1. Introduction
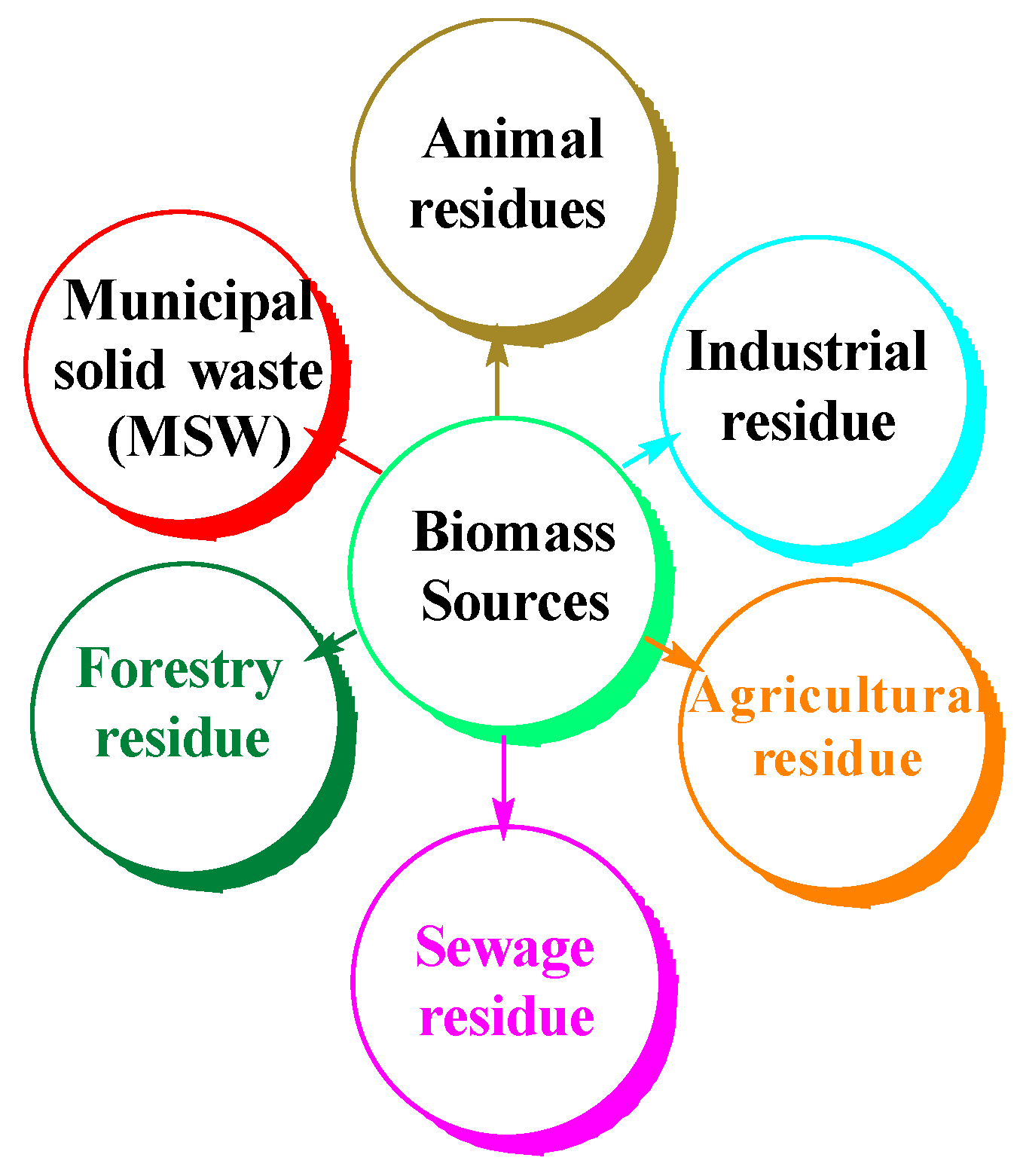
Biomass is at present the most widespread form of renewable energy, and its valorization is constantly increasing due to the concerns about the devastating impact of fossil fuel consumption, climate change, global warming, and their negative impact on human health [
1]. The most important sources of biomass are presented in
Figure 1 [
2].
