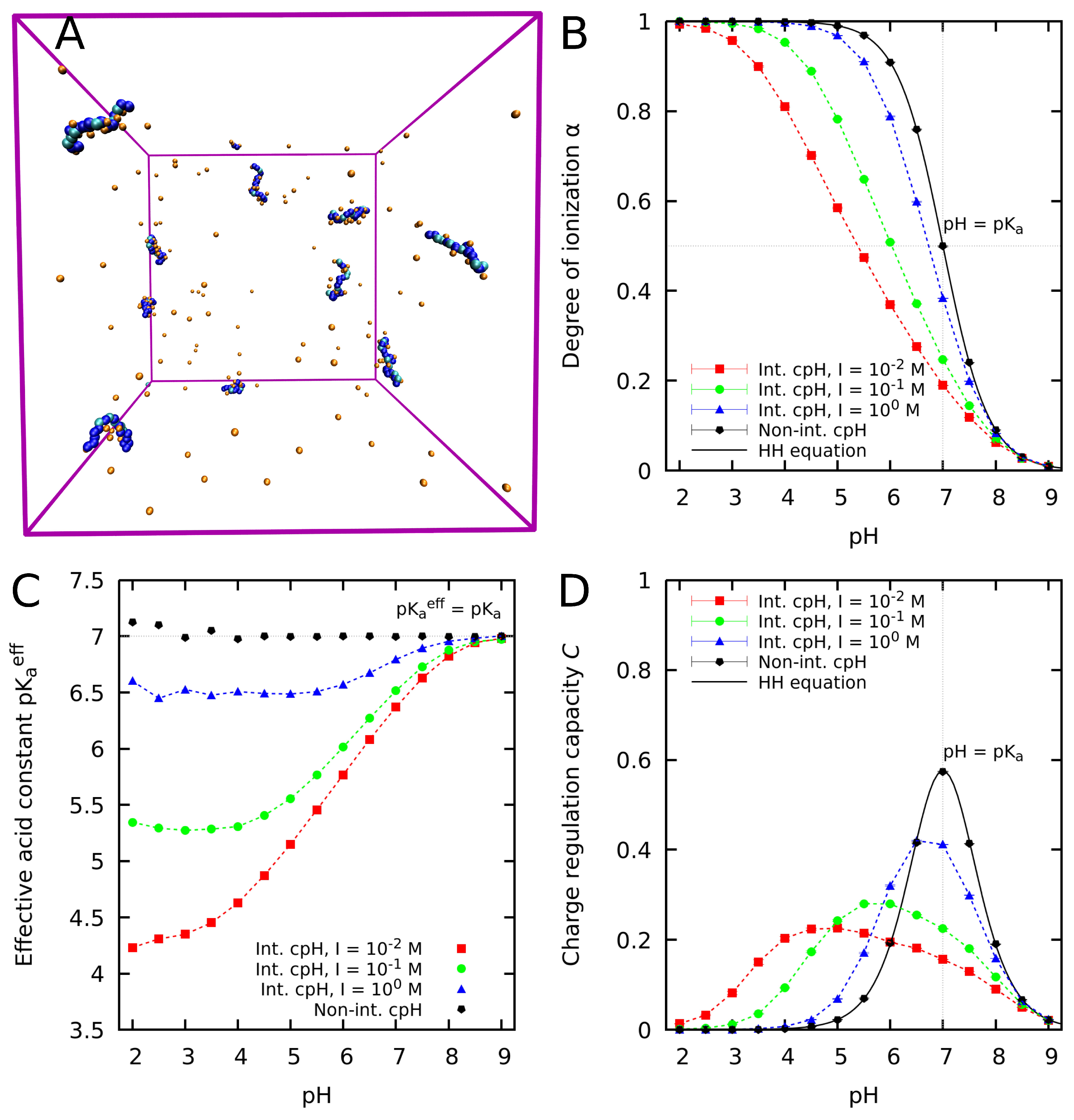
4. Computer Simulations of Weak Polyelectrolytes at Constant pH
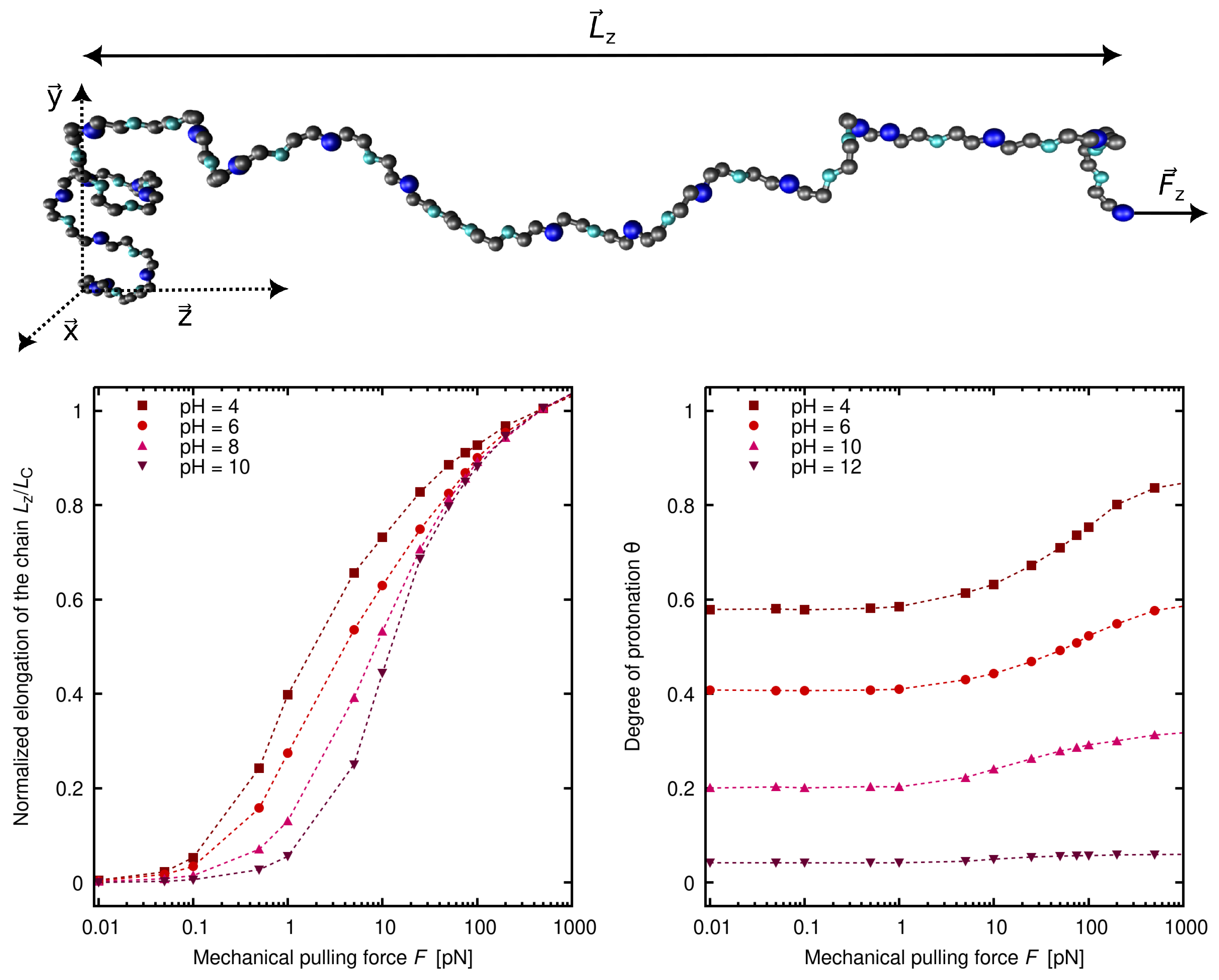
5. The Role of Charge Regulation in the Elasticity of Weak Polyelectrolytes
6. Adsorption of Weak Polyelectrolytes onto Charged Objects
6.1. Adsorption of a Single Chain
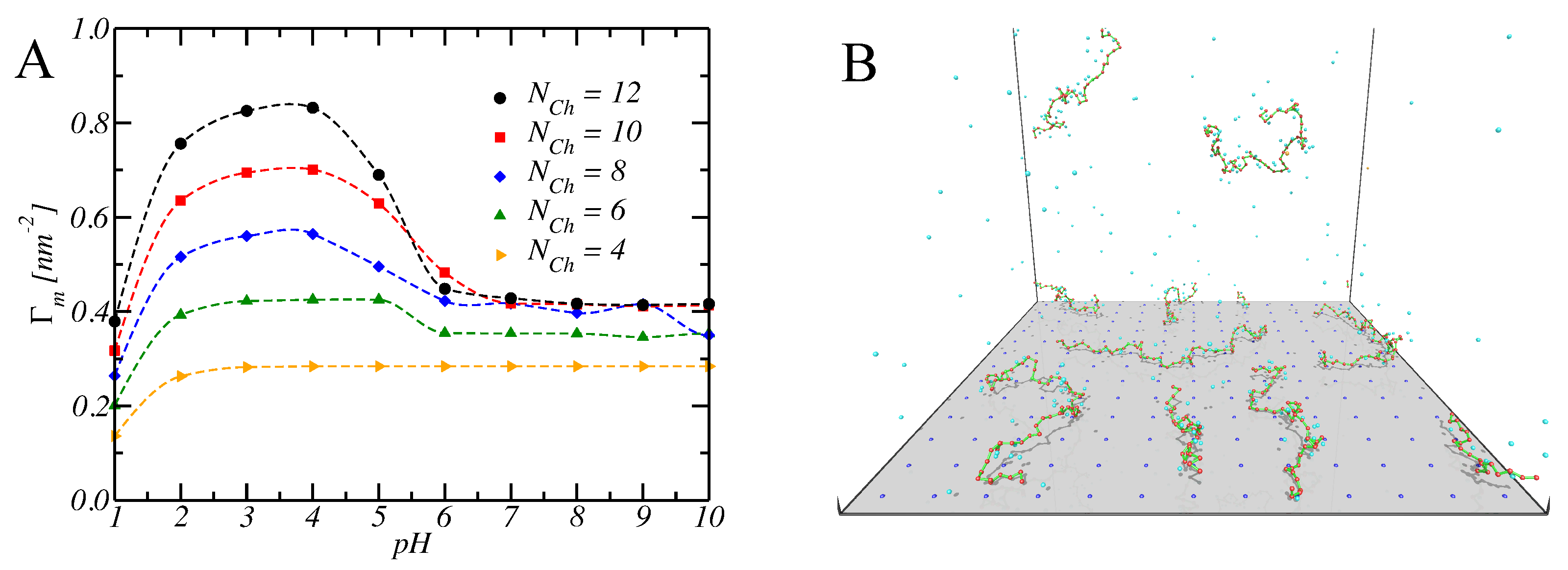
6.2. Adsorption of Multiple Chains
7. Weak Polyelectrolytes in Crowded Systems
7.1. Effect of Macromolecular Crowding in the Properties of Weak Polyelectrolytes
7.2. Charge Regulation Triggered by Macromolecular Crowding
8. Outlooks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AFM | Atomic Force Microscopy |
| CMP | Casein MacroPeptide |
| cpH | constant pH Monte Carlo (cpH) |
| CR | Charge Regulation |
| DH | Debye–Hückel |
| DLVO | Derjaguin–Landau–Verwey–Overbeek |
| DNA | DeoxyriboNucleic Acid |
| FJC | Freely Jointed Chain |
| FRC | Freely Rotating Chain |
| FTPS | Fast Proton Titration Scheme |
| FWPE | Flexible Weak PolyElectrolyte |
| HH | Henderson–Hasselbalch |
| IDP | Intrinsically Disordered Protein |
| LEIP | Local Effective Interaction Parameters |
| LPEI | Linear PolyEthileneImine |
| LR | Long Range |
| MC | Monte Carlo |
| MCr | Macromolecular Crowding |
| MD | Molecular Dynamics |
| MM | Molecular Mechanics |
| NMR | Nuclear Magnetic Resonance |
| PAA | PolyAcrylic Acid |
| PE(s) | PolyElectrolyte(s) |
| PMAA | Poly(MethAcrylic Acid) |
| QM | Quantum Mechanics |
| RIS | Rotational Isomeric State |
| SB | Site Binding |
| SBRIS | Site Binding Rotational Isomeric State |
| SR | Short Range |
| TM | Transfer Matrix |
| WSIP | Wrong Side of the Isoelectric Point |
| WLC | Worm-Like Chain |
References
- Friedsam, C.; Gaub, H.E.; Netz, R.R. Probing surfaces with single-polymer atomic force microscope experiments. Biointerphases 2006, 1, MR1–MR21. [Google Scholar] [CrossRef] [Green Version]
- Muthukumar, M. 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50, 9528–9560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkovec, M.; Jönsson, B.; Koper, G.J.M. Ionization Processes and Proton Binding in Polyprotic Systems: Small Molecules, Proteins, Interfaces, and Polyelectrolytes. In Surface and Colloid Science; Matijević, E., Ed.; Springer: Boston, MA, USA, 2001; pp. 99–339. [Google Scholar]
- Wyman, J.; Gill, S.J. Binding and Linkage: Functional Chemistry of Biological Macromolecules; University Science Books: Sausalito, CA, USA, 1990. [Google Scholar]
- Warshel, A.; Sharma, P.K.; Kato, M.; Parson, W.W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta—Proteins Proteom. 2006, 1764, 1647–1676. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Jönsson, B. Charge regulation in biomolecular solution. Q. Rev. Biophys. 2013, 46, 265–281. [Google Scholar] [CrossRef] [Green Version]
- Lipfert, J.; Doniach, S.; Das, R.; Herschlag, D. Understanding Nucleic Acid–Ion Interactions. Annu. Rev. Biochem. 2014, 83, 813–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartig, S.M.; Greene, R.R.; Dikov, M.M.; Prokop, A.; Davidson, J.M. Multifunctional nanoparticulate polyelectrolyte complexes. Pharm. Res. 2007, 24, 2353–2369. [Google Scholar] [CrossRef] [PubMed]
- Borkovec, M.; Koper, G.J.M.; Piguet, C. Ion binding to polyelectrolytes. Curr. Opin. Colloid Interface Sci. 2006, 11, 280–289. [Google Scholar] [CrossRef]
- Hamacek, J.; Borkovec, M.; Piguet, C. Simple thermodynamics for unravelling sophisticated self-assembly processes. Dalton Trans. 2006, 12, 1473. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, T.; Wang, C.; Lin, Z.; Huang, G.; Sumer, B.D.; Gao, J. Molecular basis of cooperativity in pH-triggered supramolecular self-assembly. Nat. Commun. 2016, 7, 13214. [Google Scholar] [CrossRef] [Green Version]
- Buffle, J. Complexation Reactions in Aquatic Systems: An Analytical Approach; Ellis Horwood Ltd.: Chichester, UK, 1988. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; Wiley: Hoboken, NJ, USA, 1996. [Google Scholar]
- Companys, E.; Garcés, J.L.; Salvador, J.; Galceran, J.; Puy, J.; Mas, F. Electrostatic and specific binding to macromolecular ligands. A general analytical expression for the Donnan volume. Colloids Surf. A Physicochem. Eng. Asp. 2007, 306, 2–13. [Google Scholar] [CrossRef] [Green Version]
- David, C.; Companys, E.; Galceran, J.; Garcés, J.L.; Mas, F.; Rey-Castro, C.; Salvador, J.; Puy, J. Competitive ion complexation to polyelectrolytes: Determination of the stepwise stability constants. the Ca2+/H+/polyacrylate system. J. Phys. Chem. B 2007, 111, 10421–10430. [Google Scholar] [CrossRef] [PubMed]
- David, C.; Companys, E.; Galceran, J.; Garcés, J.L.; Mas, F.; Rey-Castro, C.; Salvador, J.; Puy, J. Competitive Cd2+/H+ complexation to polyacrylic acid described by the stepwise and intrinsic stability constants. J. Phys. Chem. B 2008, 112, 10092–10100. [Google Scholar] [CrossRef]
- Rey-Castro, C.; Mongin, S.; Huidobro, C.; David, C.; Salvador, J.; Garcés, J.L.; Galceran, J.; Mas, F.; Puy, J. Effective Affinity Distribution for the Binding of Metal Ions to a Generic Fulvic Acid in Natural Waters. Environ. Sci. Technol. 2009, 43, 7184–7191. [Google Scholar] [CrossRef] [PubMed]
- Trefalt, G.; Behrens, S.H.; Borkovec, M. Charge Regulation in the Electrical Double Layer: Ion Adsorption and Surface Interactions. Langmuir 2016, 32, 380–400. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.M.; Achetoni, M.M.; Garcés, J.L.; Madurga, S.; Mas, F.; Baieli, M.F.; Narambuena, C.F. Adsorption of flexible proteins in the ‘wrong side’ of the isoelectric point: Casein macropeptide as a model system. Colloids Surf. B Biointerfaces 2022, 217, 112617. [Google Scholar] [CrossRef]
- Lund, M.; Jönsson, B. On the Charge Regulation of Proteins. Biochemistry 2005, 44, 5722–5727. [Google Scholar] [CrossRef] [PubMed]
- Landsgesell, J.; Nová, L.; Rud, O.; Uhlík, F.; Sean, D.; Hebbeker, P.; Holm, C.; Košovan, P. Simulations of ionization equilibria in weak polyelectrolyte solutions and gels. Soft Matter 2019, 15, 1155–1185. [Google Scholar] [CrossRef]
- Koper, G.J.; Borkovec, M. Proton binding by linear, branched, and hyperbranched polyelectrolytes. Polymer 2010, 51, 5649–5662. [Google Scholar] [CrossRef] [Green Version]
- Panagiotopoulos, A.Z. Charge correlation effects on ionization of weak polyelectrolytes. J. Phys. Condens. Matter 2009, 21, 424113. [Google Scholar] [CrossRef]
- Uyaver, S.; Seidel, C. Effect of Varying Salt Concentration on the Behavior of Weak Polyelectrolytes in a Poor Solvent. Macromolecules 2009, 42, 1352–1361. [Google Scholar] [CrossRef]
- Garcés, J.L.; Madurga, S.; Borkovec, M. Coupling of conformational and ionization equilibria in linear poly(ethylenimine): A study based on the site binding/rotational isomeric state (SBRIS) model. Phys. Chem. Chem. Phys. 2014, 16, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Uhlík, F.; Košovan, P.; Limpouchová, Z.; Procházka, K.; Borisov, O.V.; Leermakers, F.A.M. Modeling of Ionization and Conformations of Starlike Weak Polyelectrolytes. Macromolecules 2014, 47, 4004–4016. [Google Scholar] [CrossRef]
- Nová, L.; Uhlík, F.; Košovan, P. Local pH and effective pKA of weak polyelectrolytes – insights from computer simulations. Phys. Chem. Chem. Phys. 2017, 19, 14376–14387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landsgesell, J.; Holm, C.; Smiatek, J. Simulation of weak polyelectrolytes: A comparison between the constant pH and the reaction ensemble method. Eur. Phys. J. Spec. Top. 2017, 226, 725–736. [Google Scholar] [CrossRef]
