

Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins


Abstract
:1. Introduction
2. Main Elements of Virtual Free-Radical Polymerization
3. Digital Twins Concept, Samples, Virtual Device, and Intellectual Product of Virtual Free-Radical Polymerization of Vinyl Monomers
4. Initiation of Virtual Free-Radical Polymerization
4.1. Monomer Radical Generation
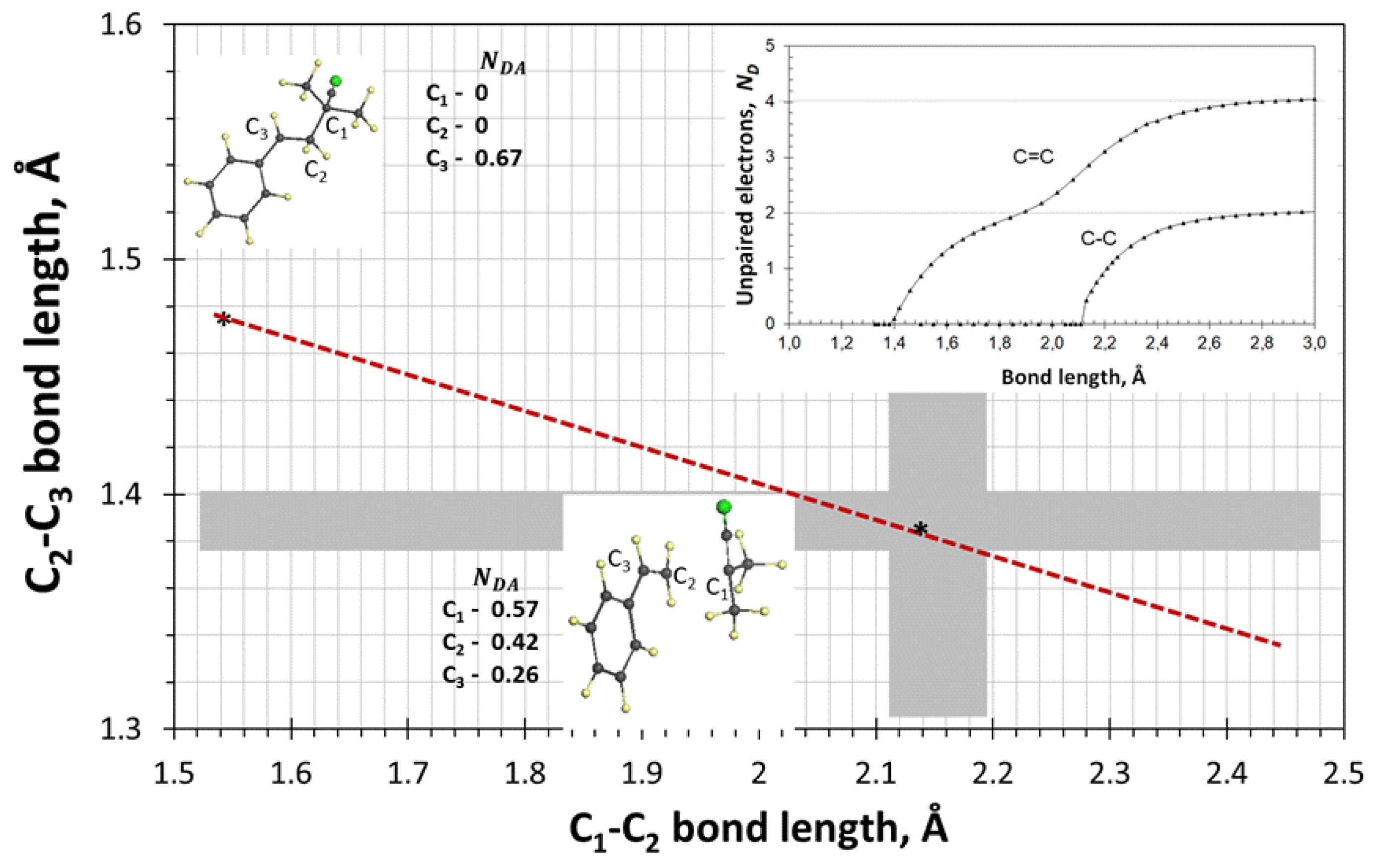
4.2. Transition State of Monomer Radicals
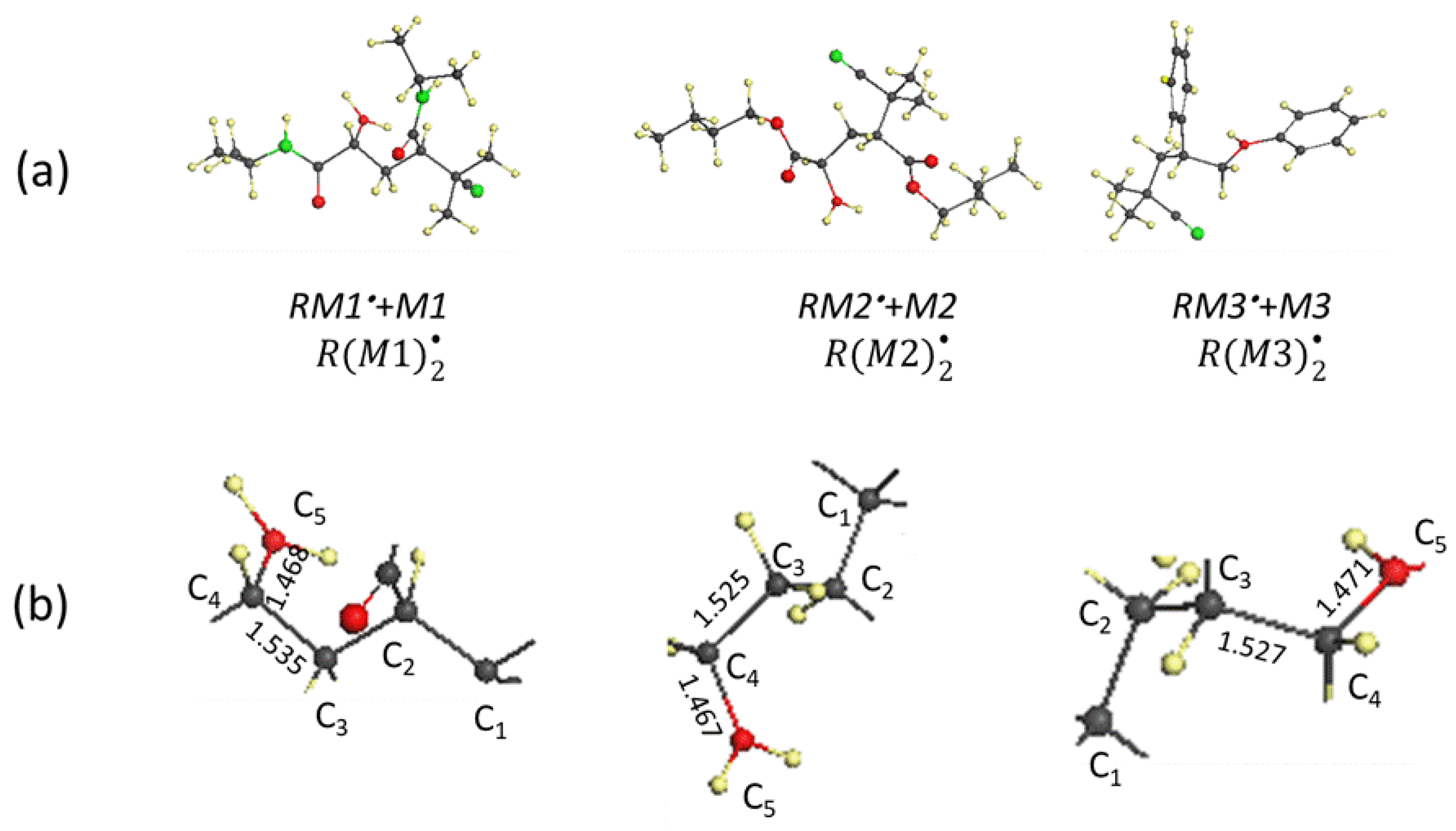
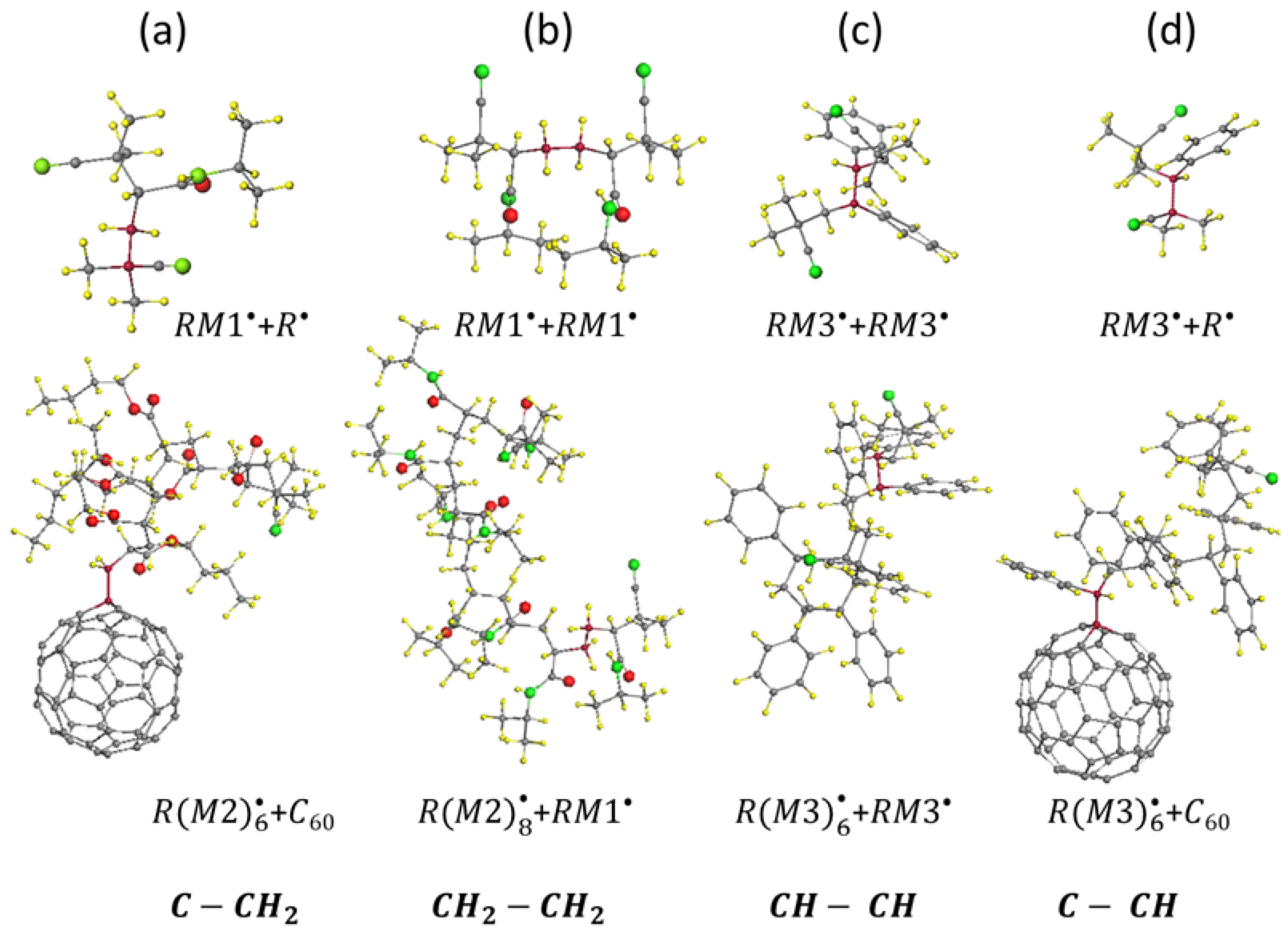
5. First-Step Propagation of Virtual Free-Radical Polymerization—The Generation of Dimer Radicals
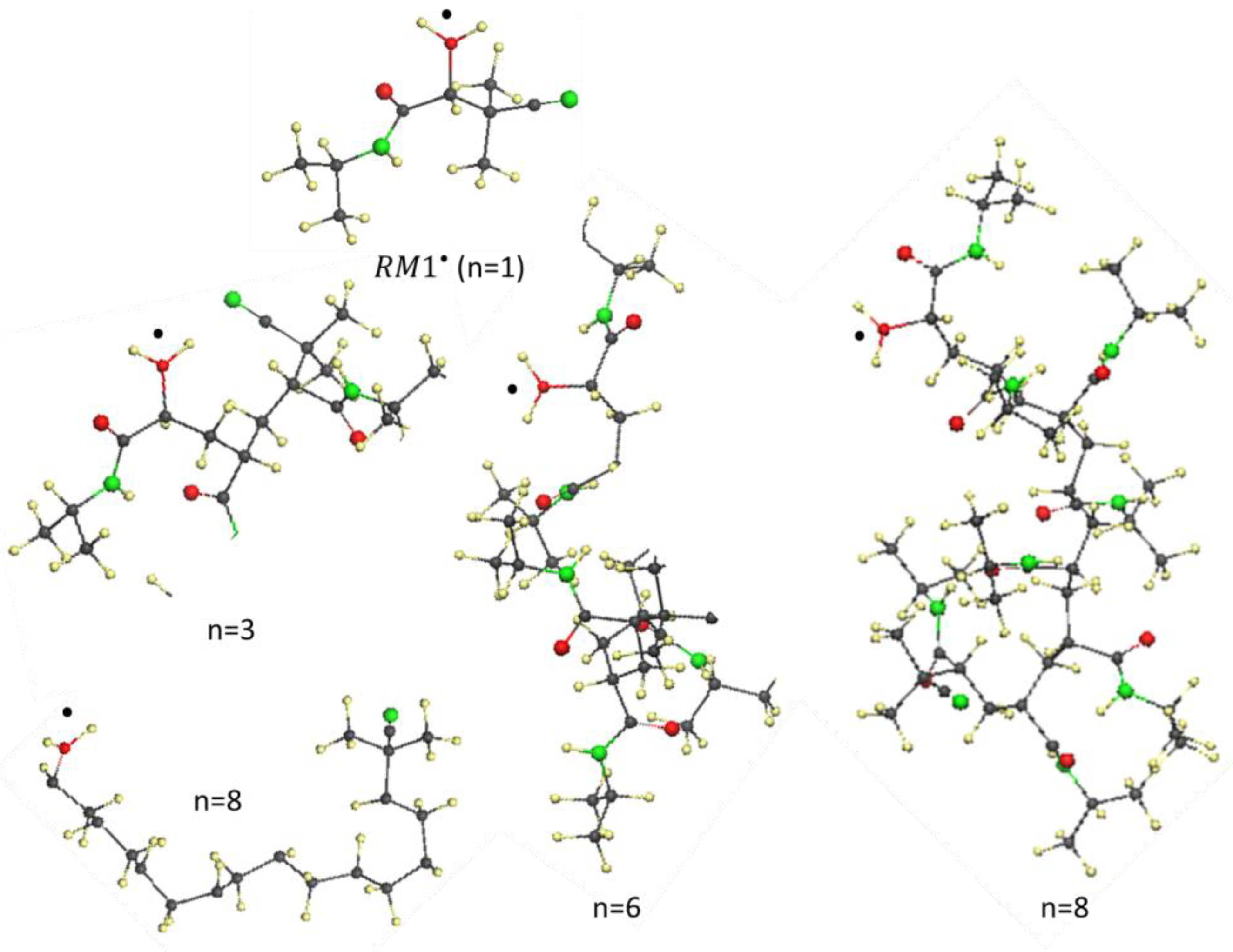
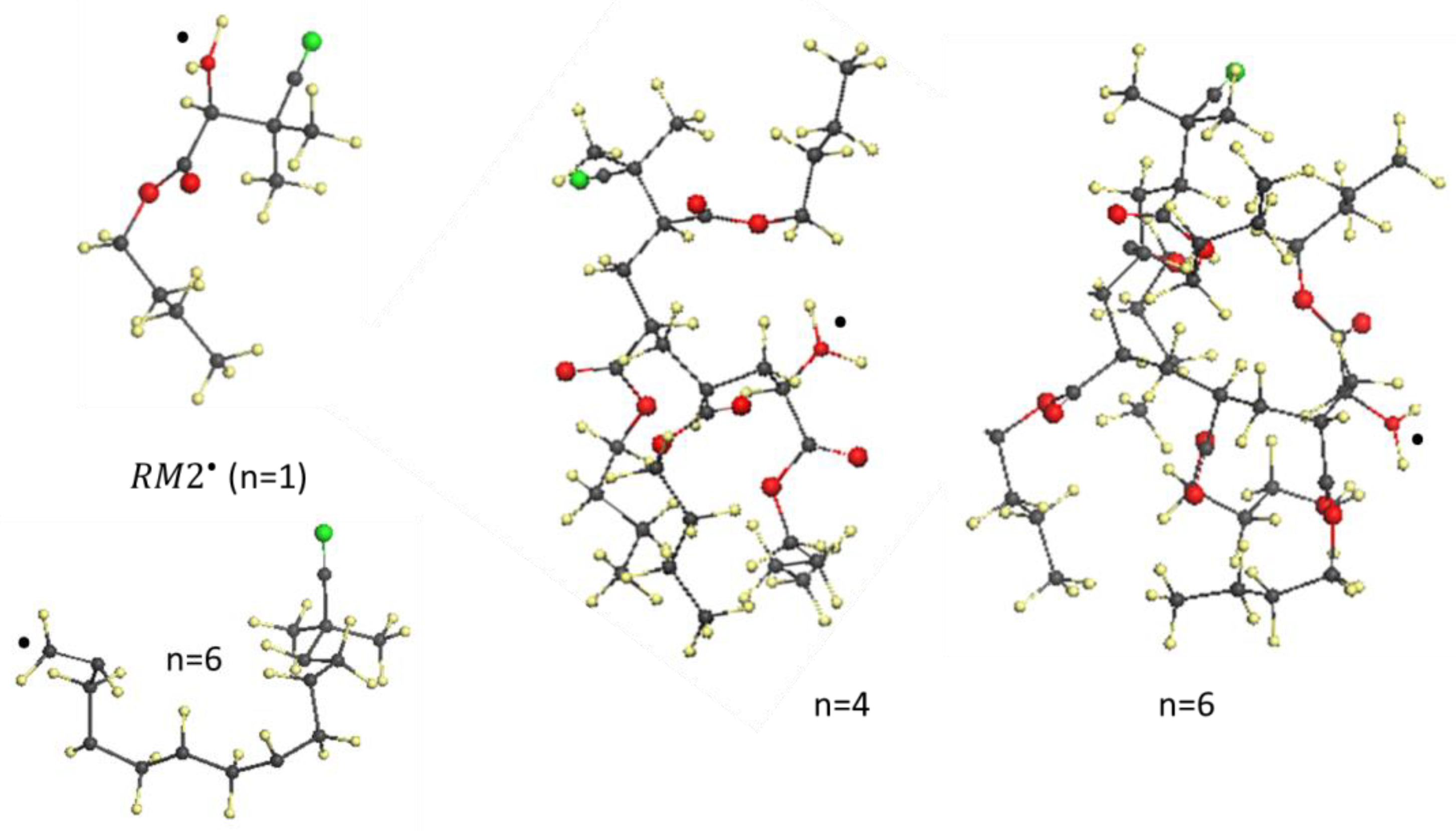
6. Vinyl Oligomer Radicals of Free-Radical Polymerization
7. Termination of the Polymer Chain Growth
8. Conclusions
- Oligomer radicals , starting with monomer one, constitute the successive links of the monomers;
- The first link RM• is produced in the stage of the polymerization initiation, while all the rest occur in due course during the gradual polymerization propagation;
- All the studied vinyl monomers form stable oligomer radicals characterized with a significant ;
- Spin-density and energy parameters can distinguish monomers by the number of the involved vinyl groups, thus dividing them into single-group and set-group monomers;
- The structural composition of all the -oligomer radicals is subordinated to the spin-density algorithm of formation and is configured around a basic ‘clothesline’ consisting of intermolecular junctions, − 1 of which are composed of one intermolecular and one intramolecular sp3C-C bond, accompanied by methine and methylene units, while the remaining currently working one involves an elongated intramolecular sp2C-C bond, thus providing a target for radical attack;
- A consequent sp2sp3 transformation of the covalent C-C bonds, accompanying the clothesline elongation, combined with the resistance to emerging steric hindrances, has resulted in a bizarre, non-straight line image of the clothesline.
- Characterizing the transition state by is the central point allowing the direct determination of ;
