
Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental and Computational Methods to Investigate the Mechanisms
3. Examples of Mechanistic Studies of Palladium and Nickel-Catalyzed Ethylene Polymerizations
3.1. Brookhart-Type Catalysts
3.2. Grubbs-Type Catalysts
3.3. Drent Type Catalysts
3.4. Other Type of Catalysts
4. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Annual Production of Plastics Worldwide from 1950 to 2021. Available online: https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/ (accessed on 11 October 2023).
- Dong, J.Y.; Hu, Y. Design and synthesis of structurally well-defined functional polyolefins via transition metal-mediated olefin polymerization chemistry. Coord. Chem. Rev. 2006, 250, 47–65. [Google Scholar] [CrossRef]
- Rünzi, T.; Mecking, S. Saturated Polar-Substituted Polyethylene Elastomers from Insertion Polymerization. Adv. Funct. Mater. 2014, 24, 387–395. [Google Scholar] [CrossRef]
- Dai, S.; Chen, C. Palladium-Catalyzed Direct Synthesis of Various Branched, Carboxylic Acid-Functionalized Polyolefins: Characterization, Derivatization, and Properties. Macromolecules 2018, 51, 6818–6824. [Google Scholar] [CrossRef]
- Na, Y.; Dai, S.; Chen, C. Direct Synthesis of Polar-Functionalized Linear Low-Density Polyethylene (LLDPE) and Low-Density Polyethylene (LDPE). Macromolecules 2018, 51, 4040–4048. [Google Scholar] [CrossRef]
- Sui, X.; Hong, C.; Pang, W.; Chen, C. Unsymmetrical a-diimine palladium catalysts and their properties in olefin (co)polymerization. Mater. Chem. Front. 2017, 1, 967–972. [Google Scholar] [CrossRef]
- Boaen, N.K.; Hillmyer, M.A. Post-polymerization functionalization of polyolefins. Chem. Soc. Rev. 2005, 34, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.E.; Wagener, K.B.; Baugh, L.S.; Rucker, S.P.; Schulz, D.N.; Varma-Nair, M.; Berluche, E. Linear Copolymers of Ethylene and Polar Vinyl Monomers via Olefin Metathesis-Hydrogenation: Synthesis, Characterization, and Comparison to Branched Analogues. Macromolecules 2007, 40, 2643–2656. [Google Scholar] [CrossRef]
