
Modeling Coil–Globule–Helix Transition in Polymers by Self-Interacting Random Walks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Method
2.1. Self-Interacting Walks
2.2. Numerical Simulations
2.3. Evaluation Metrics
3. Results and Discussion
3.1. Structural Properties at Low Temperature
3.2. Implications in Protein Folding
3.3. Comparison with Other Models
3.4. Impact of the Temperature T
3.4.1. Globule–Coil Transition
3.4.2. Helix–Globule–Coil Transition
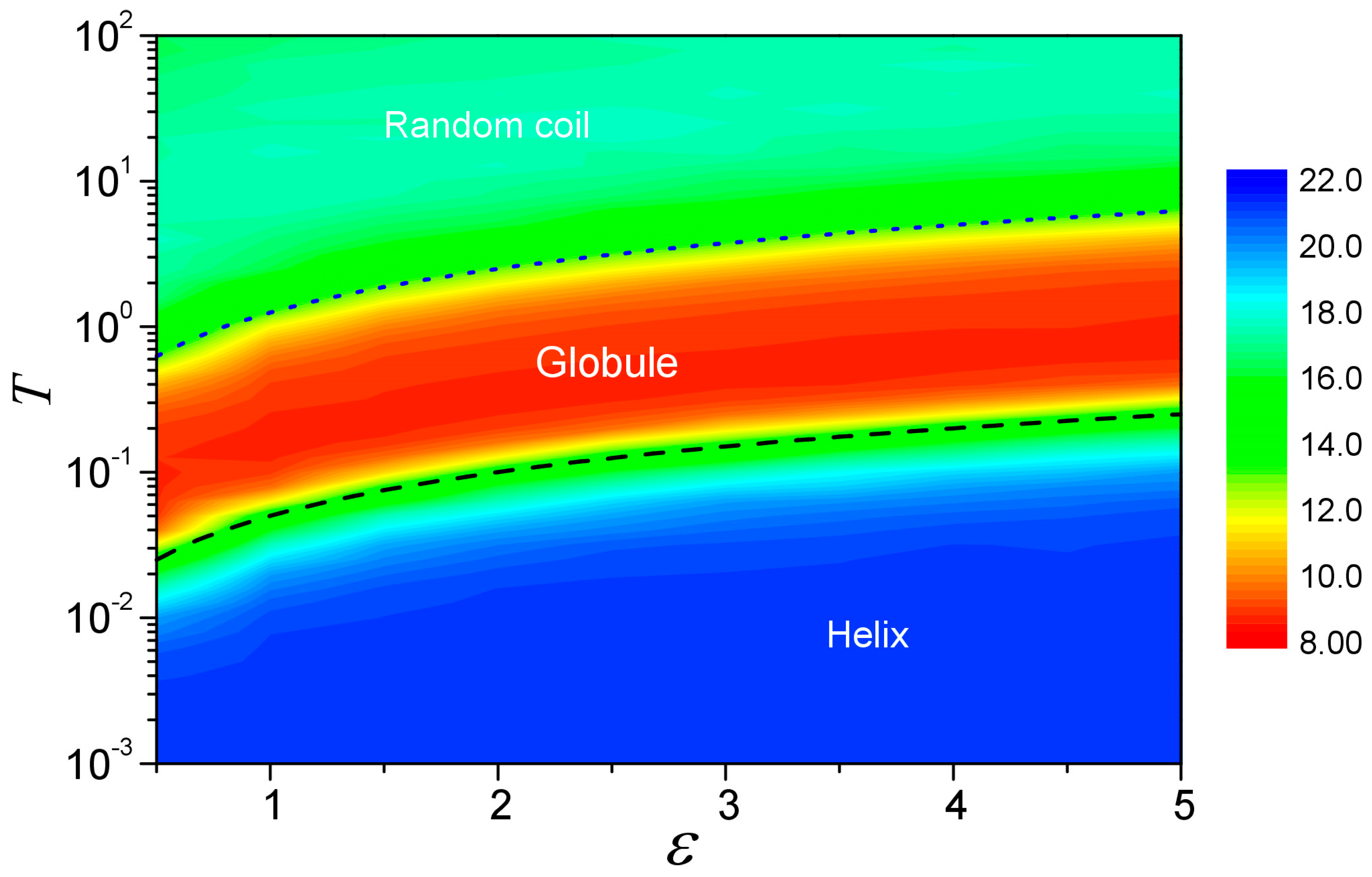
3.5. Diagram of Phase Transition
3.6. Structure Transitions at Other Equilibrium Distances
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Weiss, G.H.; Weiss, G.H. Aspects and Applications of the Random Walk; Elsevier Science & Technology: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Bressloff, P.C.; Newby, J.M. Stochastic models of intracellular transport. Rev. Mod. Phys. 2013, 85, 135. [Google Scholar] [CrossRef]
- Łopuszański, K.; Miękisz, J. Random walks with asymmetric time delays. Phys. Rev. E 2022, 105, 064131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, W.; Zhang, X.; Long, G.; Fan, J.; Chen, H.; Deng, L. A novel fractal solution for permeability and Kozeny-Carman constant of fibrous porous media made up of solid particles and porous fibers. Powder Technol. 2019, 349, 92–98. [Google Scholar] [CrossRef]
- Xiao, B.; Zhu, H.; Chen, F.; Long, G.; Li, Y. A fractal analytical model for Kozeny-Carman constant and permeability of roughened porous media composed of particles and converging-diverging capillaries. Powder Technol. 2023, 420, 118256. [Google Scholar] [CrossRef]
- Herranz, M.; Benito, J.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Polymorph Stability and Free Energy of Crystallization of Freely-Jointed Polymers of Hard Spheres. Polymers 2023, 15, 1335. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Hsieh, Y.H.; Chen, C.N. Exact Enumeration Approach to Estimate the Theta Temperature of Interacting Self-Avoiding Walks on the Simple Cubic Lattice. Polymers 2022, 14, 4536. [Google Scholar] [CrossRef] [PubMed]
- Régnier, L.; Bénichou, O.; Krapivsky, P.L. Range-Controlled Random Walks. Phys. Rev. Lett. 2023, 130, 227101. [Google Scholar] [CrossRef]
- Ivanova, A.S.; Polotsky, A.A. Random copolymer adsorption onto a periodic heterogeneous surface: A partially directed walk model. Phys. Rev. E 2022, 106, 034501. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X.-W.; Jin, Z.-Z. Directed random walks in continuous space. Phys. Rev. E 2002, 65, 052105. [Google Scholar] [CrossRef]
- Madras, N.; Slade, G. The Self-Avoiding Walk; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Ordemann, A.; Berkolaiko, G.; Havlin, S.; Bunde, A. Swelling-collapse transition of self-attracting walks. Phys. Rev. E 2000, 61, R1005. [Google Scholar] [CrossRef]
- Tan, Z.J.; Zou, X.W.; Zhang, W.; Huang, S.Y.; Jin, Z.Z. Self-Attracting Walk on Non-Uniform Substrates. Mod. Phys. Lett. B 2002, 16, 449–457. [Google Scholar] [CrossRef]
- De Gennes, P.-G.; Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Ordemann, A.; Tomer, E.; Berkolaiko, G.; Havlin, S.; Bunde, A. Structural properties of self-attracting walks. Phys. Rev. E 2001, 64, 046117. [Google Scholar] [CrossRef]