- was determined when constructing barrier profiles for two pairs of oligomer radicals involving RM1• and RM3• as well as and that represent elementary reactions of types (1) and (2), respectively;
- The obtained profiles clearly distinguish single-group and set-group monomers, thus pointing to a different kinetics of the latter;
- On the background of this difference, the transition state reveals a particular common feature related to the identical position of the profile maximum on the reaction coordinate, which is the intermolecular sp3C-C bond of the current junction, at 2.1 Å in all the cases;
- Simultaneously, with a critical behavior of this sp3 bond, the length of the junction intramolecular sp2C-C bond takes a value of 1.378 Å, which is that fixes the beginning of the radicalization of the sp2 bonds under elongation;
- Framing the energy parameter with two critical values, related to covalent sp3 and sp2 bonds, simultaneously indicates the spin-determining nature of the transition state of the vinyl oligomer radicals;
- The obtained data, determined as the difference between and , combined with the data of these oligomer radicals, allows the restoration of the EPS relations connecting the two sets of values for both pairs, attributing them to two different elementary reactions;
- The obtained EPS relation, concerning the elementary reaction of type (2), laid the foundations for obtaining values for all the rest of the oligomer radicals;
- The -based kinetic behavior of single-group and set-group oligomer radicals is different with respect to the initiation and propagation stages of FRP: the former is greatly favorable for the set-group ones, while the latter is slightly more favorable for the single-group ones.
- The discussed trends are related to the initiation and propagation stages of free-radical polymerization, while revealing similar trends for the termination of polymer chains is still waiting for a particular algorithm.
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ladik, J.; André, J.M.; Seel, M. (Eds.) Quantum Chemistry of Polymers—Solid State Aspects; NATO ASI Series; Springer: Dordrecht, The Netherlands, 1984; Volume 123. [Google Scholar]