- Yang, H.; Islam, M.; Budde, C.; Rowan, S.J. Ring-Opening Metathesis Polymerization as a Route to Controlled Copolymers of Ethylene and Polar Monomers: Synthesis of Ethylene–Vinyl Chloride-Like Copolymers. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 2107–2116. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Laredo, W.R.; Grubbs, R.H. Ring-Opening Metathesis Polymerization of Functionalized Cyclooctenes by a Ruthenium-Based Metathesis Catalyst? Macromolecules 1995, 28, 6311–6316. [Google Scholar] [CrossRef]
- Mutlu, H.; de Espinosa, L.M.; Meier, M.R. Acyclic diene metathesis: A versatile tool for the construction of defined polymer architectures. Chem. Soc. Rev. 2011, 40, 1404–1445. [Google Scholar] [CrossRef] [PubMed]
- Boz, E.; Nemeth, A.J.; Ghiviriga, I.; Jeon, K.; Alamo, R.G.; Wagener, K.B. Precision Ethylene/Vinyl Chloride Polymers via Condensation Polymerization. Macromolecules 2007, 40, 6545–6551. [Google Scholar] [CrossRef]
- Boz, E.; Nemeth, A.J.; Alamo, R.G.; Wagener, K.B. Precision Ethylene/Vinyl Bromide Polymers. Adv. Synth. Catal. 2007, 349, 137–141. [Google Scholar] [CrossRef]
- Boz, E.; Wagener, K.B.; Ghosal, A.; Fu, R.; Alamo, R.G. Synthesis and Crystallization of Precision ADMET Polyolefins Containing Halogens. Macromolecules 2006, 39, 4437–4447. [Google Scholar] [CrossRef]
- Nakamura, A.; Ito, S.; Nozaki, K. Coordination-Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Y.; Marks, T.J. Early Transition Metal Catalysis for Olefin-Polar Monomer Copolymerization. Angew. Chem. Int. Ed. 2020, 59, 14726–14735. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and α-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Younkin, T.R.; Connor, E.F.; Henderson, J.I.; Friedrich, S.K.; Grubbs, R.H.; Bansleben, D.A. Neutral, Single-Component Nickel (II) Polyolefin Catalysts that Tolerate Heteroatoms. Science 2000, 297, 460–462. [Google Scholar] [CrossRef]
- Drent, E.; van Dijk, R.; van Ginkel, R.; van Oort, B.; Pugh, R.I. Palladium catalysed copolymerisation of ethene with alkylacrylates: Polar comonomer built into the linear polymer chain. Chem. Commun. 2002, 7, 744–745. [Google Scholar] [CrossRef]
- Wucher, P.; Goldbach, V.; Mecking, S. Electronic Influences in Phosphinesulfonato Palladium(II) Polymerization Catalysts. Organometallics 2013, 32, 4516–4522. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C. Rational Design of High-Performance Phosphine Sulfonate Nickel Catalysts for Ethylene Polymerization and Copolymerization with Polar Monomers. ACS Catal. 2017, 7, 1308–1312. [Google Scholar] [CrossRef]