- Blanco, P.M.; Madurga, S.; Mas, F.; Garcés, J.L. Effect of Charge Regulation and Conformational Equilibria in the Stretching Properties of Weak Polyelectrolytes. Macromolecules 2019, 52, 8017–8031. [Google Scholar] [CrossRef]
- Ullner, M.; Jonsson, B. Monte Carlo study of titrating polyelectrolytes in the presence of salt. Macromolecules 1996, 29, 6645–6655. [Google Scholar] [CrossRef]
- Ullner, M.; Jönsson, B.; Söderberg, B.; Peterson, C. A Monte Carlo study of titrating polyelectrolytes. J. Chem. Phys. 1996, 104, 3048–3057. [Google Scholar] [CrossRef] [Green Version]
- Garcés, J.L.; Koper, G.J.; Borkovec, M. Ionization Equilibria and Conformational Transitions in Polyprotic Molecules and Polyelectrolytes. J. Phys. Chem. B 2006, 110, 10937–10950. [Google Scholar] [CrossRef]
- Noszal, B.; Sandor, P. Rota-microspeciation of aspartic acid and asparagine. Anal. Chem. 1989, 61, 2631–2637. [Google Scholar] [CrossRef]
- Borkovec, M.; Koper, G.J. Affinity Distributions of Polyampholytes with Interacting Acid-Base Groups. Langmuir 1994, 10, 2863–2865. [Google Scholar] [CrossRef]
- Burak, Y.; Netz, R.R. Charge Regulation of Interacting Weak Polyelectrolytes. J. Phys. Chem. B 2004, 108, 4840–4849. [Google Scholar] [CrossRef] [Green Version]
- Koper, G.; Borkovec, M. Binding of metal ions to polyelectrolytes and their oligomeric counterparts: An application of a generalized potts model. J. Phys. Chem. B 2001, 105, 6666–6674. [Google Scholar] [CrossRef]
- da Silva, F.L.B.; Boström, M.; Persson, C. Effect of Charge Regulation and Ion–Dipole Interactions on the Selectivity of Protein–Nanoparticle Binding. Langmuir 2014, 30, 4078–4083. [Google Scholar] [CrossRef]
- Landsgesell, J.; Hebbeker, P.; Rud, O.; Lunkad, R.; Košovan, P.; Holm, C. Grand-Reaction Method for Simulations of Ionization Equilibria Coupled to Ion Partitioning. Macromolecules 2020, 53, 3007–3020. [Google Scholar] [CrossRef] [Green Version]
- Baptista, A.M.; Martel, P.J.; Petersen, S.B. Simulation of protein conformational freedom as a function of pH: Constant-pH molecular dynamics using implicit titration. Proteins 1997, 27, 523–544. [Google Scholar] [CrossRef]
- Bürgi, R.; Kollman, P.A.; Van Gunsteren, W.F. Simulating proteins at constant pH: An approach combining molecular dynamics and Monte Carlo simulation. Proteins 2002, 47, 469–480. [Google Scholar] [CrossRef]
- Tanford, C. Physical Chemistry of Macromolecules; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1961. [Google Scholar]
- Katchalsky, A. Polye1ectrolytes. Pure Appl. Chem. 1971, 26, 327–374. [Google Scholar] [CrossRef]
- Oosawa, F. Polyelectrolytes; M. Dekker: New York City, NY, USA, 1971. [Google Scholar]
- Dubin, P.; Bock, J.; Davis, R.; Schulz, D.N.; Thies, C. Macromolecular Complexes in Chemistry and Biology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1994. [Google Scholar]
- Förster, S.; Schmidt, M. Polyelectrolytes in solution. In Physical Properties of Polymers; Abe, A., Albertsson, A., Coates, G.W., Genzer, J., Kobayashi, S., Lee, K., Leibler, L., Long, T.E., Möller, M., Okay, O., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; pp. 51–133. [Google Scholar]
- Barrat, J.L.; Joanny, J.F. Theory of polyelectrolyte solutions. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1997; Volume 94, pp. 1–66. [Google Scholar]
- Holm, C.; Joanny, J.; Kremer, K.; Netz, R.; Reineker, P.; Seidel, C.; Vilgis, T.A.; Winkler, R. Polyelectrolyte Theory; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Muthukumar, M. Theory of counter-ion condensation on flexible polyelectrolytes: Adsorption mechanism. J. Chem. Phys. 2004, 120, 9343–9350. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Theory of polyelectrolytes in solutions and at surfaces. Prog. Polym. Sci. 2005, 30, 1049–1118. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Theory and simulations of charged polymers: From solution properties to polymeric nanomaterials. Curr. Opin. Colloid Interface Sci. 2008, 13, 376–388. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Polyelectrolytes: On the doorsteps of the second century. Polymer 2020, 202, 122714. [Google Scholar] [CrossRef]
- Borkovec, M.; Koper, G.J. A cluster expansion method for the complete resolution of microscopic ionization equilibria from NMR titrations. Anal. Chem. 2000, 72, 3272–3279. [Google Scholar] [CrossRef]
- Madurga, S.; Nedyalkova, M.; Mas, F.; Garcés, J.L. Ionization and Conformational Equilibria of Citric Acid: Delocalized Proton Binding in Solution. J. Phys. Chem. A 2017, 121, 5894–5906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakács, Z.; Kraszni, M.; Noszál, B. Determination of microscopic acid-base parameters from NMR-pH titrations. Anal. Bioanal. Chem. 2002, 378, 1428–1448. [Google Scholar] [CrossRef]
- Nagasawa, M.; Murase, T.; Kondo, K. Potentiometric titration of stereoregular polyelectrolytes. J. Phys. Chem. 1965, 69, 4005–4012. [Google Scholar] [CrossRef]
- Madurga, S.; Garcés, J.L.; Companys, E.; Rey-Castro, C.; Salvador, J.; Galceran, J.; Vilaseca, E.; Puy, J.; Mas, F. Ion binding to polyelectrolytes: Monte Carlo simulations versus classical mean field theories. Theor. Chem. Acc. 2009, 123, 127–135. [Google Scholar] [CrossRef]
- Madurga, S.; Rey-Castro, C.; Pastor, I.; Vilaseca, E.; David, C.; Garcés, J.L.; Puy, J.; Mas, F. A semi-grand canonical Monte Carlo simulation model for ion binding to ionizable surfaces: Proton binding of carboxylated latex particles as a case study. J. Chem. Phys. 2011, 135, 184103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunkad, R.; Murmiliuk, A.; Tošner, Z.; Štěpánek, M.; Košovan, P. Role of pKA in Charge Regulation and Conformation of Various Peptide Sequences. Polymers 2021, 13, 214. [Google Scholar] [CrossRef]
- Acerenza, L.; Mizraji, E. Cooperativity: A unified view. Biochim. Biophys. Acta 1997, 1339, 155–166. [Google Scholar] [CrossRef]
- Garcés, J.L.; Mas, F.; Puy, J.; Galceran, J.; Salvador, J. Use of activity coefficients for bound and free sites to describe metal–macromolecule complexation. J. Chem. Soc. Faraday Trans. 1998, 94, 2783–2794. [Google Scholar] [CrossRef]
- Garcés, J.L.; Mas, F.; Cecília, J.; Companys, E.; Galceran, J.; Salvador, J.; Puy, J.; Galceran, J. Complexation isotherms in metal speciation studies at trace concentration levels. Voltammetric techniques in environmental samples. Phys. Chem. Chem. Phys. 2002, 4, 3764–3773. [Google Scholar] [CrossRef]
- Garcés, J.L.; Acerenza, L.; Mizraji, E.; Mas, F. A hierarchical approach to cooperativity in macromolecular and self-assembling binding systems. J. Biol. Phys. 2008, 34, 213–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cera, E. Thermodynamic Theory of Site-Specific Binding Processes in Biological Macromolecules; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Hill, T.L. Approximate calculation of the electrostatic free energy of nucleic acids and other cylindrical macromolecules. Arch. Biochem. Biophys. 1955, 57, 229–239. [Google Scholar] [CrossRef]
- Murmiliuk, A.; Košovan, P.; Janata, M.; Procházka, K.; Uhlík, F.; Štěpánek, M. Local pH and Effective pK of a Polyelectrolyte Chain: Two Names for One Quantity? ACS Macro Lett. 2018, 7, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Lunkad, R.; Biehl, P.; Murmiliuk, A.; Blanco, P.M.; Mons, P.; Štěpánek, M.; Schacher, F.H.; Košovan, P. Simulations and Potentiometric Titrations Enable Reliable Determination of Effective pKa Values of Various Polyzwitterions. Macromolecules 2022, 55, 7775–7784. [Google Scholar] [CrossRef]