- Ladik, J. Quantum Theory of Polymers as Solids; Plenum Press: New York, NY, USA, 1988. [Google Scholar]
- Tosy, C. (Ed.) Proceedings of the First Donegani Scientific Workshop on Strategies for Computer Chemistry: October 12–13, 1987; Kluwer Academic Publishing: New York, NY, USA; Boston, MA, USA; Dordrecht, The Netherlands; London, UK; Moscow, Russia, 1989. [Google Scholar]
- Andre, J.-M.; Delhalle, J.; Bredas, J.-L. (Eds.) Quantum Chemistry Aided Design of Organic Polymers: An Introduction to the Quantum Chemistry of Polymers and Its Applications; World Sci. Publ.: Singapore, 1991. [Google Scholar]
- Bicerano, J. (Ed.) Computational Modeling of Polymers; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Fischer, H.; Radom, L. Factors controlling the addition of carbon-centered radicals to alkenes. An experimental and theoretical perspective. Angew. Chem. Int. Ed. 2001, 40, 1340–1371. [Google Scholar] [CrossRef]
- Heuts, J.P.A. Theory of radical reactions. In Handbook of Radical Polymerization; Matyjaszewski, K., Davis, T.P., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2002; pp. 1–76. [Google Scholar]
- Dykstra, C.; Frenking, G.; Kim, K.; Scuseria, G. (Eds.) Theory and Applications of Computational Chemistry: The First Forty Years; Elsevier B.V.: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Andre, J.-M. Computational quantum chemistry on polymer chains: Aspects of the last half century. In Theory and Applications of Computational Chemistry: The First Forty Years; Dykstra, C., Frenking, G., Kim, K., Scuseria, G., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2005; pp. 1011–1045. [Google Scholar]
- Coote, M.L. Quantum-chemical modeling of free-radical polymerization. Macromol. Theory Simul. 2009, 18, 388–400. [Google Scholar] [CrossRef]
- Gigmes, D.; Bertin, D.; Lefay, C.; Guillaneuf, Y. Kinetic modeling of nitroxide-mediated polymerization: Conditions for living and controlled polymerization. Macromol. Theory Simul. 2009, 18, 402–419. [Google Scholar] [CrossRef]