- Tan, Z.J.; Zou, X.W.; Huang, S.Y.; Zhang, W.; Jin, Z.Z. Random walk with memory enhancement and decay. Phys. Rev. E 2002, 65, 041101. [Google Scholar] [CrossRef] [PubMed]
- Perra, N.; Baronchelli, A.; Mocanu, D.; Gonçalves, B.; Pastor-Satorras, R.; Vespignani, A. Random walks and search in time-varying networks. Phys. Rev. Lett. 2012, 109, 238701. [Google Scholar] [CrossRef] [PubMed]
- Haran, G. How, when and why proteins collapse: The relation to folding. Curr. Opin. Struct. Biol. 2012, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, S.; Lou, E.; Tan, Y.L.; Tan, Z.J. RNA 3D Structure Prediction: Progress and Perspective. Molecules 2023, 28, 5532. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, Y.L.; Yu, S.; Shi, Y.Z.; Tan, Z.J. Predicting 3D structures and stabilities for complex RNA pseudoknots in ion solutions. Biophys. J. 2023, 122, 1503–1516. [Google Scholar] [CrossRef]
- Tian, F.J.; Zhang, C.; Zhou, E.; Dong, H.L.; Tan, Z.J.; Zhang, X.H.; Dai, L. Universality in RNA and DNA deformations induced by salt, temperature change, stretching force, and protein binding. Proc. Natl. Acad. Sci. USA 2023, 120, e2218425120. [Google Scholar] [CrossRef]
- Tan, Y.L.; Wang, X.; Yu, S.; Zhang, B.; Tan, Z.J. cgRNASP: Coarse-grained statistical potentials with residue separation for RNA structure evaluation. NAR Genom. Bioinform. 2023, 5, lqad016. [Google Scholar] [CrossRef]
- Tan, Y.L.; Wang, X.; Shi, Y.Z.; Zhang, W.; Tan, Z.J. rsRNASP: A residue-separation-based statistical potential for RNA 3D structure evaluation. Biophys. J. 2022, 121, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.J.; Chen, S.J. RNA helix stability in mixed Na+/Mg2+ solution. Biophys. J. 2007, 92, 3615–3632. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.J.; Chen, S.J. Nucleic acid helix stability: Effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006, 90, 1175–1190. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X.W.; Zhang, W.B.; Jin, Z.Z. Random walks on a (2 + 1)-dimensional deformable medium. Phys. Rev. Lett. 2002, 88, 056102. [Google Scholar] [CrossRef] [PubMed]
- Lennard-Jones, J.E. Cohesion. Proc. Phys. Soc. (1926–1948) 1931, 43, 461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.; Thornton, J. Helix geometry in proteins. J. Mol. Biol. 1988, 201, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Varshney, V.; Carri, G.A. Coupling between Helix-Coil and Coil-Globule Transitions in Helical Polymers. Phys. Rev. Lett. 2005, 95, 168304. [Google Scholar] [CrossRef]
- Yang, Z.; Deng, Z.; Zhang, L. Coil-helix-globule transition for self-attractive semiflexible ring chains. Polymer 2017, 110, 105–113. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, E.; Tan, Z.-J. Modeling Coil–Globule–Helix Transition in Polymers by Self-Interacting Random Walks. Polymers 2023, 15, 3688. https://doi.org/10.3390/polym15183688
Huang E, Tan Z-J. Modeling Coil–Globule–Helix Transition in Polymers by Self-Interacting Random Walks. Polymers. 2023; 15(18):3688. https://doi.org/10.3390/polym15183688
Chicago/Turabian StyleHuang, Eddie, and Zhi-Jie Tan. 2023. "Modeling Coil–Globule–Helix Transition in Polymers by Self-Interacting Random Walks" Polymers 15, no. 18: 3688. https://doi.org/10.3390/polym15183688