- Podgornik, R. General theory of charge regulation and surface differential capacitance. J. Chem. Phys. 2018, 149, 104701. [Google Scholar] [CrossRef]
- Blanco, P.M.; Madurga, S.; Narambuena, C.F.; Mas, F.; Garcés, J.L. Role of Charge Regulation and Fluctuations in the Conformational and Mechanical Properties of Weak Flexible Polyelectrolytes. Polymers 2019, 11, 1962. [Google Scholar] [CrossRef] [Green Version]
- Božič, A.; Podgornik, R. Site Correlations, Capacitance, and Polarizability From Protein Protonation Fluctuations. J. Phys. Chem. B 2021, 125, 12902–12908. [Google Scholar] [CrossRef]
- Srivastava, D.; Santiso, E.; Gubbins, K.; Barroso da Silva, F.L. Computationally Mapping pKa Shifts Due to the Presence of a Polyelectrolyte Chain around Whey Proteins. Langmuir 2017, 33, 11417–11428. [Google Scholar] [CrossRef]
- Lunkad, R.; Barroso da Silva, F.L.; Košovan, P. Both Charge-Regulation and Charge-Patch Distribution Can Drive Adsorption on the Wrong Side of the Isoelectric Point. J. Am. Chem. Soc. 2022, 144, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Di Cera, E. Stochastic linkage: Effect of random fluctuations on a two state process Stochastic linkage: Effect of random fluctuations on a two-state process. J. Phys. Chem. 1991, 95, 5082–5086. [Google Scholar] [CrossRef]
- Blanco, P.M.; Madurga, S.; Garcés, J.L.; Mas, F.; Dias, R.S. Influence of macromolecular crowding on the charge regulation of intrinsically disordered proteins. Soft Matter 2021, 17, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Narambuena, C.F.; Ausar, F.S.; Bianco, I.D.; Beltramo, D.M.; Leiva, E.P. Aggregation of casein micelles by interactions with chitosans: A study by Monte Carlo simulations. J. Agric. Food Chem. 2005, 53, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Kirkwood, J.G. On the Theory of Strong Electrolyte Solutions. J. Chem. Phys. 1937, 2, 767. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Electrostatic Forces between Surfaces in Liquids. In Intermolecular and Surface Forces, 3rd ed.; Israelachvili, J.N., Ed.; Academic Press: San Diego, CA, USA, 2011; pp. 291–340. [Google Scholar]
- Ullner, M. Comments on the Scaling Behavior of Flexible Polyelectrolytes within the Debye-Huckel Approximation. J. Phys. Chem. B 2003, 107, 8097–8110. [Google Scholar] [CrossRef]
- Garcés, J.L.; Madurga, S.; Rey-Castro, C.; Mas, F. Dealing with long-range interactions in the determination of polyelectrolyte ionization properties. Extension of the transfer matrix formalism to the full range of ionic strengths. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Bokris, J.O.M.; Reddy, A.K. Modern Electrochemistry, an Introduction to a Interdisciplinary Area; Springer: New York, NY, USA, 1973. [Google Scholar]
- Kirkwood, J.G. Theory of Solutions of Molecules Containing Widely Separated Charges with Special Application to Zwitterions. J. Chem. Phys. 1934, 2, 351. [Google Scholar] [CrossRef]
- Jurado de Carvalho, S.; Ghiotto, R.C.T.; Barroso da Silva, F.L. Monte Carlo and modified Tanford-Kirkwood results for macromolecular electrostatics calculations. J. Phys. Chem. B 2006, 110, 8832–8839. [Google Scholar] [CrossRef]
- Borkovec, M.; Daicic, J.; Koper, G.J. Ionization properties of interfaces and linear polyelectrolytes: A discrete charge Ising model. Phys. A Stat. Mech. Its Appl. 2001, 298, 1–23. [Google Scholar] [CrossRef]
- Borkovec, M.; Daicic, J.; Koper, G.J. On the difference in ionization properties between planar interfaces and linear polyelectrolytes. Proc. Natl. Acad. Sci. USA 1997, 94, 3499–3503. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J. Statistical Mechanics of Chain Molecules; Wiley: Hoboken, NJ, USA, 1969. [Google Scholar]
- De Groot, J.; Koper, G.J.M.; Borkovec, M.; De Bleijser, J. Dissociation behavior of poly (maleic acid): Potentiometric titrations, viscometry, pulsed field gradient NMR, and model calculations. Macromolecules 1998, 31, 4182–4188. [Google Scholar] [CrossRef]
- Flory, P. Principles of Polymer Chemistry; Cornell University: Ithaca, NY, USA, 1953. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: New York, NY, USA, 2003; Volume 23. [Google Scholar]
- Kuhn, W. Über die gestalt fadenförmiger moleküle in lösungen. Kolloid-Zeitschrift 1934, 68, 2–15. [Google Scholar] [CrossRef]
- Kuhn, W. Beziehungen zwischen Molekülgröße, statistischer Molekülgestalt und elastischen Eigenschaften hochpolymerer Stoffe. Kolloid-Zeitschrift 1939, 87, 3. [Google Scholar] [CrossRef]
- Kuhn, W. Molekülkonstellation und Kristallitorientierung als Ursachen kautschukähnlicher Elastizität. Kolloid-Zeitschrift 1936, 76, 258. [Google Scholar] [CrossRef]
- Kratky, O.; Porod, G. Röntgenuntersuchung gelöster fadenmoleküle. Recl. Des Trav. Chim. Des Pays-Bas 1949, 68, 1106. [Google Scholar] [CrossRef]
- Porod, G. The mean separation of chain ends in threadlike molecules. Mon. Für Chem. 1949, 80, 251. [Google Scholar] [CrossRef]
- Lin, K.; Chen, M. Universal amplitude ratios for three-dimensional self-avoiding walks. J. Phys. A Math. Gen. 2002, 35, 1501. [Google Scholar]
- Tonelli, A.; Patterson, G. (Eds.) Modern Applications of Flory’s "Statistical Mechanics of Chain Molecules"; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020. [Google Scholar]
- Chandler, D. Introduction to Modern Statistical Mechanics; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Blanco, P.M. Coupling of Binding and Conformational Equilibria in Weak Polyelectrolytes. Dynamics and Charge Regulation of Biopolymers in Crowded Media. Ph.D. Thesis, University of Barcelona, Barcelona, Catalonia, Spain, 2020. [Google Scholar]
- Wang, M.D.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Stretching DNA with optical tweezers. Biophys. J. 1997, 72, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.U.; Chrisey, L.A.; Colton, R.J. Direct Measurement of the Forces Between Complementary Strands of DNA. Science 1994, 266, 771–773. [Google Scholar] [CrossRef]
- Bustamante, C.; Bryant, Z.; Smith, S.B. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camunas-Soler, J.; Ribezzi-Crivellari, M.; Ritort, F. Elastic Properties of Nucleic Acids by Single–Molecule Force Spectroscopy. Annu. Rev. Biophys. 2016, 45, 65–84. [Google Scholar] [CrossRef]
- Smith, S.B.; Cui, Y.; Bustamante, C. B-DNA: The Elastic Response of Overstretching Individual Double-Stranded and Single-Stranded. Science 1996, 271, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marszalek, P.E.; Oberhauser, A.F.; Pang, Y.P.; Fernandez, J.M. Polysaccharide elasticity governed by chair-boat transitions of the glucopyranose ring. Nature 1998, 396, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Giannotti, M.I.; Rinaudo, M.; Vancso, G.J. Force spectroscopy of hyaluronan by atomic force microscopy: From hydrogen-bonded networks toward single–chain behavior. Biomacromolecules 2007, 8, 2648–2652. [Google Scholar] [CrossRef] [PubMed]
- Valiaev, A.; Dong, W.L.; Oas, T.G.; Chilkoti, A.; Zauscher, S. Force-induced prolyl cis-trans isomerization in elastin-like polypeptides. J. Am. Chem. Soc. 2007, 129, 6491–6497. [Google Scholar] [CrossRef]