- Carrow, B.P.; Nozaki, K. Synthesis of Functional Polyolefins Using Cationic Bisphosphine Monoxide-Palladium Complexes. J. Am. Chem. Soc. 2012, 134, 8802–8805. [Google Scholar] [CrossRef]
- Mitsushige, Y.; Yasuda, H.; Carrow, B.P.; Ito, S.; Kobayashi, M.; Tayano, T.; Watanabe, Y.; Okuno, Y.; Hayashi, S.; Kuroda, J.; et al. Methylene-Bridged Bisphosphine Monoxide Ligands for Palladium-Catalyzed Copolymerization of Ethylene and Polar Monomers. ACS Macro Lett. 2018, 7, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.S.; Sato, N.; Tanna, A.; Oishi, Y.; Konishi, Y.; Shimizu, F. Nickel-Catalyzed Copolymerization of Ethylene and Alkyl Acrylates. J. Am. Chem. Soc. 2017, 139, 3611–3614. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, C. A Versatile Ligand Platform for Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Angew. Chem. Int. Ed. 2018, 57, 3094–3098. [Google Scholar] [CrossRef] [PubMed]
- Michalak, A.; Ziegler, T. DFT Studies on the Copolymerization of R-Olefins with Polar Monomers: Ethylene-Methyl Acrylate Copolymerization Catalyzed by a Pd-Based Diimine Catalyst. J. Am. Chem. Soc. 2001, 123, 12266–12278. [Google Scholar] [CrossRef] [PubMed]
- Michalak, A.; Ziegler, T. DFT Studies on the Copolymerization of α-Olefins with Polar Monomers: Comonomer Binding by Nickel- and Palladium-Based Catalysts with Brookhart and Grubbs Ligands. Organometallics 2001, 20, 1521–1532. [Google Scholar] [CrossRef]
- Deubel, D.V.; Ziegler, T. DFT Study of Olefin versus Nitrogen Bonding in the Coordination of Nitrogen-Containing Polar Monomers to Diimine and Salicylaldiminato Nickel(II) and Palladium(II) Complexes. Implications for Copolymerization of Olefins with Nitrogen-Containing Polar Monomers. Organometallics 2002, 21, 1603–1611. [Google Scholar] [CrossRef]
- Michalak, A.; Ziegler, T. A Comparison of Ni- and Pd-Diimine Complexes as Catalysts for Ethylene/Methyl Acrylate Copolymerization. A Static and Dynamic Density Functional Theory Study. Organometallics 2003, 22, 2660–2669. [Google Scholar] [CrossRef]
- Szabo, M.J.; Jordan, R.F.; Michalak, A.; Piers, W.E.; Weiss, T.; Yang, S.Y.; Ziegler, T. Polar Copolymerization by a Palladium-Diimine-Based Catalyst. Influence of the Catalyst Charge and Polar Substituent on Catalyst Poisoning and Polymerization Activity. A Density Functional Theory Study. Organometallics 2004, 23, 5565–5572. [Google Scholar] [CrossRef]