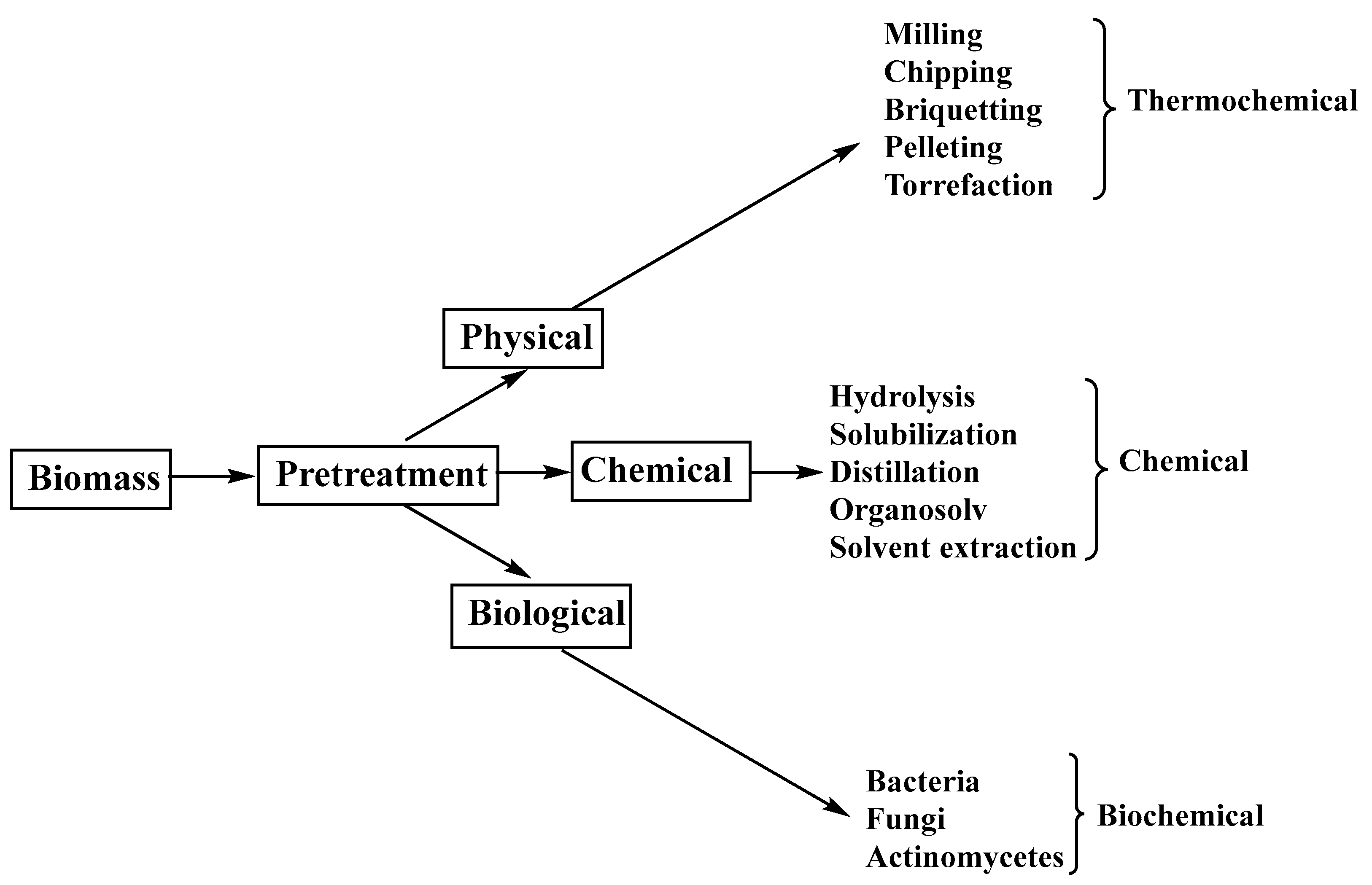
The most frequently used methods for biomass processing [
3], presented in
Figure 2, include mechanical, thermochemical, chemical, and biochemical processing.
The advantages of using biomass:
It represents a renewable energy resource. When considering the valorization of the natural residues to generate energy, it can be considered practically inexhaustible due to the continuous waste generation in nature.
The utilization can lead to the reduction of greenhouse gas emissions. The emissions during the burning process are reabsorbed during the formation process of the new biomass. However, the overall CO2 balance depends on the biomass processing strategy.
It has a low market price. Compared to conventional fuels, biomass recovered energy has the potential to offer a more economical alternative, with a reduction of the costs up to 33%.
Biomass constitutes an abundant global resource. Almost all regions on the planet can permit the generation of natural biomass waste, which is available for local use. In addition, generally, there is no need for large infrastructure to make biomass available for use.
Although there are not many, the disadvantages of using biomass-derived energy must be considered:
In some areas, due to local conditions, the recovery of biomass can be more expensive than in others. This is an important aspect when considering the biomass collection, pretreatment, and storage of different types of biomass.
It requires large land areas for the processes utilized to obtain energy from biomass, especially for storage, because the residues tend to display a low energy density.
In some cases, the overutilization of this energy resource can cause damage to ecosystems or fragmentation due to the activities during the biomass residue collection process.
Biomass residues are considered alternative sources of energy which can reduce or eliminate the dependence on fossil fuels [
4]. The use of fossil carburants has a negative impact on the environment due to the emission produced and the accumulation of CO
2 in the atmosphere which is a factor in the global warming process [
5]. Society must migrate towards the utilization of renewable energy resources such as solar, wind, or biobased fuels [
6]. The biomass type and its production process affect its usage and economic value. Literature highlights six important criteria for biomass selection: accessibility, conversion technology, production costs, process efficiency, cost of raw biomass, production costs, and amount of emission generated during its processing.
This review covers biomass conversion processes to value-added products by liquefaction using polyols or phenol and hydrothermal processes with a special interest in polyurethane-based final products. The influence of the most important parameters (temperature, catalyst, reaction time, etc.) on the above-mentioned conversion processes is analyzed. The second part of the paper is focused on the synthesis of polyurethane materials using the polyols resulting from biomass valorization.
2. Polyols
The general terminology of polyols refers to molecules that have two or more hydroxyl functional groups. From the point of view of molecular weight (M
w), they can be polyols or even polyols-polymers, which can be polyesters, polyethers, polycarbonates, or polyacrylates [
7]. The polyols with a low molecular weight, such as ethyleneglycol or glycerol, are used as raw materials in organic chemistry processes such as esterifications [
8], dehydrations [
9,
10], hydrogenations [
11,
12,
13], as well as in the macromolecular chemistry, in the synthesis of alkyd resins as cross-linking agent [
14] and as chain extender [
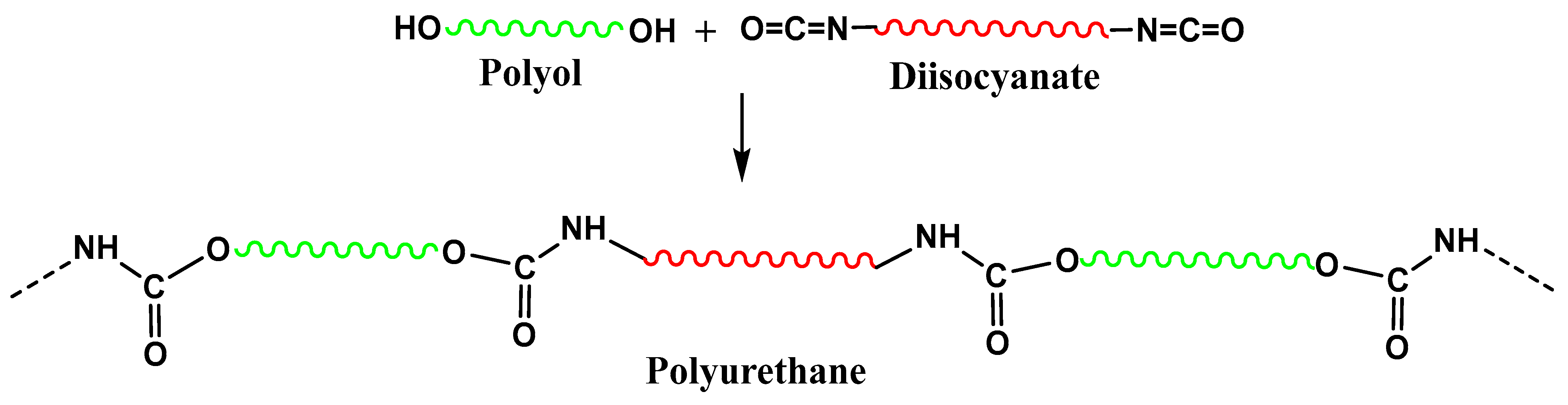
15]. Polyols are extremely important in different practical applications. Thus, polyethers polyols are mainly used in the synthesis of polyurethane foams (PU). To achieve this, a polycondensation reaction between a diisocyanate and a polyol is used (as presented in
Scheme 1).