- Radiom, M.; Kong, P.; Maroni, P.; Schäfer, M.; Kilbinger, A.F.M.; Borkovec, M. Mechanically induced cis–to–trans isomerization of carbon–carbon double bonds using atomic force microscopy. Phys. Chem. Chem. Phys. 2016, 18, 31202–31210. [Google Scholar] [CrossRef] [Green Version]
- Wiita, A.P.; Perez-Jimenez, R.; Walther, K.A.; Gräter, F.; Berne, B.J.; Holmgren, A.; Sanchez-Ruiz, J.M.; Fernandez, J.M. Probing the chemistry of thioredoxin catalysis with force. Nature 2007, 450, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Linke, W.A.; Oberhauser, A.F.; Carrion-Vazquez, M.; Kerkvliet, J.G.; Lu, H.; Marszalek, P.E.; Fernandez, J.M. Reverse engineering of the giant muscle protein titin. Nature 2002, 418, 998–1002. [Google Scholar] [CrossRef]
- Krysiak, S.; Liese, S.; Netz, R.R.; Hugel, T. Peptide desorption kinetics from single molecule force spectroscopy studies. J. Am. Chem. Soc. 2014, 136, 688–697. [Google Scholar] [CrossRef]
- Giannotti, M.I.; Vancso, G.J. Interrogation of single synthetic polymer chains and polysaccharides by AFM-based force spectroscopy. ChemPhysChem 2007, 8, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Magonov, S.; Su, C.; Quate, C.F.; Solgaard, O. An atomic force microscope tip designed to measure time-varying nanomechanical forces. Nat. Nanotechnol. 2007, 2, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Sahin, O. A nanomechanical interface to rapid single-molecule interactions. Nat. Commun. 2011, 2, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Bullerjahn, J.T.; Lallemang, M.; Kroy, K.; Balzer, B.N.; Hugel, T. Angle-dependent strength of a single chemical bond by stereographic force spectroscopy. Chem. Sci. 2022, 13, 5734–5740. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Jäger, M.; Bullerjahn, J.T.; Hugel, T.; Wolf, S.; Balzer, B.N. Anisotropic Friction in a Ligand–Protein Complex. Nano Lett. 2023, 23, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Cai, W.; Zhang, S.; Cui, S. Single-Chain Mechanics at Low Temperatures: A Study of Three Kinds of Polymers from 153 to 300 K. Macromolecules 2023, 56, 3204–3212. [Google Scholar] [CrossRef]
- Saleh, O.A. Single polymer mechanics across the force regimes. J. Chem. Phys. 2015, 142, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.B.; Finzi, L.; Bustamante, C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 1992, 258, 1122–1126. [Google Scholar] [CrossRef]
- Zhang, W.; Zou, S.; Wang, C.; Zhang, X. Single polymer chain elongation of poly (N-isopropylacrylamide) and poly (acrylamide) by atomic force microscopy. J. Phys. Chem. B 2000, 104, 10258–10264. [Google Scholar] [CrossRef]
- Bustamante, C.; Marko, J.F.; Siggia, E.D.; Smith, S. Entropic Elasticity of X-Phage DNA Explicit and Implicit Learning and Maps of Cortical Motor Output. Science 1994, 265, 1599–1600. [Google Scholar] [CrossRef] [Green Version]
- Marko, J.F.; Siggia, E.D. Stretching DNA. Macromolecules 1995, 28, 8759–8770. [Google Scholar] [CrossRef]
- Radiom, M.; Borkovec, M. Influence of ligand-receptor interactions on force-extension behavior within the freely jointed chain model. Phys. Rev. E 2017, 96, 062501. [Google Scholar] [CrossRef] [PubMed]
- Kierfeld, J.; Niamploy, O.; Sa-Yakanit, V.; Lipowsky, R. Stretching of semiflexible polymers with elastic bonds. Eur. Phys. J. E 2004, 14, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Hugel, T.; Rief, M.; Seitz, M.; Gaub, H.E.; Netz, R.R. Highly stretched single polymers: Atomic-force-microscope experiments versus ab-initio theory. Phys. Rev. Lett. 2005, 94, 048301. [Google Scholar] [CrossRef]
- Kreuzer, H.J.; Grunze, M. Stretching of single polymer strands: A first-principles theory. Europhys. Lett. (EPL) 2001, 55, 640–646. [Google Scholar] [CrossRef]
- Oesterhelt, F.; Rief, M.; Gaub, H.E. Single molecule force spectroscopy by AFM indicates helical structure of poly(ethylene-glycol) in water. New J. Phys. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- Liese, S.; Gensler, M.; Krysiak, S.; Schwarzl, R.; Achazi, A.; Paulus, B.; Hugel, T.; Rabe, J.P.; Netz, R.R. Hydration Effects Turn a Highly Stretched Polymer from an Entropic into an Energetic Spring. ACS Nano 2017, 11, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Hanke, F.; Serr, A.; Kreuzer, H.J.; Netz, R.R. Stretching single polypeptides: The effect of rotational constraints in the backbone. Europhys. Lett. (EPL) 2010, 92, 53001. [Google Scholar] [CrossRef]
- Neuert, G.; Hugel, T.; Netz, R.R.; Gaub, H.E. Elasticity of poly(azobenzene-peptides). Macromolecules 2006, 39, 789–797. [Google Scholar] [CrossRef]
- Pincus, P. Excluded Volume Effects and Stretched Polymer Chains. Macromolecules 1975, 9, 386–388. [Google Scholar] [CrossRef]
- Cui, S.; Albrecht, C.; Kühner, F.; Gaub, H.E. Weakly bound water molecules shorten single-stranded DNA. J. Am. Chem. Soc. 2006, 128, 6636–6639. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Yu, Y.; Lin, Z. Modeling single chain elasticity of single-stranded DNA: A comparison of three models. Polymer 2009, 50, 930–935. [Google Scholar] [CrossRef]
- Luo, Z.; Zhang, A.; Chen, Y.; Shen, Z.; Cui, S. How big is big enough? Effect of length and shape of side chains on the single-chain enthalpic elasticity of a macromolecule. Macromolecules 2016, 49, 3559–3565. [Google Scholar] [CrossRef]
- Wang, K.; Pang, X.; Cui, S. Inherent stretching elasticity of a single polymer chain with a carbon–carbon backbone. Langmuir 2013, 29, 4315–4319. [Google Scholar] [CrossRef]
- Cai, W.; Lu, S.; Wei, J.; Cui, S. Single-chain polymer models incorporating the effects of side groups: An approach to general polymer models. Macromolecules 2019, 52, 7324–7330. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Kitano, T.; Ito, K. Dissociation Behaviour of Poly(fumaric acid) and Poly(maleic acid). 3. Infrared and Ultraviolet Spectroscopy. Macromolecules 1992, 25, 1294–1299. [Google Scholar] [CrossRef]
- Marcus, R.A. Titration of Polyelectrolytes at Higher Ionic Strengths. J. Phys. Chem. 1954, 58, 621–623. [Google Scholar] [CrossRef]
- Smits, R.; Koper, G.; Mandel, M. The influence of nearest-and next-nearest-neighbor interactions on the potentiometric titration of linear poly (ethylenimine). J. Phys. Chem. 1993, 97, 5745–5751. [Google Scholar] [CrossRef]
- Koper, G.J.; Borkovec, M. Exact affinity distributions for linear polyampholytes and polyelectrolytes. J. Chem. Phys. 1996, 104, 4204–4213. [Google Scholar] [CrossRef]
- Reed, C.E.; Reed, W.F. Monte Carlo study of titration of linear polyelectrolytes. J. Chem. Phys. 1992, 96, 1609. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.M.; Madurga, S.; Mas, F.; Garcés, J.L. Coupling of charge regulation and conformational equilibria in linearweak polyelectrolytes: Treatment of long-range interactions via effective short-ranged and pH-dependent interaction parameters. Polymers 2018, 10, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flory, P.J.; Jernigan, R.L. Second and Fourth Moments of Chain Molecules. J. Chem. Phys. 1965, 42, 3509–3519. [Google Scholar] [CrossRef]
- Borkovec, M.; Koper, G.J. Ising models and acid-base properties of weak polyelectrolytes. Ber. Der Bunsenges. Für Phys. Chem. 1996, 100, 764–769. [Google Scholar] [CrossRef]
- Mieres-Perez, J.; Sanchez-Garcia, E. Quantum mechanics/molecular mechanics multiscale modeling of biomolecules. In Advances in Physical Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; Volume 54, pp. 143–183. [Google Scholar]