- Deglmann, P.; Mueller, I.; Becker, F.; Schaefer, A.; Hungenberg, K.-D.; Weiß, H. Prediction of propagation rate coefficients in free radical solution polymerization based on accurate quantum chemical methods: Vinylic and related monomers, including acrylates and acrylic acid. Macromol. React. Eng. 2009, 3, 496–515. [Google Scholar] [CrossRef]
- Dossi, M.; Storti, G.; Moscatelli, D. Initiation kinetics in free-radical polymerization: Prediction of thermodynamic and kinetic parameters based on ab initio calculations. Macromol. Theory Simul. 2010, 19, 170–178. [Google Scholar] [CrossRef]
- Mavroudakis, E.; Cuccato, D.; Moscatelli, D. On the use of quantum chemistry for the determination of propagation, copolymerization, and secondary reaction kinetics in free radical polymerization. Polymers 2015, 7, 1789–1819. [Google Scholar] [CrossRef] [Green Version]
- Ruipérez, F. Application of quantum chemical methods in polymer chemistry. Int. Rev. Phys. Chem. 2019, 38, 343–403. [Google Scholar] [CrossRef]
- Hayashi, Y.; Kawauchi, S. Development of a quantum chemical descriptor expressing aromatic/quinoidal character for designing narrow-bandgap π-conjugated polymers. Polym. Chem. 2019, 10, 5584–5593. [Google Scholar] [CrossRef] [Green Version]
- Gay, J. Molecular Dynamics Studies of Polymer Systems; Department of Chemical Engineering at Worcester Polytechnic Institute: Worcester, MA, USA, 2012. [Google Scholar]
- Gartne, T.E.; Jayaraman, A. Modeling and simulations of polymers: A roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, S.; Lischewski, A.; Orlov, Y.; Deglmann, P.; Daiss, A.; Schreiner, E.; Vale, H. Toward a digital polymer reaction engineering. Adv. Chem. Eng. 2020, 56, 187–230. [Google Scholar]
- Micheletti, C.; Hauke, P.; Faccioli, P. Polymer physics by quantum computing. Phys. Rev. Lett. 2021, 127, 080501. [Google Scholar] [CrossRef]
- Sheka, E.F. Digital Twins in the graphene technology. arXiv 2022, arXiv:2208.14926. [Google Scholar]
- Sheka, E.F. Digital Twins solve the mystery of Raman spectra of parental and reduced graphene oxides. Nanomaterials 2022, 12, 4209. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. (Eds.) Handbook of Radical Polymerization; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Mishra, M.; Yagci, Y. (Eds.) Handbook of Vinyl Polymers 2: Radical Polymerization, Process, and Technology, New Ed.; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Li, W.; Jiang, W. Effect of Solvent molecular size on the self-assembly of amphiphilic diblock copolymer in selective solvent. Macromol. Theory Simul. 2009, 18, 434–440. [Google Scholar] [CrossRef]