Unfortunately, a large part of the polyols used for PU is industrially produced using petrochemical derivatives, which are depletable and this justifies an increased interest in biopolyols derived from renewable resources [
16,
17]. For this reason, this review aims to present the newest, respectively, the most important methods to produce biopolyols from renewable resources, one of the most important raw materials being the lignocellulose biomass [
18,
19].
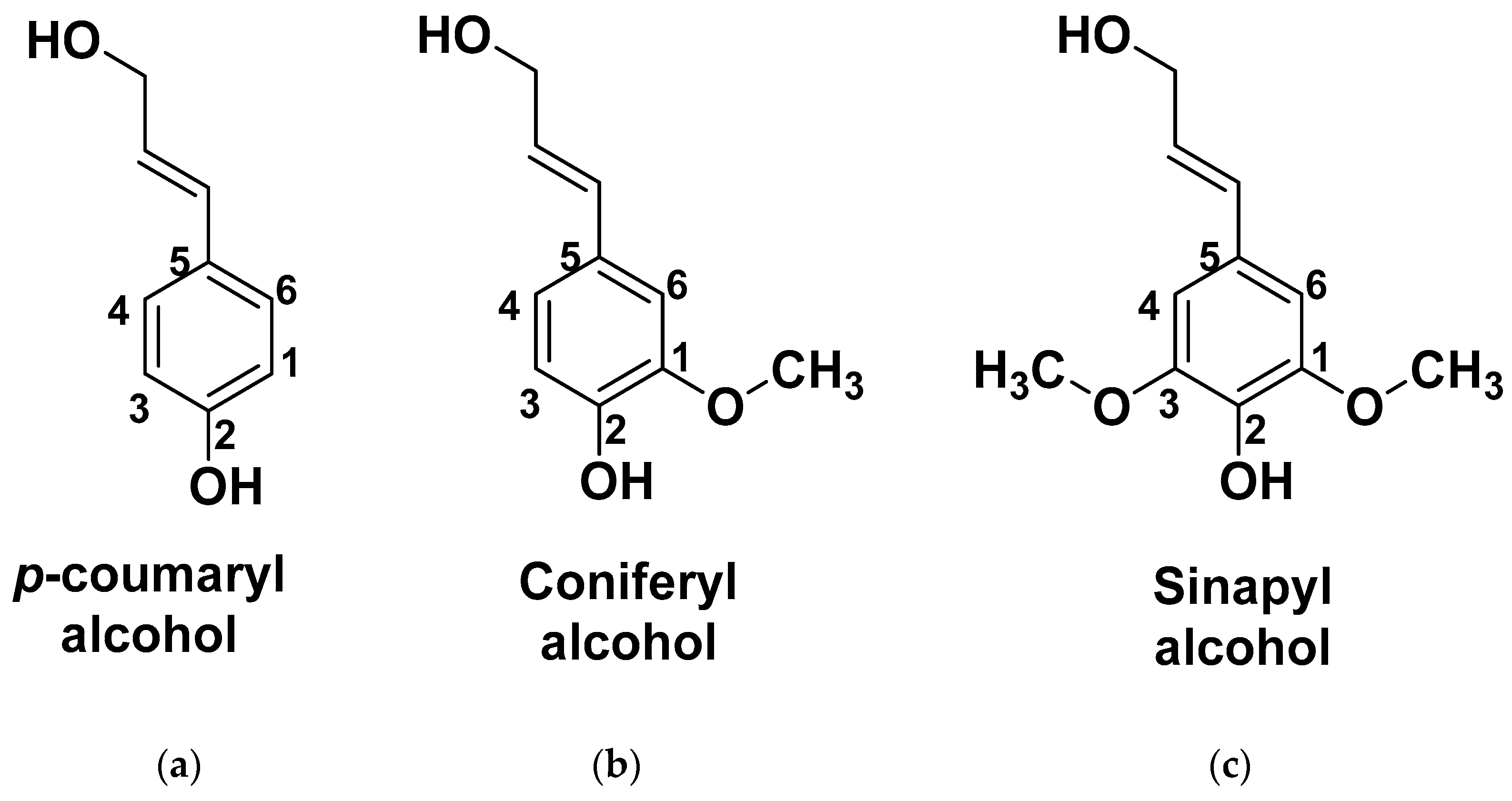
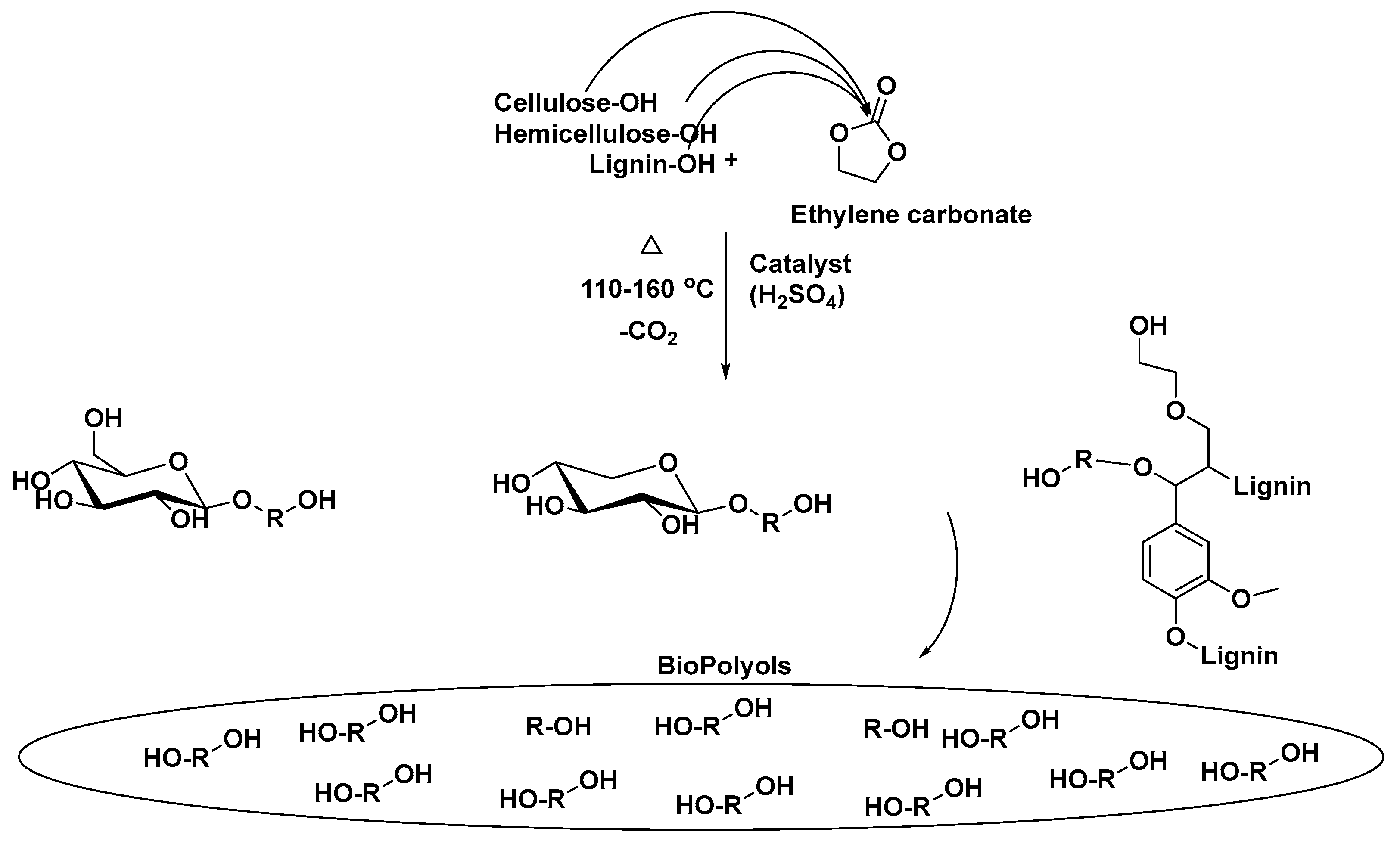
The main structural units of lignocellulosic biomass are cellulose (30–35%), hemicellulose (15–35%), and lignin (20–35%) [
20] all of which are highly functionalized materials rich in hydroxyl groups, making them promising feedstocks to produce bio-based polyols (
Figure 3). In this context, because the biomass is solid, it must be brought to a liquid state for further reactions to be facilitated.
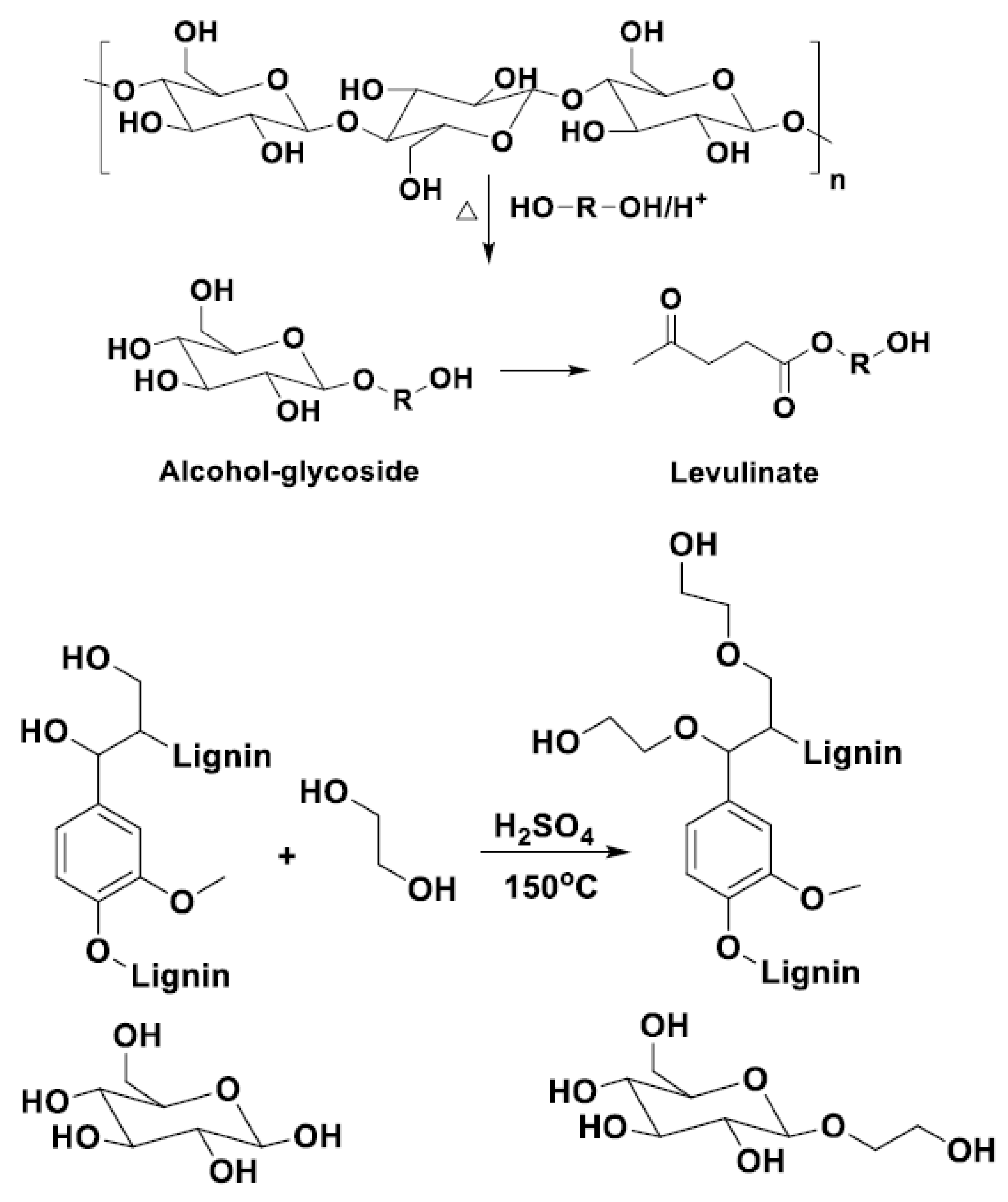
The lignocellulose biomass liquefaction can be achieved by two routes: oxypropylation [
21] and solvolysis (the actual liquefaction) [
22,
23,
24]. The first method consists of the grafting of propylene oxide oligomers on the cellulosic main polymer chain [
25,
26]. The inconvenience of this reaction is the requirement of high pressure and a rather high temperature (100–200 °C), but the products obtained consisting of grafted cellulose, propylene oxide, and unmodified biomass form a mixture that can be used directly to obtain PU foams [
26]. Presented in
Scheme 2 is the general oxypropylation reaction, respectively, the mechanism of this reaction [
26].
The second method entails atmospheric pressure and temperatures of 150–250 °C, using both basic and acidic catalysts. As solvents, glycerol and ethylene glycol are frequently used [
27,
28,
29,
30,
31].
5. Applications of Products Obtained by Biomass Liquefaction
First of all, liquefaction reactions allow access to biobased value-added products such as biofuels and biochemical biomaterials [