- Baptista, A.M.; Teixeira, V.H.; Soares, C.M.; Baptista, M.; Teixeira, V.H. Constant-pH molecular dynamics using stochastic titration Constant-p H molecular dynamics using stochastic titration. J. Chem. Phys. 2002, 117, 4184–4199. [Google Scholar] [CrossRef]
- Machuqueiro, M.; Baptista, A.M. Is the prediction of pKa values by constant-pH molecular dynamics being hindered by inherited problems? Proteins Struct. Funct. Bioinform. 2011, 79, 3437–3447. [Google Scholar] [CrossRef]
- de Oliveira, V.M.; Liu, R.; Shen, J. Constant ph molecular dynamics simulations: Current status and recent applications. Curr. Opin. Struct. Biol. 2022, 77, 102498. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Johnson, J.K.; Panagiotopoulos, A.Z.; Gubbins, K.E. Reactive canonical Monte Carlo: A new simulation technique for reacting or associating fluids. Mol. Phys. 1994, 81, 717–733. [Google Scholar] [CrossRef]
- Smith, W.R.; Triska, B. The reaction ensemble method for the computer simulation of chemical and phase equilibria. I. Theory and basic examples. J. Chem. Phys. 1994, 100, 3019–3027. [Google Scholar] [CrossRef]
- Teixeira, A.A.R.; Lund, M.; Barroso da Silva, F.L. Fast Proton Titration Scheme for Multiscale Modeling of Protein Solutions. J. Chem. Theory Comput. 2010, 6, 3259–3266. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; MacKernan, D. Benchmarking a Fast Proton Titration Scheme in Implicit Solvent for Biomolecular Simulations. J. Chem. Theory Comput. 2017, 13, 2915–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weik, F.; Weeber, R.; Szuttor, K.; Breitsprecher, K.; de Graaf, J.; Kuron, M.; Landsgesell, J.; Menke, H.; Sean, D.; Holm, C. ESPResSo 4.0—An extensible software package for simulating soft matter systems. Eur. Phys. J. Spec. Top. 2019, 227, 1789–1816. [Google Scholar] [CrossRef] [Green Version]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Jurij, R.; Per, L. MOLSIM: A modular molecular simulation software. J. Comput. Chem. 2015, 36, 1259–1274. [Google Scholar] [CrossRef]
- Lund, M.; Trulsson, M.; Persson, B. Faunus: An object oriented framework for molecular simulation. Source Code Biol. Med. 2008, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Stenqvist, B.; Thuresson, A.; Kurut, A.; Vácha, R.; Lund, M. Faunus—A flexible framework for Monte Carlo simulation. Mol. Simul. 2013, 39, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Uyaver, S.; Seidel, C. First-order conformational transition of annealed polyelectrolytes in a poor solvent. Europhys. Lett. 2003, 64, 536–542. [Google Scholar] [CrossRef]
- Carnal, F.; Ulrich, S.; Stoll, S. Influence of explicit ions on titration curves and conformations of flexible polyelectrolytes: A Monte Carlo study. Macromolecules 2010, 43, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Clavier, A.; Seijo, M.; Carnal, F.; Stoll, S. Surface charging behavior of nanoparticles by considering site distribution and density, dielectric constant and pH changes—A Monte Carlo approach. Phys. Chem. Chem. Phys. 2015, 17, 4346–4353. [Google Scholar] [CrossRef] [PubMed]
- Cerdà, J.J.; Ballenegger, V.; Lenz, O.; Holm, C. P3M algorithm for dipolar interactions. J. Chem. Phys. 2008, 129, 234104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbez, C.; Jönsson, B. A New Monte Carlo Method for the Titration of Molecules and Minerals. In Applied Parallel Computing. State of the Art in Scientific Computing; Kågström, B., Elmroth, E., Dongarra, J., Waśniewski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4699, pp. 66–72. [Google Scholar]
- Košovan, P.; Landsgesell, J.; Nová, L.; Uhlík, F.; Beyer, D.; Blanco, P.M.; Staňo, R.; Holm, C. Reply to the ‘Comment on “Simulations of ionization equilibria in weak polyelectrolyte solutions and gels”’ by J. Landsgesell, L. Nová, O. Rud, F. Uhlík, D. Sean, P. Hebbeker, C. Holm and P. Košovan, Soft Matter, 2019, 15, 1155–1185. Soft Matter 2023, 19, 3522–3525. [Google Scholar] [CrossRef] [PubMed]
- Curk, T.; Yuan, J.; Luijten, E. Accelerated simulation method for charge regulation effects. J. Chem. Phys. 2022, 156, 044122. [Google Scholar] [CrossRef]
- Rud, O.V.; Landsgesell, J.; Holm, C.; Košovan, P. Modeling of weak polyelectrolyte hydrogels under compression—Implications for water desalination. Desalination 2021, 506, 114995. [Google Scholar] [CrossRef]
- Staňo, R.; Košovan, P.; Tagliabue, A.; Holm, C. Electrostatically Cross-Linked Reversible Gels—Effects of pH and Ionic Strength. Macromolecules 2021, 54, 4769–4781. [Google Scholar] [CrossRef]
- Beyer, D.; Košovan, P.; Holm, C. Simulations Explain the Swelling Behavior of Hydrogels with Alternating Neutral and Weakly Acidic Blocks. Macromolecules 2022, 55, 10751–10760. [Google Scholar] [CrossRef]
- Landsgesell, J.; Beyer, D.; Hebbeker, P.; Košovan, P.; Holm, C. The pH-Dependent Swelling of Weak Polyelectrolyte Hydrogels Modeled at Different Levels of Resolution. Macromolecules 2022, 55, 3176–3188. [Google Scholar] [CrossRef]
- Jacobson, D.R.; McIntosh, D.B.; Saleh, O.A. The snakelike chain character of unstructured RNA. Biophys. J. 2013, 105, 2569–2576. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, D.R.; Mcintosh, D.B.; Stevens, M.J.; Rubinstein, M.; Saleh, O.A. Single-stranded nucleic acid elasticity arises from internal electrostatic tension. Proc. Natl. Acad. Sci. USA 2017, 114, 5095–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netz, R.R. Strongly stretched semiflexible extensible polyelectrolytes and DNA. Macromolecules 2001, 34, 7522–7529. [Google Scholar] [CrossRef]
- Dessinges, M.N.; Maier, B.; Zhang, Y.; Peliti, M.; Bensimon, D.; Croquette, V. Stretching Single Stranded DNA, a Model Polyelectrolyte. Phys. Rev. Lett. 2002, 89, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Seol, Y.; Skinner, G.M.; Visscher, K. Elastic properties of a single-stranded charged homopolymeric ribonucleotide. Phys. Rev. Lett. 2004, 93, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Saleh, O.A.; McIntosh, D.B.; Pincus, P.; Ribeck, N. Nonlinear low-force elasticity of single-stranded DNA molecules. Phys. Rev. Lett. 2009, 102, 068301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcintosh, D.B.; Saleh, O.A. Salt Species-Dependent Electrostatic Effects on ssDNA Elasticity. Macromolecules 2011, 44, 2328–2333. [Google Scholar] [CrossRef]
- Bosco, A.; Camunas-Soler, J.; Ritort, F. Elastic properties and secondary structure formation of single–stranded DNA at monovalent and divalent salt conditions. Nucleic Acids Res. 2014, 42, 2064–2074. [Google Scholar] [CrossRef]
- Ullner, M.; Jönsson, B.; Peterson, C.; Sommelius, O.; Söderberg, B. The electrostatic persistence length calculated from Monte Carlo, variational and perturbation methods. J. Chem. Phys. 1997, 107, 1279–1287. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.; McIntosh, D.; Saleh, O. Simulations of stretching a strong, flexible polyelectrolyte. Macromolecules 2012, 45, 5757–5765. [Google Scholar] [CrossRef]
- Stevens, M.J.; McIntosh, D.B.; Saleh, O.A. Simulations of stretching a strong, flexible polyelectrolyte: Using Long chains to access the pincus scaling regime. Macromolecules 2013, 46, 6369–6373. [Google Scholar] [CrossRef]
- Stevens, M.J.; Berezney, J.P.; Saleh, O.A. The effect of chain stiffness and salt on the elastic response of a polyelectrolyte. J. Chem. Phys. 2018, 149, 163328. [Google Scholar] [CrossRef]