- Williams, B.S.; Leatherman, M.D.; White, P.S.; Brookhart, M. Reactions of Vinyl Acetate and Vinyl Trifluoroacetate with Cationic Diimine Pd(II) and Ni(II) Alkyl Complexes: Identification of Problems Connected with Copolymerizations of These Monomers with Ethylene. J. Am. Chem. Soc. 2005, 127, 5132–5146. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, W.; Daugulis, O.; Brookhart, M. Mechanistic Studies of Pd(II)-Catalyzed Copolymerization of Ethylene and Vinylalkoxysilanes: Evidence for a β Silyl Elimination Chain Transfer Mechanism. J. Am. Chem. Soc. 2016, 138, 16120–16129. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Luo, Y.; Chen, C. A Second-Coordination-Sphere Strategy to Modulate Nickel- and Palladium-Catalyzed Olefin Polymerization and Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 11604–11609. [Google Scholar] [CrossRef] [PubMed]
- Michalak, A.; Ziegler, T. DFT Studies on Substituent Effects in Palladium-Catalyzed Olefin Polymerization. Organometallics 2000, 19, 1850–1858. [Google Scholar] [CrossRef]
- Michalak, A.; Ziegler, T. Polymerization of Ethylene Catalyzed by a Nickel(+2) Anilinotropone-Based Catalyst: DFT and Stochastic Studies on the Elementary Reactions and the Mechanism of Polyethylene Branching. Organometallics 2003, 22, 2069–2079. [Google Scholar] [CrossRef]
- Waltman, A.W.; Younkin, T.R.; Grubbs, R.H. Insights into the Deactivation of Neutral Nickel Ethylene Polymerization Catalysts in the Presence of Functionalized Olefins. Organometallics 2004, 23, 5121–5123. [Google Scholar] [CrossRef]
- Berkefeld, A.; Drexler, M.; Moller, H.M.; Mecking, S. Mechanistic Insights on the Copolymerization of Polar Vinyl Monomers with Neutral Ni(II) Catalysts. J. Am. Chem. Soc. 2009, 131, 12613–12622. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, A.; Mecking, S. Deactivation Pathways of Neutral Ni(II) Polymerization Catalysts. J. Am. Chem. Soc. 2009, 131, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Olscher, F.; Schnetmann, I.G.; Monteil, V.; Mecking, S. Role of Radical Species in Salicylaldiminato Ni(II) Mediated Polymer Chain Growth: A Case Study for the Migratory Insertion Polymerization of Ethylene in the Presence of Methyl Methacrylate. J. Am. Chem. Soc. 2015, 137, 14819–14828. [Google Scholar] [CrossRef]