80]. One of the most important applications consists in the synthesis of biopolyols and their utilization for polyurethane products fabrication, such as polyurethane foams with high thermal and compression resistance, also presenting biodegradability [
81]. In addition to foams, membranes for gas separation (CO
2/CH
4) were also obtained [
82]. As expected, wood adhesives represent a direct application of bio-based polyurethanes [
22]. Following a hydrothermal liquefaction process, biofuels with limited emission of SO
x and NO
x are also obtained [
83].
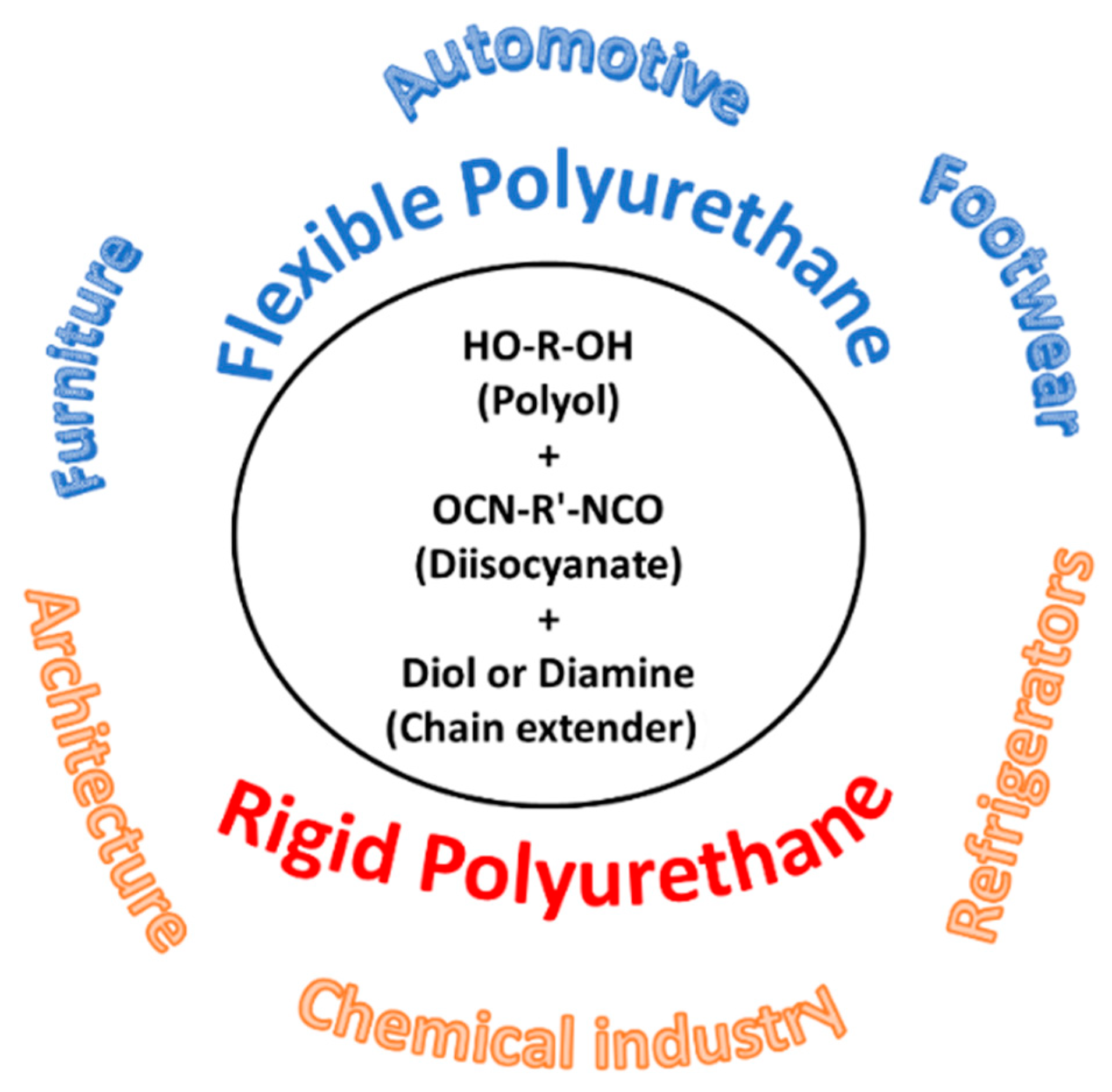
Polyurethanes find applications in a wide variety of products, such as mattresses in furniture (flexible); cushioning, bumpers, sound insulation in automotive, components (flexible, semi-flexible, rigid PUFs); thermal insulation in refrigerators, refrigerated transport, cold storage and piping (rigid PUFs); soles in shoes (PU elastomers and elastomeric foams); and adhesives, coatings, and sealants (thermoset/thermoplastic PUs) [
84,
85]. The development of biobased products with similar or enhanced properties also presenting biodegradability can therefore bring significant economical, societal, and environmental benefits.
Depending on their physical properties, polyurethane foams can be classified into two major groups: flexible and rigid polyurethane foams. The Young modulus and the yield strength are very different for these types of polyurethane. These materials possess good thermal parameters being applicable over a wide range of temperatures (from −200 °C to +135 °C). The average thermal conductivity coefficient of polyurethane foams is 0.026 W/m2, and the most favorable apparent density after the curing of rigid foam is usually 35–50 kg/m3.
Flexible and rigid foams are low-density foams and semi-rigid foams have medium density [
86]. The flexibility depends on the chemical composition as well as matrix polymer characteristics such as the degree of crystallinity and the degree of crosslinking. Polyurethane foams are the main type of cellular plastic. A wide range of materials such as soft flexible foams and tough rigid foams can be synthesized thanks to the versatile chemical characteristics of polyurethane chemistry [