- Berezney, J.P.; Saleh, O.A. Electrostatic Effects on the Conformation and Elasticity of Hyaluronic Acid, a Moderately Flexible Polyelectrolyte. Macromolecules 2017, 50, 1085–1089. [Google Scholar] [CrossRef]
- Sodric, S. Stretching of Weak Polyelectrolytes at the Single-Molecule Level. Bachelor’s Thesis, University of Barcelona, Barcelona, Catalonia, Spain, 2019. [Google Scholar]
- Orradre, J. Computational Study of Charge Regulation Effect on the Stretching of Weak Polyelectrolytes. Bachelor’s Thesis, University of Barcelona, Barcelona, Catalonia, Spain, 2020. [Google Scholar]
- Jarillo, J.; Morín, J.A.; Beltrán-Heredia, E.; Villaluenga, J.P.G.; Ibarra, B.; Cao, F.J. Mechanics, thermodynamics, and kinetics of ligand binding to biopolymers. PLoS ONE 2017, 12, e0174830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Marko, J.F. Maxwell relations for single-DNA experiments: Monitoring protein binding and double-helix torque with force-extension measurements. Phys. Rev. E 2008, 77, 031916. [Google Scholar] [CrossRef] [PubMed]
- Marko, J.F.; Siggia, E.D. Driving Proteins Off DNA Using Applied Tension. Biophys. J. 1997, 73, 2173–2178. [Google Scholar] [CrossRef] [Green Version]
- Stoll, A.L.S. Adsorption of a weakly charged polymer on an oppositely charged colloidal particle: Monte Carlo simulations investigation. Polymer 2005, 46, 1359–1372. [Google Scholar]
- Netz, R.R.; Orland, H. Variational charge renormalization in charged systems. Eur. Phys. J. E 2003, 11, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Dobrynin, A.V. Electrostatic Persistence Length of Semiflexible and Flexible Polyelectrolytes. Macromolecules 2005, 38, 9304–9314. [Google Scholar] [CrossRef]
- de Vries, R.; Stuart, M.C. Theory and Simulations of Macroion Complexation. Curr. Opin. Colloid Interface Sci. 2006, 11, 295–301. [Google Scholar] [CrossRef]
- Ulrich, S.; Seijo, M.; Stoll, S. The Many Facets of Polyelectrolytes and Oppositely Charged Macroions Complex Formation. Curr. Opin. Colloid Interface Sci. 2006, 11, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Borkovec, M.; Papastavrou, G. Interactions between solid surfaces with adsorbed polyelectrolytes of opposite charge. Curr. Opin. Colloid Interface Sci. 2008, 13, 429–437. [Google Scholar] [CrossRef]
- Messina, R. Electrostatics in Soft Matter. J. Phys. Condens. Matter 2009, 21, 113102. [Google Scholar] [CrossRef] [Green Version]
- Szilagyi, I.; Trefalt, G.; Tiraferri, A.; Maroni, P.; Borkovec, M. Polyelectrolyte Adsorption, Interparticle Forces, and Colloidal Aggregation. Soft Matter 2014, 10, 2479–2502. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Krause, P.; Chiappisi, L.; Noirez, L.; Gradzielski, M. Structural Control of Polyelectrolyte/microemulsion Droplet Complexes (pemecs) With Different Polyacrylates. Chem. Sci. 2019, 10, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, S.; Laguecir, A.; Stoll, S. Complexation of a Weak Polyelectrolyte with a Charged Nanoparticle. Solution Properties and Polyelectrolyte Stiffness Influences. Macromolecules 2005, 38, 8939–8949. [Google Scholar] [CrossRef]
- Stornes, M.; Linse, P.; Dias, R.S. Monte Carlo Simulations of Complexation between Weak Polyelectrolytes and a Charged Nanoparticle. Influence of Polyelectrolyte Chain Length and Concentration. Macromolecules 2017, 50, 5978–5988. [Google Scholar] [CrossRef]
- Carnal, F.; Stoll, S. Chain stiffness, salt valency, and concentration influences on titration curves of polyelectrolytes: Monte Carlo simulations. J. Chem. Phys. 2011, 134, 044904. [Google Scholar] [CrossRef]
- de Oliveira, V.M.; de Carvalho, S.J. Adsorption of pH-Responsive Polyelectrolyte Chains Onto Spherical Macroions. Eur. Phys. J. E 2014, 37, 75. [Google Scholar] [CrossRef]
- Carnal, F.; Clavier, A.; Stoll, S. Polypeptide-nanoparticle interactions and corona formation investigated by monte carlo simulations. Polymers 2016, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, S.J.; Metzler, R.; Cherstvy, A.G. Critical Adsorption of Polyelectrolytes Onto Charged Janus Nanospheres. Phys. Chem. Chem. Phys. 2014, 16, 15539–15550. [Google Scholar] [CrossRef] [Green Version]
- Božič, A.L.; Podgornik, R. Anomalous Multipole Expansion: Charge Regulation of Patchy Inhomogeneously Charged Spherical Particles. J. Chem. Phys. 2018, 149, 163307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avni, Y.; Andelman, D.; Podgornik, R. Charge Regulation With Fixed and Mobile Charged Macromolecules. Curr. Opin. Electrochem. 2019, 13, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Yigit, C.; Heyda, J.; Dzubiella, J. Charged patchy particle models in explicit salt: Ion distributions, electrostatic potentials, and effective interactions. J. Chem. Phys. 2015, 143, 064904. [Google Scholar] [CrossRef] [PubMed]
- Yigit, C.; Kanduc, M.; Ballau, M.; Dzubiella, J. Interaction of Charged Patchy Protein Models with Like-Charged Polyelectrolyte Brushes. Langmuir 2017, 33, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, P.; Bojanich, L.; Sanchez-Varretti, F.; Ramirez-Pastor, A.J.; Quiroga, E.; Boeris, V.; Narambuena, C.F. Protonation of β-lactoglobulin in the presence of strong polyelectrolyte chains: A study using Monte Carlo simulation. Colloids Surf. B Biointerfaces 2017, 160, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Torres, P.B.; Quiroga, E.; Ramirez-Pastor, A.J.; Boeris, V.; Narambuena, C.F. Interaction between β-Lactoglobuline and Weak Polyelectrolyte Chains: A Study Using Monte Carlo Simulation. J. Phys. Chem. B 2019, 123, 8617–8627. [Google Scholar] [CrossRef]
- Stornes, M.; Blanco, P.M.; Dias, R.S. Polyelectrolyte-nanoparticle mutual charge regulation and its influence on their complexation. Colloids Surf. A Physicochem. Eng. Asp. 2021, 628, 127258. [Google Scholar] [CrossRef]
- Zhukouskaya, H.; Blanco, P.M.; Černochová, Z.; Čtveráčková, L.; Staňo, R.; Pavlova, E.; Vetrík, M.; Černoch, P.; Filipová, M.; Šlouf, M.; et al. Anionically Functionalized Glycogen Encapsulates Melittin by Multivalent Interaction. Biomacromolecules 2022, 23, 3371–3382. [Google Scholar] [CrossRef]
- Lunkad, R.; Murmiliuk, A.; Hebbeker, P.; Boublík, M.; Tošner, Z.; Štěpánek, M.; Košovan, P. Quantitative prediction of charge regulation in oligopeptides. Mol. Syst. Des. Eng. 2021, 6, 122–131. [Google Scholar] [CrossRef]
- Hattori, T.; Hallberg, R.; Dubin, P.L. Roles of Electrostatic Interaction and Polymer Structure in the Binding of β-Lactoglobulin to Anionic Polyelectrolytes: Measurement of Binding Constants by Frontal Analysis Continuous Capillary Electrophoresis. Langmuir 2000, 16, 9738–9743. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Lund, M.; Jönsson, B.; Åkesson, T. On the Complexation of Proteins and Polyelectrolytes. J. Phys. Chem. B 2006, 110, 4459–4464. [Google Scholar] [CrossRef] [PubMed]
- Mattison, K.W.; Dubin, P.L.; Brittain, I.J. Complex formation between bovine serum albumin and strong polyelectrolytes: Effect of polymer charge density. J. Phys. Chem. B 1998, 102, 3830–3836. [Google Scholar] [CrossRef] [Green Version]
- de Vries, R.; Weinbreck, F.; de Kruif, C.G. Theory of polyelectrolyte adsorption on heterogeneously charged surfaces applied to soluble protein-polyelectrolyte complexes. J. Chem. Phys. 2003, 118, 4649–4659. [Google Scholar] [CrossRef]