- Bagdasar’yan, K.S. Teoriya Radikal’noi Polimerizatsii (Free_Radical Polymerization Theory); Nauka: Moskwa, Rosja, 1966. (In Russian) [Google Scholar]
- Starkweather, H.W.; Taylor, G.B. The kinetics of the polymerization of vinyl acetate. J. Am. Chem. Soc. 1930, 52, 4708–4714. [Google Scholar] [CrossRef]
- Semenov, N.N. Tsepnyie Reakcii (Chain Reactions); Goschimizdat: Moskva, Rosja, 1934. (In Russian) [Google Scholar]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. Npj Comput. Mater. 2016, 2, 15011. [Google Scholar] [CrossRef] [Green Version]
- Vashisth, A.; Ashraf, C.; Bakis, C.E.; van Duin, A.C.T. Effect of chemical structure on thermo-mechanical properties of epoxy polymers: Comparison of accelerated ReaxFF simulations and experiments. Polymer 2018, 158, 354–363. [Google Scholar] [CrossRef]
- Denisov, E.T.; Sarkisov, O.M.; Likhtenshtein, G.I. Chemical Kinetics: Fundamentals and Recent Developments; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Sheka, E.F.; Shaymardanova, L.K. C60-based composites in view of topochemical reactions. J. Mater. Chem. 2011, 21, 17128. [Google Scholar] [CrossRef]
- Zayets, V.A. CLUSTER-Z1: Quantum-Chemical Software for Calculations in the s,p-Basis; Institute of Surface Chemistry, National Academy of Sciences of Ukraine: Kyiv, Ukraine, 1990. (In Russian) [Google Scholar]
- Berzigiyarov, P.K.; Zayets, V.A.; Ginzburg, I.Y.; Razumov, V.F.; Sheka, E.F. NANOPACK: Parallel codes for semiempirical quantum chemical calculations of large systems in the sp- and spd-basis. Int. J. Quantum Chem. 2002, 88, 449–462. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healey, E.F.; Stewart, J.J.P. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Thiel, W. Semiempirical quantum chemical methods in computational chemistry. In Theory and Applications of Computational Chemistry: The First Forty Years; Dykstra, C., Frenking, G., Kim, K., Scuseria, G., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2005; pp. 559–580. [Google Scholar]