87,
88].
The polyurethane foams derived from the lignocellulose biomass liquefaction are used to obtain rigid and semirigid foams [
89]. In the case of acidic catalysis, the polyols are initially neutralized with a base NaOH or MgO. Because the glycolysis process is usually conducted down to low molecular weight polyols, the resulting foams do not display flexibility, which leads to the formation of rigid foams or the need to add commercial polyols to obtain semirigid foams. The applications of polyurethane foams are presented in
Figure 9.
Examples of rigid foams were obtained by P. Kosmela et al. [
90] via partial substitution of petrochemical-derived polyols with bio-based ones, which were characterized by slightly increased apparent density and average cell size compared to unmodified materials. The best mechanical performance was observed for the sample containing 35% (weight %) of biopolyol in the polyol mixture, which indicates a synergistic effect between the applied polyols. The applied modification delayed the thermal degradation of foams due to changes in the thermal decomposition process.
The use of bio-based polyol for polyurethane foam also leads to an increase in the average cell size [
90]. This aspect can be explained by the higher water content of the biopolyol (0.63 wt%) compared to the non-renewable biopolyol (<0.1 wt%) used in the study [
90], which leads to a slightly higher generation of carbon dioxide during the reaction with isocyanate. In addition, the lower viscosity of the bio-polyol-petrochemical polyol mixture (7600 compared to 9200 mPa∙s) can be a contributing factor that facilitates the diffusion of CO
2 and hydrofluorocarbon used [
90]. Similar results related to the increase of cell size during the incorporation of bio-based polyols into polyurethane foams have been reported by others [
87,
91,
92].