- Haras, A.; Anderson, G.W.; Michalak, A.; Rieger, B.; Ziegler, T. Computational Insight into Catalytic Control of Poly(ethylene-methyl acrylate) Topology. Organometallics 2006, 25, 4491–4497. [Google Scholar] [CrossRef]
- Noda, S.; Nakamura, A.; Kochi, T.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Formation of Linear Polyethylene Chain Catalyzed by Palladium Phosphine-Sulfonate Complexes: Experiment and Theoretical Studies. J. Am. Chem. Soc. 2009, 131, 14088–14100. [Google Scholar] [CrossRef]
- Nozaki, K.; Kusumoto, S.; Noda, S.; Kochi, T.; Chung, L.W.; Morokuma, K. Why Did Incorporation of Acrylonitrile to a Linear Polyethylene Become Possible? Comparison of Phosphine-Sulfonate Ligand with Diphosphine and Imine-Phenolate Ligands in the Pd-Catalyzed Ethylene/Acrylonitrile Copolymerization. J. Am. Chem. Soc. 2010, 132, 16030–16042. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, F.P.; Caporaso, L.; Cavallo, L.; Mecking, S.; Falivene, L. Mechanism of Insertion Polymerization of Allyl Ethers. Macromolecules 2018, 51, 4525–4531. [Google Scholar] [CrossRef]
- Sun, J.; Chen, M.; Luo, G.; Chen, C.; Luo, Y. Diphosphazane-monoxide and Phosphine-sulfonate Palladium Catalyzed Ethylene Copolymerization with Polar Monomers: A Computational Study. Organometallics 2019, 38, 638–646. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.; Raza, W.; Kim, K.H.; Luo, Y. Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers, Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers 2020, 12, 2401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, S.; Wang, F.; Wang, F.; Cao, J.; Zheng, H.; Chen, X.; Ren, A. Density Functional Theory Analysis of the Copolymerization of Cyclopropenone with Ethylene Using a Palladium Catalyst. Polymers 2022, 14, 5273. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Li, Y.; Chen, Y.; Hu, N. Copolymerization of Ethylene with Methyl Methacrylate with Neutral Nickel(II) Complexes Bearing β-Ketoiminato Chelate Ligands. Organometallics 2005, 24, 2502–2510. [Google Scholar] [CrossRef]