- Sheka, E.F. Fullerenes: Nanochemistry, Nanomagnetism, Nanomedicine, Nanophotonics; CRC Press, Taylor & Francis Group: Boka Raton, FL, USA, 2011. [Google Scholar]
- Sheka, E.F. Spin Chemical Physics of Graphene; Pan Stanford: Singapore, 2018. [Google Scholar]
- Sheka, E.F. Virtual vibrational spectrometry of stable radicals—Necklaced graphene molecules. Nanomaterials 2022, 12, 597. [Google Scholar] [CrossRef]
- Zipse, H. Radical stability—A theoretical perspective. Top Curr. Chem. 2006, 263, 163–189. [Google Scholar] [CrossRef]
- Krylov, A.I. The quantum chemistry of open-shell species. In Reviews in Computational Chemistry, 1st ed.; Parrill, A.L., Lipkowitz, K.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; Volume 30, pp. 151–224. [Google Scholar]
- Huang, Y.; Egap, E. Open-shell organic semiconductors: An emerging class of materials with novel properties. Polym. J. 2018, 50, 603–614. [Google Scholar] [CrossRef]
- Chen, Z.X.; Li, Y.; Huang, F. Persistent and stable organic radicals: Design, synthesis, and applications. Chem 2021, 7, 288–332. [Google Scholar] [CrossRef]
- Kaplan, I.G. Symmetry properties of the electron density and following from it limits on the KS-DFT applications. Mol. Phys. 2018, 116, 658–665. [Google Scholar] [CrossRef]
- Sheka, E.F.; Popova, N.A.; Popova, V.A. Physics and chemistry of graphene. Emergentness, magnetism, mechanophysics and mechanochemistry. Phys. Usp. 2018, 61, 645–691. [Google Scholar] [CrossRef]
- Sheka, E.F. Chemical susceptibility of fullerenes in view of Hartree–Fock approach. Int. J. Quant. Chem. 2007, 107, 2803–2816. [Google Scholar] [CrossRef]
- Sheka, E.F. Stretching and breaking of chemical bonds, correlation of electrons, and radical properties of covalent species. Adv. Quant. Chem. 2015, 70, 111–161. [Google Scholar]
- Barner-Kowollik, C.; Gunzler, F.; Junkers, T. Pushing the Limit: Pulsed laser polymerization of n-butyl acrylate at 500 Hz. Macromolecules 2008, 41, 8971–8973. [Google Scholar] [CrossRef]