Previous studies confirmed that the viscosity of the biopolyols is an important parameter when considering the use of polyurethane foams [
93] besides the molecular weight and the hydroxyl number [
93].
The mechanical properties of polyurethane determine their use. For example, the compression resistance depends on the porosity and the interconnectivity of the network (the solid that forms the foam cell membrane). A high content of closed cells determines a higher compression resistance of the polyurethane foam [
93].
A recent study [
94] presents the synthesis of polyurethanes that contain 90% polyol derived from lignocellulose degradation. Polyurethanes are known as thermally stable materials due to the urethane linkage in their structure. The breaking down of the polyurethane bond involves three stages: the formation of a diisocyanate and alcohol (240–310 °C), a primary amine, an olefin (320–470 °C), and a secondary amine (over 470 °C).
The glass transition temperature (Tg) of the synthesized polyurethanes was determined by DMA analysis. From the data presented in
Table 3, it can be observed that the possibility of using the materials for both indoor and outdoor applications.
As expected, the liquefaction reaction time has an influence on the mechanical properties of the resulting foams through the molecular weight of the obtained polyols. Recently, an intense variation of the polyol molecular weights with the liquefaction reaction time was observed [
95]. The molecular weights of the polyols are dependent on the reaction parameters (time) (
Table 4).
From an economic point of view, the focus is directed toward the reduction of energy requirements and costs, which determines the utilization of the product directly after the liquefaction reaction without de removal of the residual product [
96]. The presence of residue leads to the higher thermal stability of the polyurethanes, resulting in the increase of both the initial and maximum degradation temperature, but it also has a negative effect on the formation of urethane bonds.