- Xiong, S.; Shoshani, M.M.; Zhang, X.; Spinney, H.A.; Nett, A.J.; Henderson, B.S.; Miller, T.F.; Agapie, T. Efficient Copolymerization of Acrylate and Ethylene with Neutral P, O-Chelated Nickel Catalysts: Mechanistic Investigations of Monomer Insertion and Chelate Formation. J. Am. Chem. Soc. 2021, 143, 6516–6527. [Google Scholar] [CrossRef] [PubMed]
- Voccia, M.; Odenwald, L.; Baur, M.; Lin, F.; Falivene, L.; Mecking, S.; Caporaso, L. Mechanistic Insights into Ni(II)-Catalyzed Nonalternating Ethylene-Carbon Monoxide Copolymerization. J. Am. Chem. Soc. 2022, 144, 15111–15117. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Maity, B.; Cao, Z.; Kumawat, J.; Gupta, V.; Cavallo, L. A Multivariate Linear Regression Approach to Predict Ethene/1- Olefin Copolymerization Statistics Promoted by Group 4 Catalysts. ACS Catal. 2021, 11, 4061–4070. [Google Scholar] [CrossRef]
- Antinucci, G.; Dereli, B.; Vittoria, A.; Budzelaar, P.M.; Cipullo, R.; Goryunov, G.P.; Kulyabin, P.S.; Uborsky, D.V.; Cavallo, L.; Ehm, C.; et al. Selection of Low-Dimensional 3-D Geometric Descriptors for Accurate Enantioselectivity Prediction. ACS Catal. 2022, 12, 6934–6945. [Google Scholar] [CrossRef]
- Romano, E.; Budzelaar, P.M.; Rosa, C.D.; Talarico, G. Unconventional Stereoerror Formation Mechanisms in Nonmetallocene Propene Polymerization Systems Revealed by DFT Calculations. J. Phys. Chem. A 2022, 126, 6203–6209. [Google Scholar] [CrossRef] [PubMed]
- Zunino, R.; Luise, D.; Barone, V.; Rosa, C.D.; Talarico, G. Reversible trans-alkylation mechanisms at homogeneous catalytic systems investigated by a combined DFT and ASM-NEDA approach. Mol. Catal. 2023, 548, 113450. [Google Scholar] [CrossRef]
- D’Anania, O.; Rosa, C.D.; Talarico, G. A Computational Evaluation of the Steric and Electronic Contributions in Stereoselective Olefin Polymerization with Pyridylamido-Type Catalysts. Molecules 2023, 28, 3768. [Google Scholar] [CrossRef] [PubMed]
- Gharajedaghi, S.; Mohamadnia, Z.; Ahmadi, E.; Marefat, M.; Pareras, G.; Simon, S.; Poater, A.; Bahri-Laleh, N. Experimental and DFT study on titanium-based half-sandwich metallocene catalysts and their application for production of 1-hexene from ethylene. Mol. Catal. 2021, 509, 111636. [Google Scholar] [CrossRef]