- de Kruif, C.G.; Weinbreck, F.; de Vries, R. Complex coacervation of proteins and anionic polysaccharides. Curr. Opin. Colloid Interface Sci. 2004, 9, 340–349. [Google Scholar] [CrossRef]
- Wittemann, A.; Ballauff, M. Interaction of proteins with linear polyelectrolytes and spherical polyelectrolyte brushes in aqueous solution. Phys. Chem. Chem. Phys. 2006, 8, 5269–5275. [Google Scholar] [CrossRef]
- Xu, Y.; Mazzawi, M.; Chen, K.; Sun, L.; Dubin, P.L. Protein Purification by Polyelectrolyte Coacervation: Influence of Protein Charge Anisotropy on Selectivity. Biomacromolecules 2011, 12, 1512–1522. [Google Scholar] [CrossRef]
- Lošdorfer Božič, A.; Podgornik, R. pH Dependence of Charge Multipole Moments in Proteins. Biophys. J. 2017, 113, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- De Vos, W.M.; Leermakers, F.A.; De Keizer, A.; Cohen Stuart, M.A.; Kleijn, J.M. Field theoretical analysis of driving forces for the uptake of proteins by like-charged polyelectrolyte brushes: Effects of charge regulation and patchiness. Langmuir 2010, 26, 249–259. [Google Scholar] [CrossRef]
- Boubeta, F.M.; Soler-Illia, G.J.A.A.; Tagliazucchi, M. Electrostatically Driven Protein Adsorption: Charge Patches versus Charge Regulation. Langmuir 2018, 34, 15727–15738. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical Properties of Cells and Their Effects on Intrinsically Disordered Proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef]
- Kreub, M.; Strixner, T.; Kulozik, U. The effect of glycosylation on the interfacial properties of bovine caseinomacropeptide. Food Hydrocoll. 2009, 23, 1818–1826. [Google Scholar]
- Naji, A.; Kanduc, M.; Netz, R.R.; Podgornik, R. Exotic Electrostatics: Unusual Features of Electrostatic Interactions between Macroions. In Series in Soft Condensed Matter: Understanding Soft Condensed Matter via Modeling and Computation; Andelman, D., Reiter, G., Eds.; World Scientific: Singapore, 2010; Volume 3, pp. 265–295. [Google Scholar]
- Tong, C.; Zhu, Y.; Zhang, H.; Qiu, F.; Tang, P.; Yang, Y. The Self-Consistent Field Study of the Adsorption of Flexible Polyelectrolytes onto Two Charged Nano-objects. J. Phys. Chem. B 2011, 115, 11307–11317. [Google Scholar] [CrossRef] [PubMed]
- Forsman, J.; Nordholm, S. Polyelectrolyte Mediated Interactions in Colloidal Dispersions: Hierarchical Screening, Simulations, and a New Classical Density Functional Theory. Langmuir 2012, 28, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Forsman, J. Surface forces in electrolytes containing polyions and oppositely charged surfaces. Curr. Opin. Colloid Interface Sci. 2017, 27, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Stornes, M.; Shrestha, B.; Dias, R.S. PH-Dependent Polyelectrolyte Bridging of Charged Nanoparticles. J. Phys. Chem. B 2018, 122, 10237–10246. [Google Scholar] [CrossRef]
- Mella, M.; Tagliabue, A.; Mollica, L.; Izzo, L. Monte Carlo study of the effects of macroion charge distribution on the ionization and adsorption of weak polyelectrolytes and concurrent counterion release. J. Colloid Interface Sci. 2020, 560, 667–680. [Google Scholar] [CrossRef]
- Blaakmeer, J.; Bohmer, M.R.; Stuart, M.A.C.; Fleer, G.J. Adsorption of weak polyelectrolytes on highly charged surfaces. Poly(acrylic acid) on polystyrene latex with strong cationic groups. Macromolecules 1990, 23, 2301–2309. [Google Scholar] [CrossRef]
- Narambuena, C.F.; Blanco, P.M.; Rodriguez, A.; Rodriguez, D.E.; Madurga, S.; Garcés, J.L.; Mas, F. Non-monotonic behavior of weak-polyelectrolytes adsorption on a cationic surface: A Monte Carlo simulation study. Polymer 2021, 212, 123170. [Google Scholar] [CrossRef]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef]
- Hung, J.J.; Borwankar, A.U.; Dear, B.J.; Truskett, T.M.; Johnston, K.P. High concentration tangential flow ultrafiltration of stable monoclonal antibody solutions with low viscosities. J. Membr. Sci. 2016, 508, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Garidel, P.; Kuhn, A.B.; Schäfer, L.V.; Karow-Zwick, A.R.; Blech, M. High-concentration protein formulations: How high is high? Eur. J. Pharm. Biopharm. 2017, 119, 353–360. [Google Scholar] [CrossRef]
- Briskot, T.; Hillebrandt, N.; Kluters, S.; Wang, G.; Studts, J.; Hahn, T.; Huuk, T.; Hubbuch, J. Modeling the Gibbs–Donnan effect during ultrafiltration and diafiltration processes using the Poisson–Boltzmann theory in combination with a basic Stern model. J. Membr. Sci. 2022, 648, 120333. [Google Scholar] [CrossRef]
- Minton, A.P. Excluded volume as a determinant of protein structure and stability. Biopolymers 1981, 20, 2093–2120. [Google Scholar] [CrossRef]
- Milner, S.T.; Witten, T.A.; Cates, M.E. Theory of the Grafted Polymer Brush. Macromolecules 1988, 21, 2610–2619. [Google Scholar] [CrossRef]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Zhou, H.X.; Rivas, G.; Minton, A.P. Macromolecular Crowding and Confinement: Biochemical, Biophysical, and Potential Physiological Consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are infl uenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Konopka, M.C.; Shkel, I.A.; Cayley, S.; Record, M.T.; Weisshaar, J.C. Crowding and confinement effects on protein diffusion in vivo. J. Bacteriol. 2006, 188, 6115–6123. [Google Scholar] [CrossRef] [Green Version]
- Pastor, I.; Vilaseca, E.; Madurga, S.; Cascante, M.; Mas, F. Diffusion of alpha-Chymiotrypsin in Solution-Crowded Media. J. Phys. Chem. B 2010, 114, 4028–4034, Erratum in J. Phys. Chem. B 2010, 114, 12182. [Google Scholar] [CrossRef] [Green Version]
- Vilaseca, P.; Franzese, G. Softness dependence of the anomalies for the continuous shouldered well potential. J. Chem. Phys. 2010, 133, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnain, S.; McClendon, C.L.; Hsu, M.T.; Jacobson, M.P.; Bandyopadhyay, P. A new coarse-grained model for E. coli cytoplasm: Accurate calculation of the diffusion coefficient of proteins and observation of anomalous diffusion. PLoS ONE 2014, 9, e106466. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Klumpp, S. Biochemical reactions in crowded environments: Revisiting the effects of volume exclusion with simulations. Front. Phys. 2015, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5, e19274. [Google Scholar] [CrossRef]
- Walhout, P.K.; He, Z.; Dutagaci, B.; Nawrocki, G.; Feig, M. Molecular Dynamics Simulations of Rhodamine B Zwitterion Diffusion in Polyelectrolyte Solutions. J. Phys. Chem. B 2022, 126, 10256–10272. [Google Scholar] [CrossRef]
- Mereghetti, P.; Wade, R.C. Atomic detail brownian dynamics simulations of concentrated protein solutions with a mean field treatment of hydrodynamic interactions. J. Phys. Chem. B 2012, 116, 8523–8533. [Google Scholar] [CrossRef]
- Balbo, J.; Mereghetti, P.; Herten, D.P.; Wade, R.C. The shape of protein crowders is a major determinant of protein diffusion. Biophys. J. 2013, 104, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- Kondrat, S.; Zimmermann, O.; Wiechert, W.; Lieres, E.V. The effect of composition on diffusion of macromolecules in a crowded environment. Phys. Biol. 2015, 12, 046003. [Google Scholar] [CrossRef]
- Saxton, M.J. Anomalous diffusion due to obstacles: A Monte Carlo study. Biophys. J. 1994, 66, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Vilaseca, E.; Isvoran, A.; Madurga, S.; Pastor, I.; Garcés, J.L.; Mas, F. New insights into diffusion in 3D crowded media by Monte Carlo simulations: Effect of size, mobility and spatial distribution of obstacles. Phys. Chem. Chem. Phys. 2011, 13, 7396–7407. [Google Scholar] [CrossRef] [Green Version]