As previously presented, the viscosity of the polyols influences the nature of the pores of the polyurethane foams. The average diameter of the pores decreased from 350 to 100 µm (
Figure 10) with the increase in the liquefaction reaction time [
88], respectively with the decrease in the viscosity.
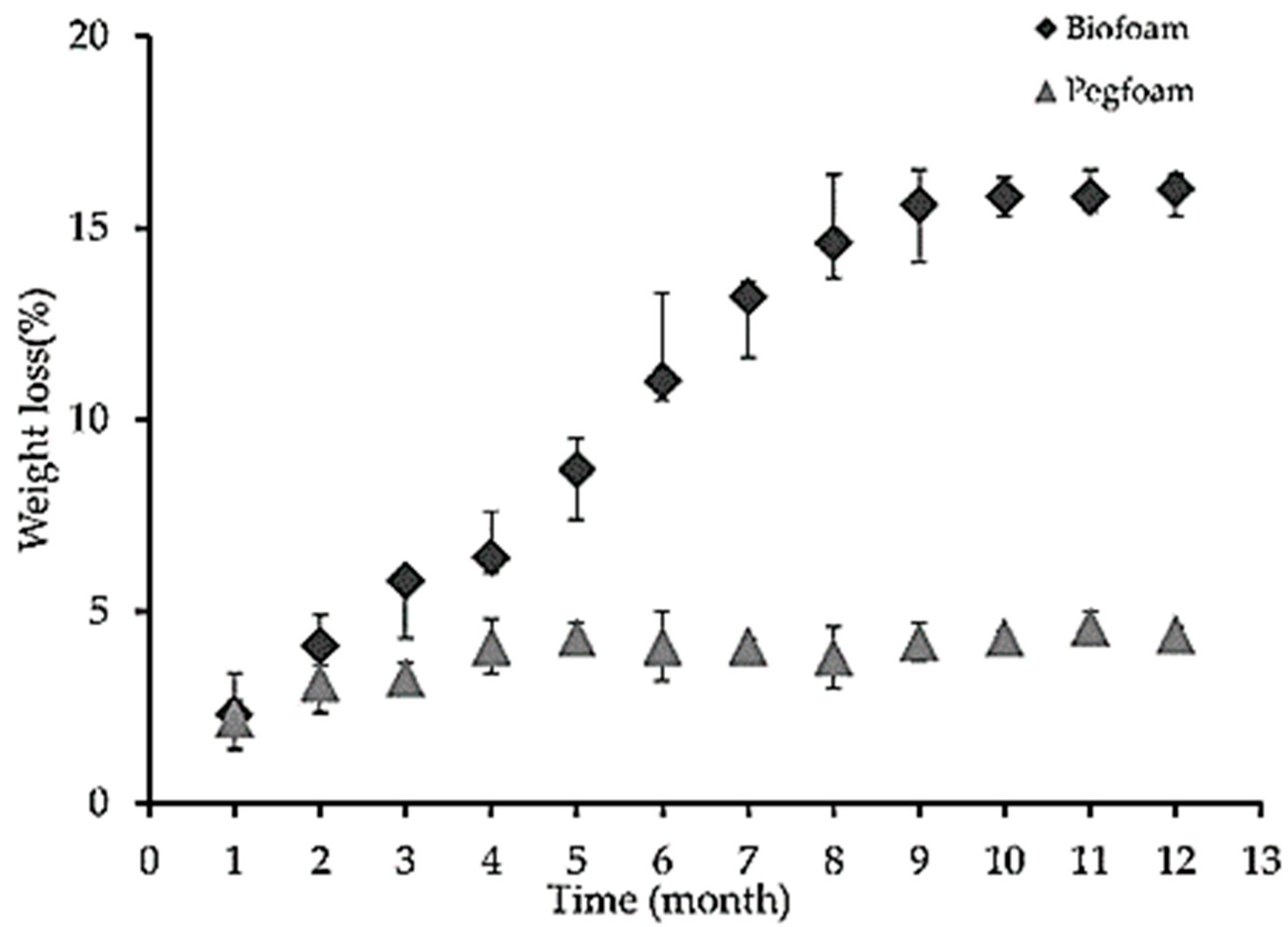
What is specific for this type of polyurethane foam is the biodegradability, measured as a weight-loss of the foam samples buried in the soil. Thus, a weight loss of around 17% was observed for 12 months (
Figure 11).
Rigid polyurethane foams are obtained by using biopolyols obtained by the liquefaction of wood meal (southern pine) [
97] in the presence of PEG400 and glycerol. This depolymerization was performed in acidic catalysis (H
2SO
4) using microwaves [
97]. Before the synthesis of the foams, the polyols were neutralized with a NaOH solution, followed by homogenization with catalyst, surfactants, water, and lastly with methylene diphenyl diisocyanate (MDI). The foams formulations employed are presented in
Table 5.
The mechanical properties of the polyurethane foams were assessed, and it was determined that a longer liquefaction reaction time leads to inferior properties, which can be explained by the decrease in the hydroxyl index value.