- Gilbert, R.G. Critically-evaluated propagation rate coefficients in free radical polymerizations i. Styrene and methyl methacrylate. Pure Appl. Chem. 1996, 68, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- Dossi, M.; Storti, G.; Moscatelli, D. Quantum chemistry: A powerful tool in polymer reaction engineering. Macromol. Symp. 2011, 302, 16–25. [Google Scholar] [CrossRef]
- Wang, C.; Guo, Z.-X.; Fu, S.; Wu, W.; Zhu, D. Polymers containing fullerene or carbon nanotube structures. Prog. Polym. Sci. 2004, 29, 1079–1141. [Google Scholar] [CrossRef]
- Hirsch, A.; Brettreich, M. Fullerenes Chemistry and Reactions; Wiley-VCH: London, UK, 2004. [Google Scholar]
- Troshin, P.A.; Troshina, O.A.; Lyubovskaya, R.N.; Razumov, V.F. Functional Derivatives of Fullerenes. Synthesis and Applications to Organic Electronics and Biomedicine; Ivanovo State University: Ivanovo, Russian, 2009; 310p. (In Russian) [Google Scholar]
- Hirsh, A. The Chemistry of Fullerenes; Thieme: Stuttgart, Germany, 1994. [Google Scholar]
- Ford, W.T.; Nishioka, T.; McCleskey, S.C.; Mourey, T.H.; Kahol, P. Structure and radical mechanism of formation of copolymers of C60 with styrene and with methyl methacrylate. Macromolecules 2000, 33, 2413–2423. [Google Scholar] [CrossRef]
- Vinogradova, L.V. Star-shaped polymers with the fullerene C60 branching center. Russ. Chem. Bull. 2012, 61, 907–922. [Google Scholar] [CrossRef]
- Atovmyan, E.G.; Grishchuk, A.A.; Estrina, G.A.; Estrin, Y.I. Formation of star-like water-soluble polymeric structures in the process of radical polymerization of N-isopropylacrylamide in the presence of C60. Russ. Chem. Bull. Int. Ed. 2016, 65, 2082–2088. [Google Scholar] [CrossRef]
- Atovmyan, E.G. On the relationship between the fullerene reactivity and degree of substitution. Russ. Chem. Bull. Int. Ed. 2017, 66, 567–570. [Google Scholar] [CrossRef]
- Pross, A. Theoretical and Physical Principles of Organic Reactivity; Wiley: New York, NY, USA, 1995. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | , e | , e (1) | , Å (2) | (3) | |
|---|---|---|---|---|---|
| ( | 1.072 | 0.807 | - | 30.73 | - |
| (NIPA) | 0 | 0 | 1.342 | −31.85 | 67.52 |
| (n BA) | 0 | 0 | 1.342 | −89.14 | 61.56 |
| (St) | 0.747 | 0.107 | 1.344 | 38.14 | 24.31 |