- Vilaseca, E.; Pastor, I.; Isvoran, A.; Madurga, S.; Garcés, J.L.; Mas, F. Diffusion in macromolecular crowded media: Monte Carlo simulation of obstructed diffusion vs. FRAP experiments. Theor. Chem. Acc. 2011, 128, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.M.; Via, M.; Garcés, J.L.; Madurga, S.; Mas, F. Brownian dynamics computational model of protein diffusion in crowded media with dextran macromolecules as obstacles. Entropy 2017, 19, 105. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.M.; Garcés, J.L.; Madurga, S.; Mas, F. Macromolecular diffusion in crowded media beyond the hard-sphere model. Soft Matter 2018, 14, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Skóra, T.; Vaghefikia, F.; Fitter, J.; Kondrat, S. Macromolecular Crowding: How Shape and Interactions Affect Diffusion. J. Phys. Chem. B 2020, 124, 7537–7543. [Google Scholar] [CrossRef]
- Słyk, E.; Skóra, T.; Kondrat, S. How macromolecules softness affects diffusion under crowding. Soft Matter 2022, 18, 5366–5370. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Povarova, O.I.; Sulatskaya, A.I.; Ferreira, L.A.; Zaslavsky, B.Y.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Protein unfolding in crowded milieu: What crowding can do to a protein undergoing unfolding? J. Biomol. Struct. Dyn. 2016, 34, 2155–2170. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Intrinsically disordered proteins in crowded milieu: When chaos prevails within the cellular gumbo. Cell. Mol. Life Sci. 2018, 75, 3907–3929. [Google Scholar] [CrossRef]
- Cino, E.A.; Karttunen, M.; Choy, W.Y. Effects of Molecular Crowding on the Dynamics of Intrinsically Disordered Proteins. PLoS ONE 2012, 7, e49876. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.; Qin, S.; Weiss, K.L.; Stanley, C.B.; Zhou, H.X. Intrinsically Disordered Protein Exhibits Both Compaction and Expansion under Macromolecular Crowding. Biophys. J. 2018, 114, 1067–1079. [Google Scholar] [CrossRef]
- Zegarra, F.C.; Homouz, D.; Gasic, A.G.; Babel, L.; Kovermann, M.; Wittung-Stafshede, P.; Cheung, M.S. Crowding-Induced Elongated Conformation of Urea-Unfolded Apoazurin: Investigating the Role of Crowder Shape in Silico. J. Phys. Chem. B 2019, 123, 3607–3617. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, I.; Vilaseca, E.; Madurga, S.; Garcés, J.L.; Cascante, M.; Mas, F. Effect of Crowding by Dextrans on the Hydrolysis of N-Succinyl-l-phenyl-Ala-p-nitroanilide Catalyzed by α-Chymotrypsin. J. Phys. Chem. B 2011, 115, 1115–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitulice, L.; Pastor, I.; Vilaseca i Font, E.; Madurga Díez, S.; Isvoran, A.; Cascante i Serratosa, M.; Mas i Pujadas, F. Influence of macromolecular crowding on the oxidation of ABTS by hydrogen peroxide catalyzed by HRP. J. Biocatal. Biotransformation 2013, 2, 1000107. [Google Scholar]
- Pastor, I.; Pitulice, L.; Balcells, C.; Vilaseca, E.; Madurga, S.; Isvoran, A.; Cascante, M.; Mas, F. Effect of crowding by Dextrans in enzymatic reactions. Biophys. Chem. 2014, 185, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Balcells, C.; Pastor, I.; Pitulice, L.; Via, M.; Madurga, S.; Vilaseca, E.; Isvoran, A.; Cascante, M.; Mas, F. Macromolecular crowding upon in-vivo-like enzyme-kinetics: Effect of enzyme-obstacle size ratio. New Front. Chem. 2015, 24, 3–16. [Google Scholar]
- Kuzmak, A.; Carmali, S.; von Lieres, E.; Russell, A.J.; Kondrat, S. Can enzyme proximity accelerate cascade reactions? Sci. Rep. 2019, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Laurent, T.C.; Ogston, A. The interaction between polysaccharides and other macromolecules. 4. The osmotic pressure of mixtures of serum albumin and hyaluronic acid. Biochem. J. 1963, 89, 249. [Google Scholar] [CrossRef] [Green Version]
- Laurent, T.C. Enzyme Reactions in Polymer Media. Eur. J. Biochem. 1971, 21, 498–506. [Google Scholar] [CrossRef]
- Laidler, K.J. Chemical Kinetics, 3rd ed.; Harper Collins: New York, NY, USA, 1987. [Google Scholar]
- Minton, A. Confinement as a determinant of macromolecular structure and reactivity. Biophys. J. 1992, 63, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Schnell, S.; Turner, T. Reaction kinetics in intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophys. Mol. Biol. 2004, 85, 235–260. [Google Scholar] [CrossRef]
- Balcells, C.; Pastor, I.; Vilaseca, E.; Madurga, S.; Cascante, M.; Mas, F. Macromolecular crowding effect upon in vitro enzyme kinetics: Mixed activation-diffusion control of the oxidation of NADH by pyruvate catalyzed by lactate dehydrogenase. J. Phys. Chem. B 2014, 118, 4062–4068. [Google Scholar] [CrossRef] [Green Version]
- Yekymov, E.; Attia, D.; Levi-Kalisman, Y.; Bitton, R.; Yerushalmi-Rozen, R. Effects of Non-Ionic Micelles on the Acid-Base Equilibria of a Weak Polyelectrolyte. Polymers 2022, 14, 1926. [Google Scholar] [CrossRef]
- Yekymov, E.; Attia, D.; Levi-Kalisman, Y.; Bitton, R.; Yerushalmi-Rozen, R. Charge Regulation of Poly (acrylic acid) in Solutions of Non-Charged Polymer and Colloids. Polymers 2023, 15, 1121. [Google Scholar] [CrossRef]
- Salehi, A.; Larson, R.G. A Molecular Thermodynamic Model of Complexation in Mixtures of Oppositely Charged Polyelectrolytes with Explicit Account of Charge Association/Dissociation. Macromolecules 2016, 49, 9706–9719. [Google Scholar] [CrossRef]
- Friedowitz, S.; Salehi, A.; Larson, R.G.; Qin, J. Role of electrostatic correlations in polyelectrolyte charge association. J. Chem. Phys. 2018, 149, 163335. [Google Scholar] [CrossRef]
- Rathee, V.S.; Sidky, H.; Sikora, B.J.; Whitmer, J.K. Role of Associative Charging in the Entropy–Energy Balance of Polyelectrolyte Complexes. J. Am. Chem. Soc. 2018, 140, 15319–15328. [Google Scholar] [CrossRef]
- Rathee, V.S.; Zervoudakis, A.J.; Sidky, H.; Sikora, B.J.; Whitmer, J.K. Weak polyelectrolyte complexation driven by associative charging. J. Chem. Phys. 2018, 148, 114901. [Google Scholar] [CrossRef] [PubMed]
- Knoerdel, A.R.; Blocher McTigue, W.C.; Sing, C.E. Transfer Matrix Model of pH Effects in Polymeric Complex Coacervation. J. Phys. Chem. B 2021, 125, 8965–8980. [Google Scholar] [CrossRef] [PubMed]
- Ďorďovič, V.; Vojtová, J.; Jana, S.; Uchman, M. Charge reversal and swelling in saccharide binding polyzwitterionic phenylboronic acid-modified poly (4-vinylpyridine) nanoparticles. Polym. Chem. 2019, 10, 5522–5533. [Google Scholar] [CrossRef]
- Marková, P.; Uchman, M. Synthesis and self-assembly of polyzwitterionic phenylboronic acid-containing double hydrophilic block copolymers. Eur. Polym. J. 2021, 151, 110439. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, P.M.; Narambuena, C.F.; Madurga, S.; Mas, F.; Garcés, J.L. Unusual Aspects of Charge Regulation in Flexible Weak Polyelectrolytes. Polymers 2023, 15, 2680. https://doi.org/10.3390/polym15122680
Blanco PM, Narambuena CF, Madurga S, Mas F, Garcés JL. Unusual Aspects of Charge Regulation in Flexible Weak Polyelectrolytes. Polymers. 2023; 15(12):2680. https://doi.org/10.3390/polym15122680
Chicago/Turabian StyleBlanco, Pablo M., Claudio F. Narambuena, Sergio Madurga, Francesc Mas, and Josep L. Garcés. 2023. "Unusual Aspects of Charge Regulation in Flexible Weak Polyelectrolytes" Polymers 15, no. 12: 2680. https://doi.org/10.3390/polym15122680