- Masoori, M.; Nekoomanesh, M.; Posada-Pérez, S.; Rashedi, R.; Bahri-Laleh, N. Exploring cocatalyst type effect on the Ziegler–Natta catalyzed ethylene polymerizations: Experimental and DFT studies. J. Polym. Res. 2022, 29, 197. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sanchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 721–794. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Simon, L.; Goodman, J.M. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GGA functionals. Org. Biomol. Chem. 2011, 9, 689–700. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sanchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef]
- Pribram-Jones, A.; Gross, D.A.; Burke, K. DFT: A theory full of holes? Annu. Rev. Phys. Chem. 2015, 66, 283–304. [Google Scholar] [CrossRef]
- Yu, H.S.; Li, S.L.; Truhlar, D.G. Perspective: Kohn-Sham density functional theory descending a staircase. J. Chem. Phys. 2016, 145, 130901. [Google Scholar] [CrossRef]
- TeVelde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; Van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T.J. Chemistry with ADF. Comput. Chem. 2001, 22, 931. Available online: https://www.scm.com (accessed on 18 October 2023). [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hicks, F.A.; Brookhart, M. A Highly Active Anilinotropone-Based Neutral Nickel(II) Catalyst for Ethylene Polymerization. Organometallics 2001, 20, 3217–3219. [Google Scholar] [CrossRef]
- Chan, M.W.; Deng, L.; Ziegler, T. Density Functional Study of Neutral Salicylaldiminato Nickel(II) Complexes as Olefin Polymerization Catalysts. Organometallics 2000, 19, 2741–2750. [Google Scholar] [CrossRef]
- Kochi, T.; Noda, S.; Yoshimura, K.; Nozaki, K. Formation of Linear Copolymers of Ethylene and Acrylonitrile Catalyzed by Phosphine Sulfonate Palladium Complexes. J. Am. Chem. Soc. 2007, 129, 8948–8949. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Mecking, S. Insertion Homo- and Copolymerization of Diallyl Ether. Angew. Chem. Int. Ed. 2015, 54, 15845–15849. [Google Scholar] [CrossRef]
- Runzi, T.; Guironnet, D.; Schnetmann, I.G.; Mecking, S. Reactivity of Methacrylates in Insertion Polymerization. J. Am. Chem. Soc. 2010, 132, 16623–16630. [Google Scholar] [CrossRef]