| Radical | (1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1.024 | 0.963 | −1.12 | −6.79 | −5.67 | 24.76 | 19.09 | 10.0 [13] | ||
| 1.024 | 0.963 | −58.41 | −64.36 | −5.85 | 18.96 (3) (2) | ||||
| 1.925 | 0.67 | 68.87 | 46.36 | −22.51 | 31.01 | 8.5 | 8.9 [13] | ||
| 1.025 | 0.96 | −38.64 | −62.9 | −24.26 | 32.65 | 8.39 | 4.37 [48] | 6.26 [48] | |
| 1.024 | 0.965 | −153.5 | −176.34 | −22.84 | 9.07 (4) (2) | ||||
| 1.928 | 0.668 | 84.5 | 67.89 | −16.61 | 28.67 | 12.06 | 7.79 [49] | 8.32 [50] 11.30 [13] |
| Radical | , e | , e | , kcal/mol | , Å (1) | , Å (2) |
|---|---|---|---|---|---|
| ; ; | - | - | |||
| 1.424 | 0.54; 0.55; 0.19 | 17.97 | 2.069 | 1.383 | |
| 2.369 | 0.57; 0.42; 0.26 | 77.37 | 2.129 | 1.387 | |
| ; ; | - | - | |||
| 1.412 | 0.67; 0.49; 0.19 | −30.25 | 2.123 | 1.373 | |
| 3.157 | 0.54; 0.41; 0.27 | 96.56 | 2.107 | 1.388 |
| Radical | , e | , e | |||
|---|---|---|---|---|---|
| 1.024 | 0.964 | −118.26 | −23.51 | 7.74 | |
| 1.024 | 0.964 | −174.16 | −24.05 | 8.49 | |
| 1.024 | 0.96 | −233.93 | −27.92 | 6.3 | |
| 1.024 | 0.961 | −289.62 | −23.84 | 8.59 | |
| 1.024 | 0.965 | −346.97 | −25.5 | 7.79 | |
| 1.024 | 0.965 | −401.97 | −23.15 | 8.92 | |
| 1.024 | 0.962 | −289.71 | −24.23 | 8.4 | |
| 1.024 | 0.963 | −402.76 | −23.91 | 8.55 | |
| 1.024 | 0.961 | −514.52 | −22.62 | 9.17 | |
| 1.024 | 0.964 | −622.18 | −18.52 | 11.14 | |
| 1.963 | 0.668 | 86.11 | −19.92 | 10.47 | |
| 2.146 | 0.67 | 105.35 | −18.9 | 10.96 | |
| 2.16 | 0.669 | 124.26 | −19.23 | 10.8 | |
| 2.376 | 0.667 | 155.02 | −7.38 | 16.49 | |
| 3.073 | 0.667 | 164.16 | −29 | 6.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheka, E.F. Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins. Polymers 2023, 15, 2999. https://doi.org/10.3390/polym15142999
Sheka EF. Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins. Polymers. 2023; 15(14):2999. https://doi.org/10.3390/polym15142999
Chicago/Turabian StyleSheka, Elena F. 2023. "Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins" Polymers 15, no. 14: 2999. https://doi.org/10.3390/polym15142999
APA StyleSheka, E. F. (2023). Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins. Polymers, 15(14), 2999. https://doi.org/10.3390/polym15142999