- Luo, S.; Vela, J.; Lief, G.R.; Jordan, R.F. Copolymerization of Ethylene and Alkyl Vinyl Ethers by a (Phosphinesulfonate)PdMe Catalyst. J. Am. Chem. Soc. 2007, 129, 8946–8947. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seidel, F.W.; Nozaki, K. Synthesis of Polyethylene with in-chain α,β-Unsaturated Ketone and Isolated Ketone Units by Pd-catalyzed Ring-opening Copolymerization of Cyclopropenones with Ethylene. Angew. Chem. Int. Ed. 2019, 58, 12955–12959. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.K.; Mecking, S.; Brookhart, M. Copolymerization of Ethylene and Propylene with Functionalized Vinyl Monomers by Palladium(II) Catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. [Google Scholar] [CrossRef]
- Mecking, S.; Johnson, L.K.; Wang, L.; Brookhart, M. Mechanistic Studies of the Palladium-Catalyzed Copolymerization of Ethylene and R-Olefins with Methyl Acrylate. J. Am. Chem. Soc. 1998, 120, 888–889. [Google Scholar] [CrossRef]
- Tian, G.; Boone, H.W.; Novak, B.M. Neutral Palladium Complexes as Catalysts for Olefin-Methyl Acrylate Copolymerization: A Cautionary Tale. Macromolecules 2001, 34, 7656–7663. [Google Scholar] [CrossRef]
- Elia, C.; Barad, S.E.; Sen, A. Palladium-Based System for the Polymerization of Acrylates. Scope and Mechanism. Organometallics 2002, 21, 4249–4256. [Google Scholar] [CrossRef]




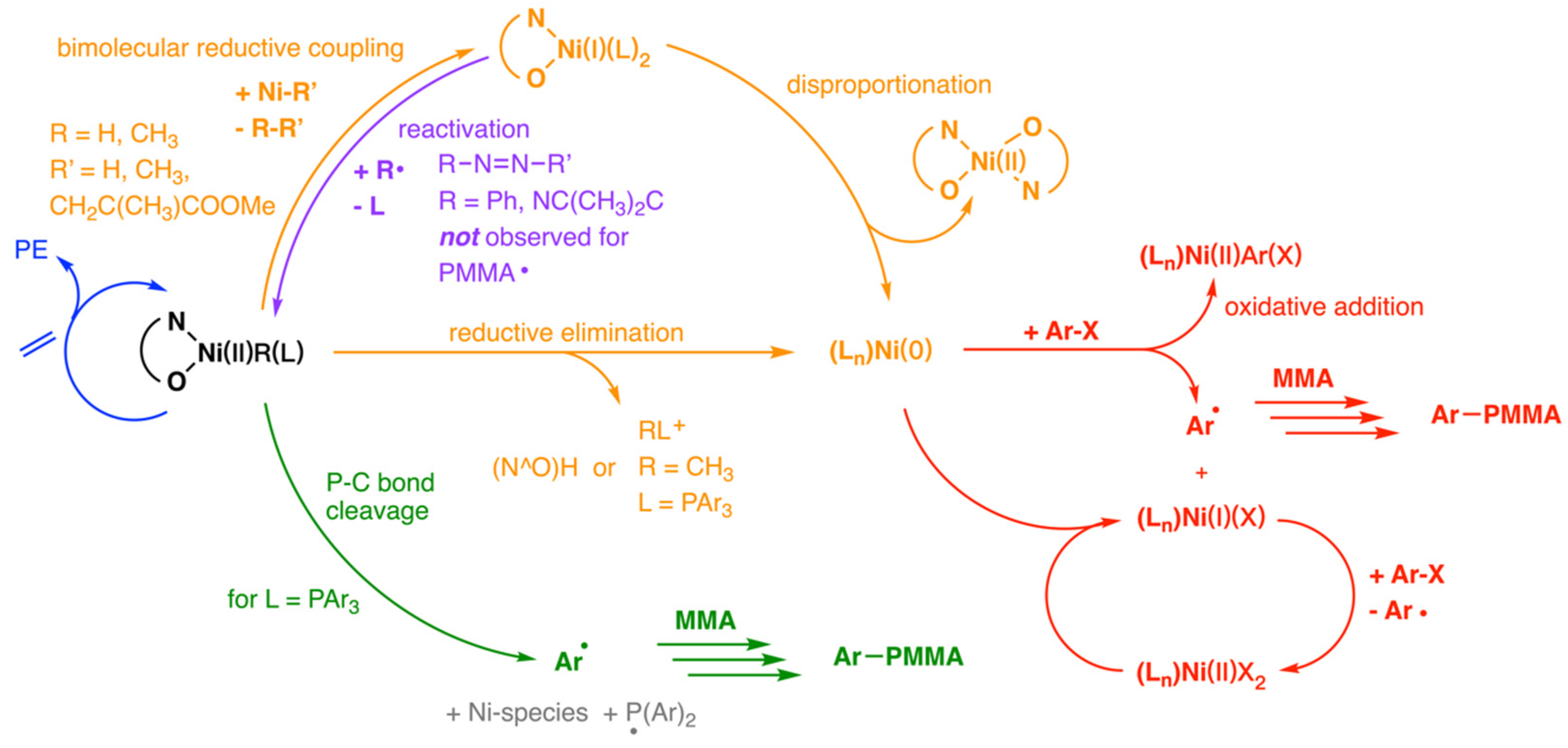
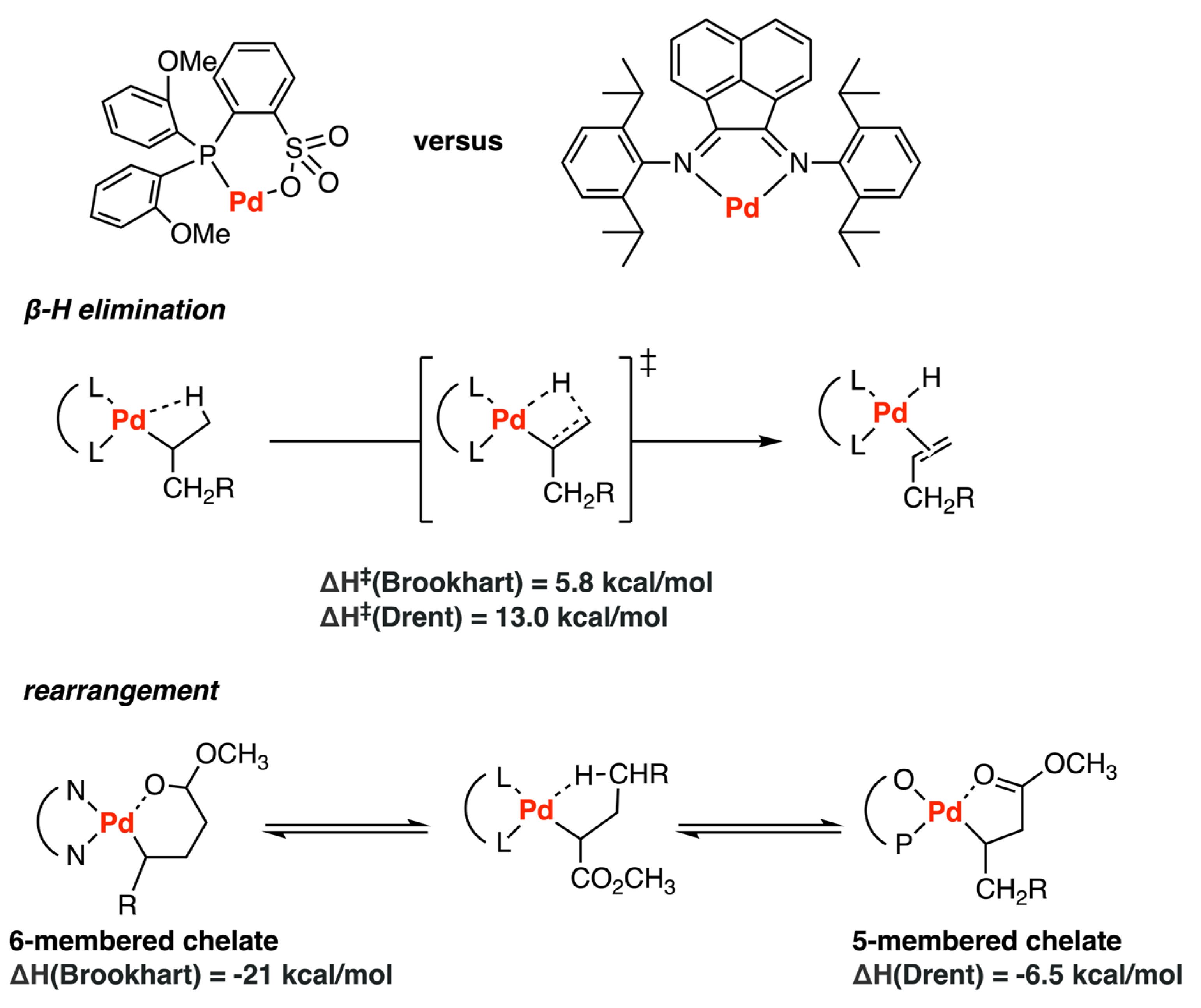
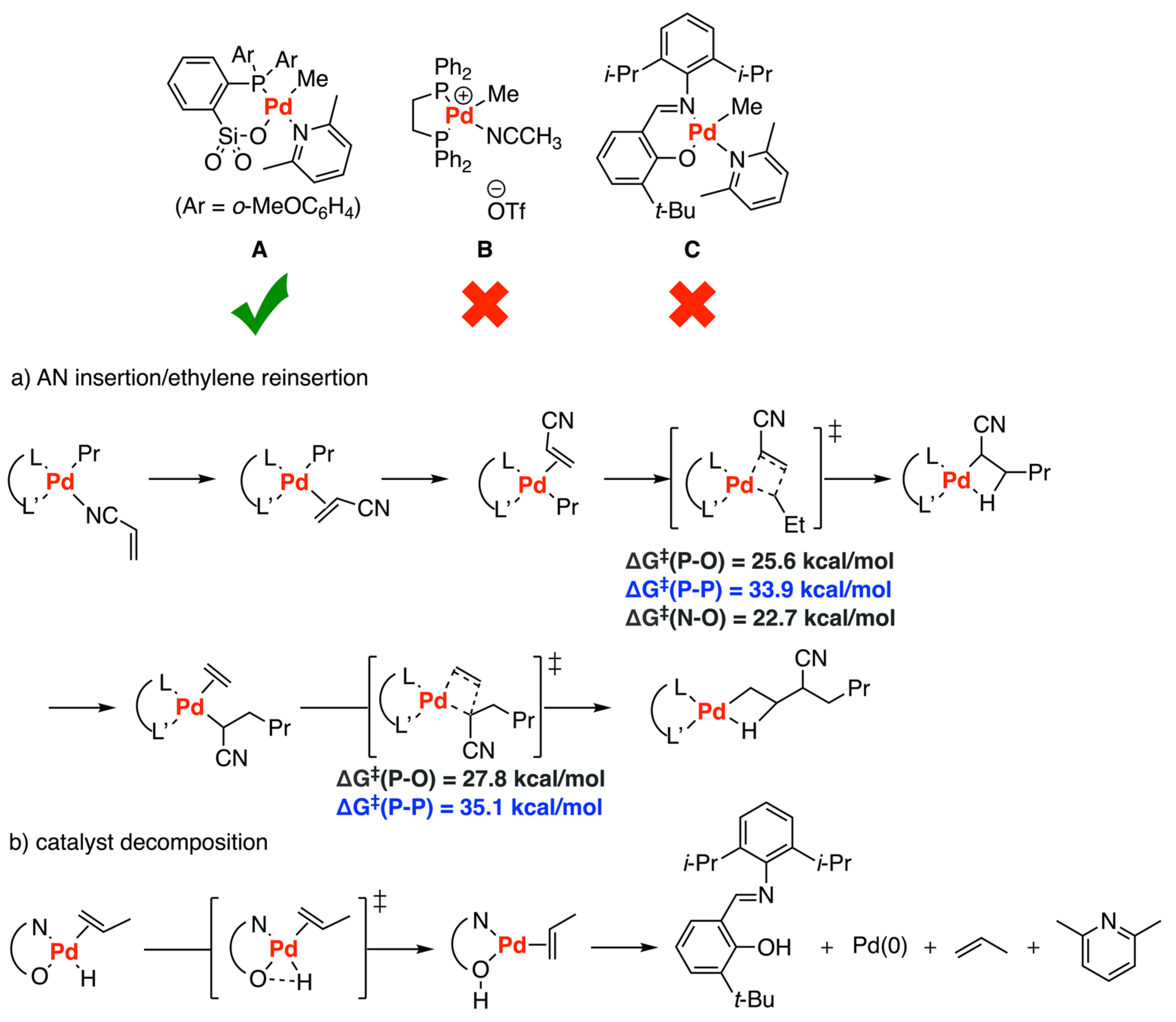

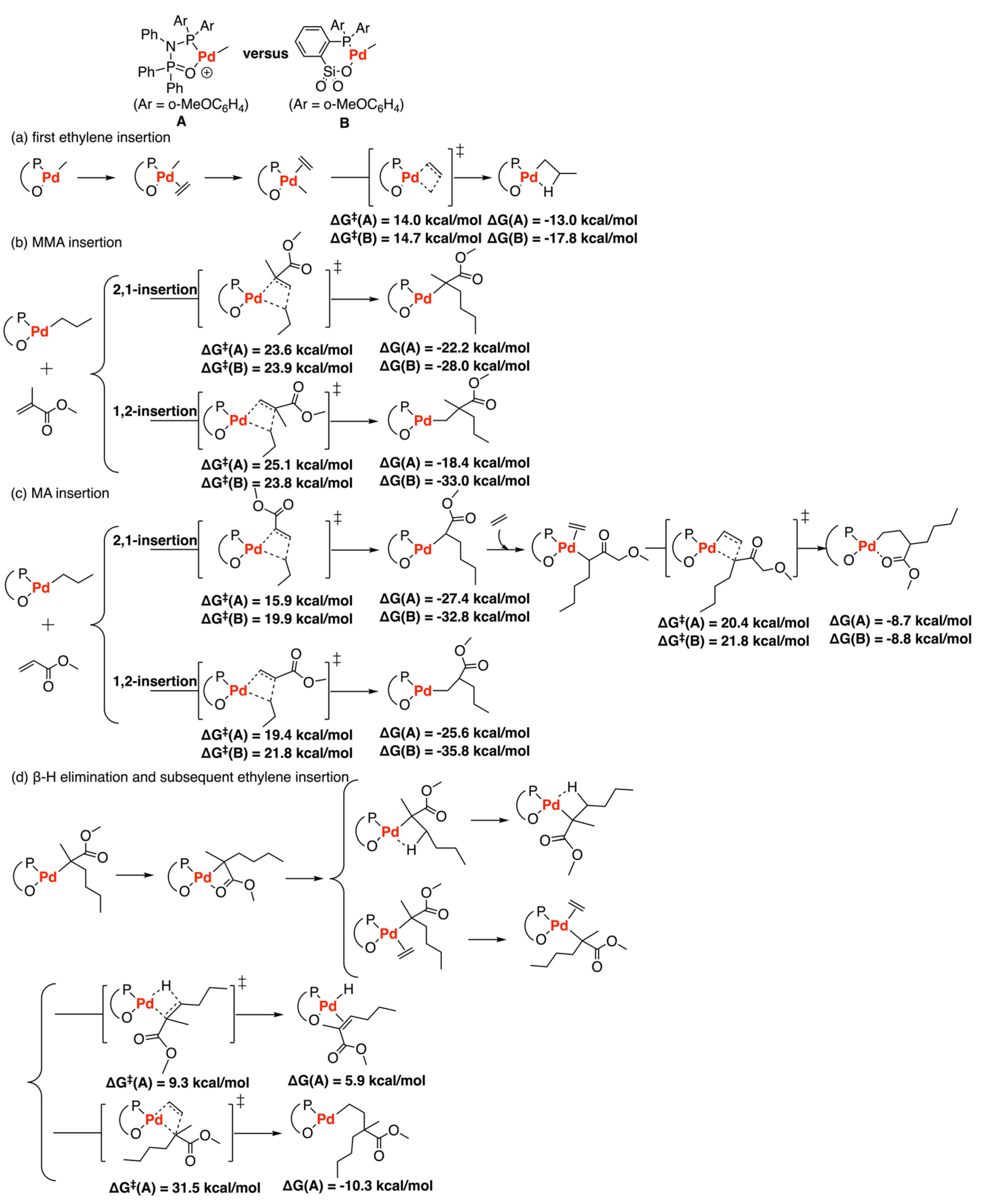




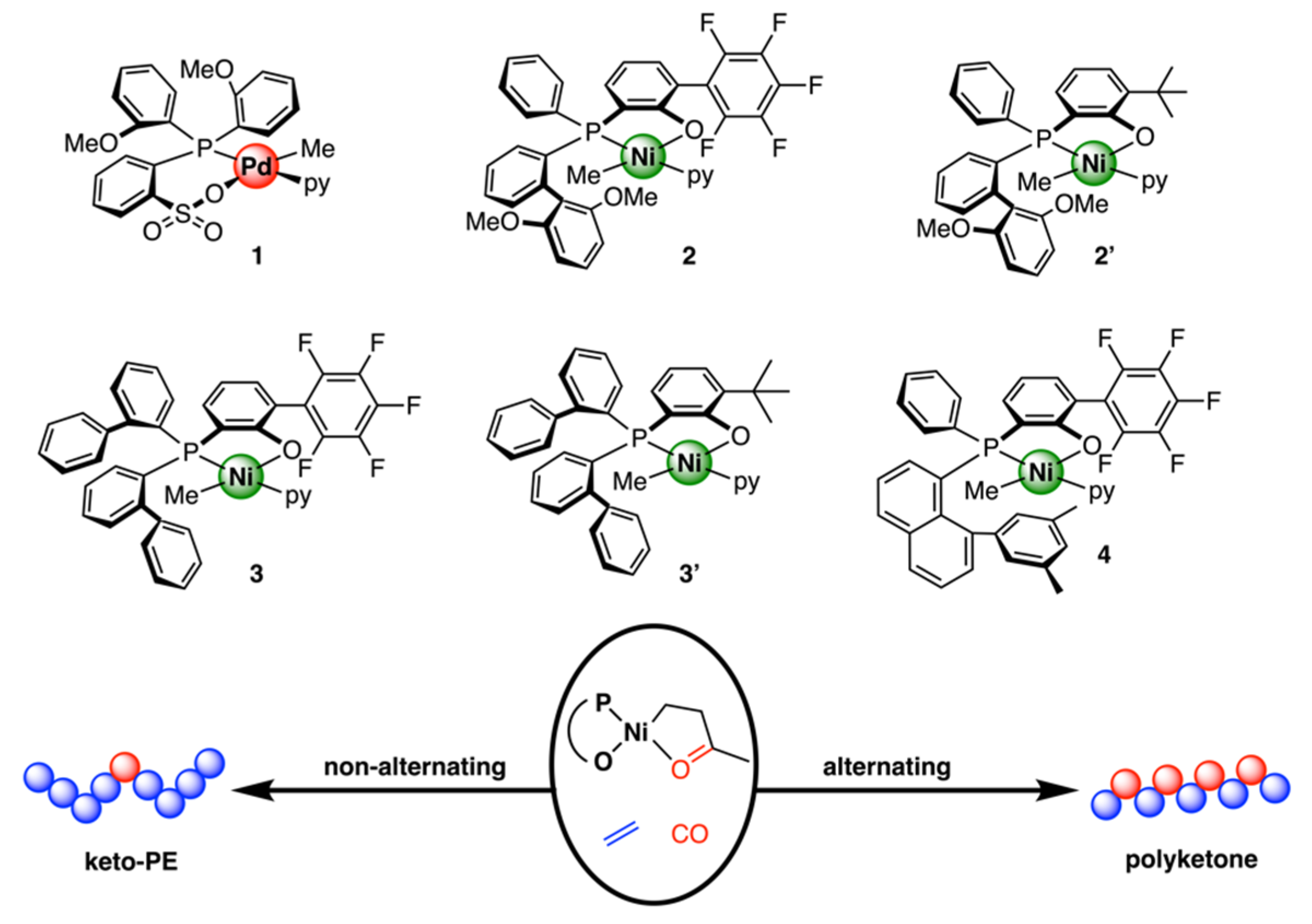

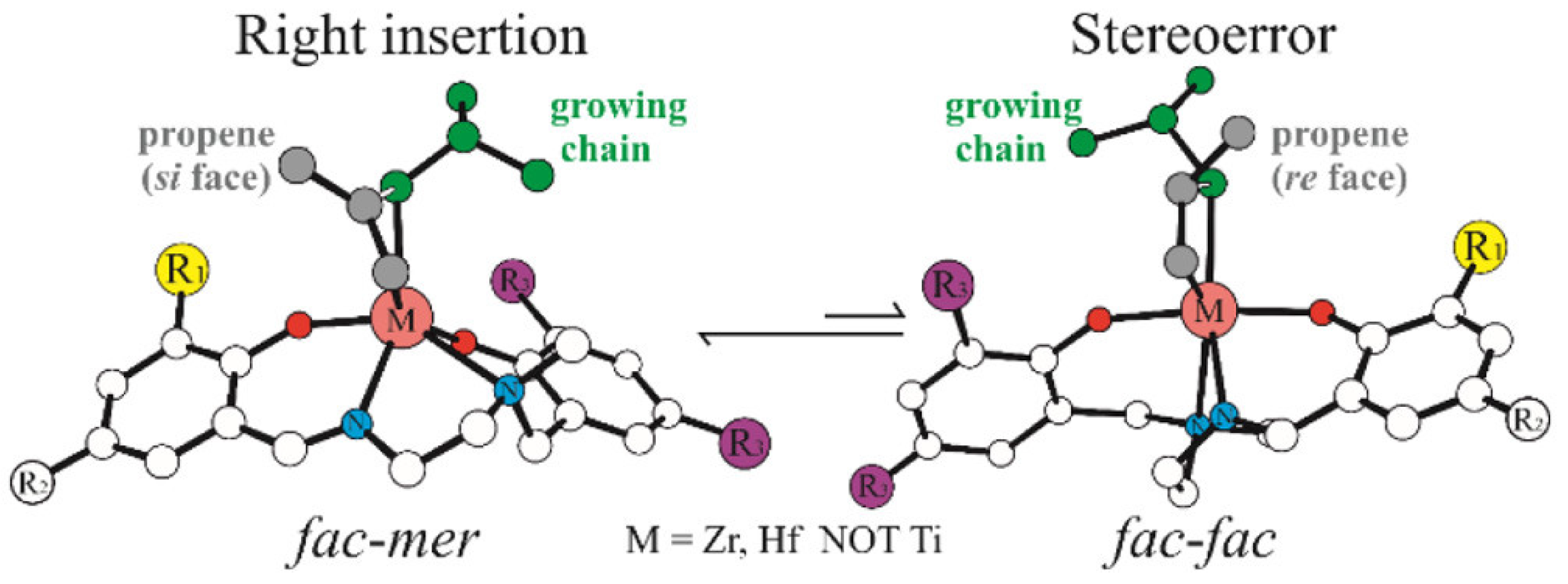
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Wang, S.; Gao, R.; Wang, Y.; Gou, Q.; Zheng, G.; Feng, H.; Fan, G.; Lai, J. Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Polymers 2023, 15, 4343. https://doi.org/10.3390/polym15224343
Song Z, Wang S, Gao R, Wang Y, Gou Q, Zheng G, Feng H, Fan G, Lai J. Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Polymers. 2023; 15(22):4343. https://doi.org/10.3390/polym15224343
Chicago/Turabian StyleSong, Zhihui, Shaochi Wang, Rong Gao, Ying Wang, Qingqiang Gou, Gang Zheng, Huasheng Feng, Guoqiang Fan, and Jingjing Lai. 2023. "Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers" Polymers 15, no. 22: 4343. https://doi.org/10.3390/polym15224343
APA StyleSong, Z., Wang, S., Gao, R., Wang, Y., Gou, Q., Zheng, G., Feng, H., Fan, G., & Lai, J. (2023). Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Polymers, 15(22), 4343. https://doi.org/10.3390/polym15224343