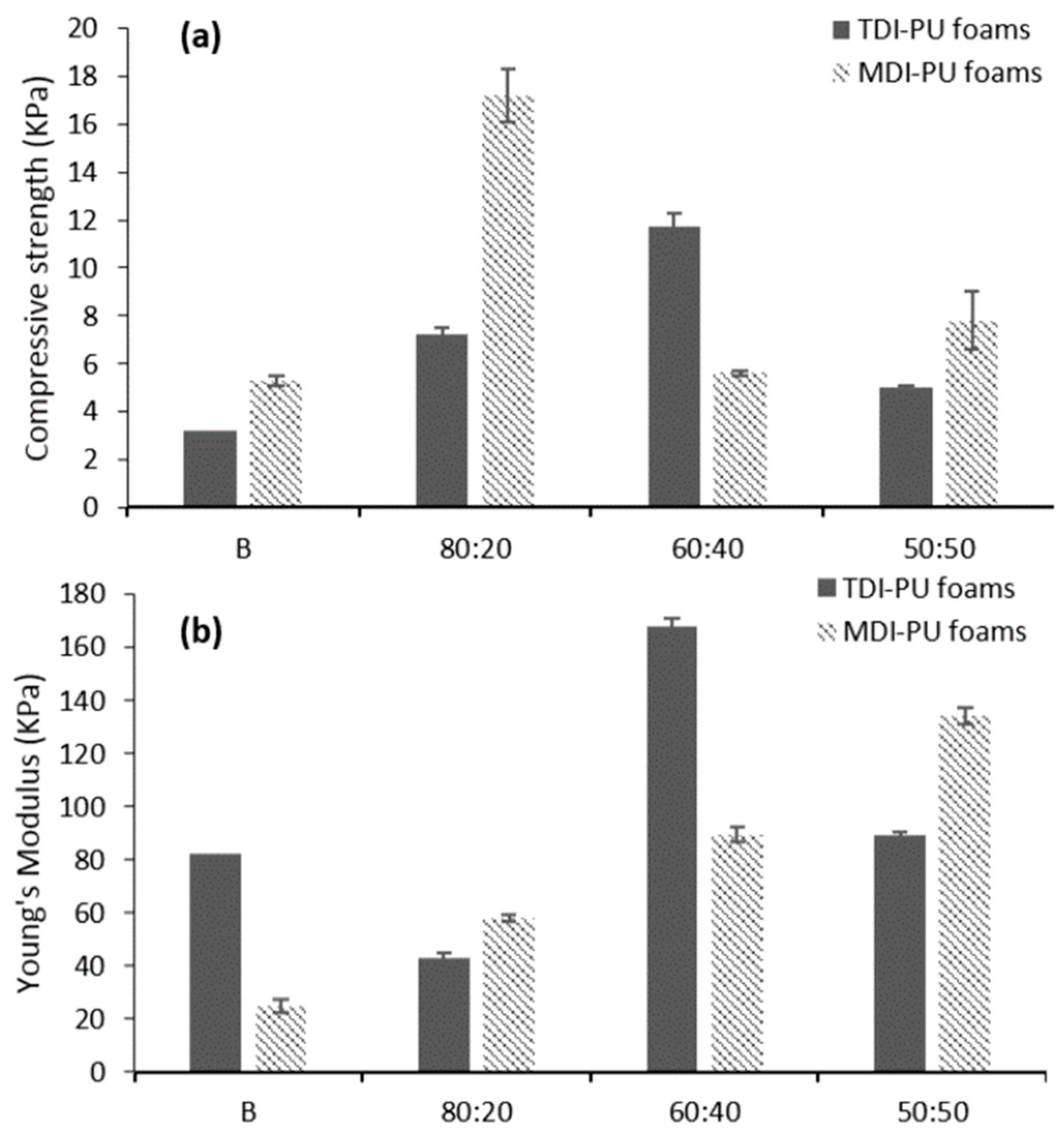
To reduce the amount of wheat straw residue, a liquefaction process using glycerol and H
2SO
4 was employed to produce polyols capable of replacing castor oil for the production of polyurethane foams [
98]. The castor oil was replaced up to 50% (weight %) and as diisocyanate both MDI and toluene-2,4-diisocyanate (TDI) were used for a better comparison. Both the mechanical and thermal properties of the polyurethane foams were improved at a replacement value for castor oil of 40% [
98]. At this optimum value, the obtained polyurethane foams display a compact and ordered morphology (
Figure 12). In addition, the mechanical properties are dependent on the amount of biopolyol introduced with an optimum for the formulation containing 40% (
Figure 13).
Additionally, the PU based on the 40% wheat straw-derived biopolyol presented an excellent biodegradability. At 30 days interval, the weight loss registered was 5.6 and 7.31% for the TDI and MDI formulations, respectively [
98].
Polyurethane (foams) nanomaterials composites with cellulose nanocrystals (CNCs) as nanofillers are also presented in the literature [
99]. The first step in their preparation was the liquefaction of rape straw under microwave irradiation using methanol and 3% H
2SO
4 as a catalyst. The nonrenewable commercial polyol was replaced up to 40% (PU40) and nanocellulose was added as filler 1 to 6% [
99]. Utilizing GC-MS analysis, the composition of the polyols obtained under microwave irradiation was determined [
99] (
Figure 14). Both bio-polyol and cellulose nanocrystals were prepared from the liquid portion and solid residue of microwave-liquefied rape straw [
99]. GC–MS,
1H NMR, and FTIR demonstrated that the bio-polyol is suitable to be employed for polyurethane foam due to the hydroxyl content. By replacing 20% of the non-renewable polyol with bio-polyol, the foam cell became more homogenous and finer, resulting in low thermal conductivity and high mechanical performance, while, further increasing bio-polyol content from 20 to 40%, the cell diameter was increased by 460% and both Young’s modulus and compressive stress decreased by 70%. However, the 40% bio-polyol foam could be remarkably reinforced by 4% CNCs because the hydroxyl-rich structure in CNCs increased the crosslinking density, resulting in the increase of physicomechanical performance of bio-foam [
99].
The properties of the obtained polyurethane foams and nanocomposites are presented in
Table 6 and their morphology in
Figure 15 [
99].
The possible reactions and chemical structure of the cellulose nanocrystals (CNC) reinforced bio-foams are presented in
Scheme 7.
The reinforcement behavior of CNCs in the preparation of bio-polyol foams was evidenced by solid-state
13C NMR and FTIR analysis. As compared with the reference (PU40), Young’s modulus and compressive stress in the optimal 4% CNCs reinforced bio-foam increased by 590% and 150%, respectively [
99].
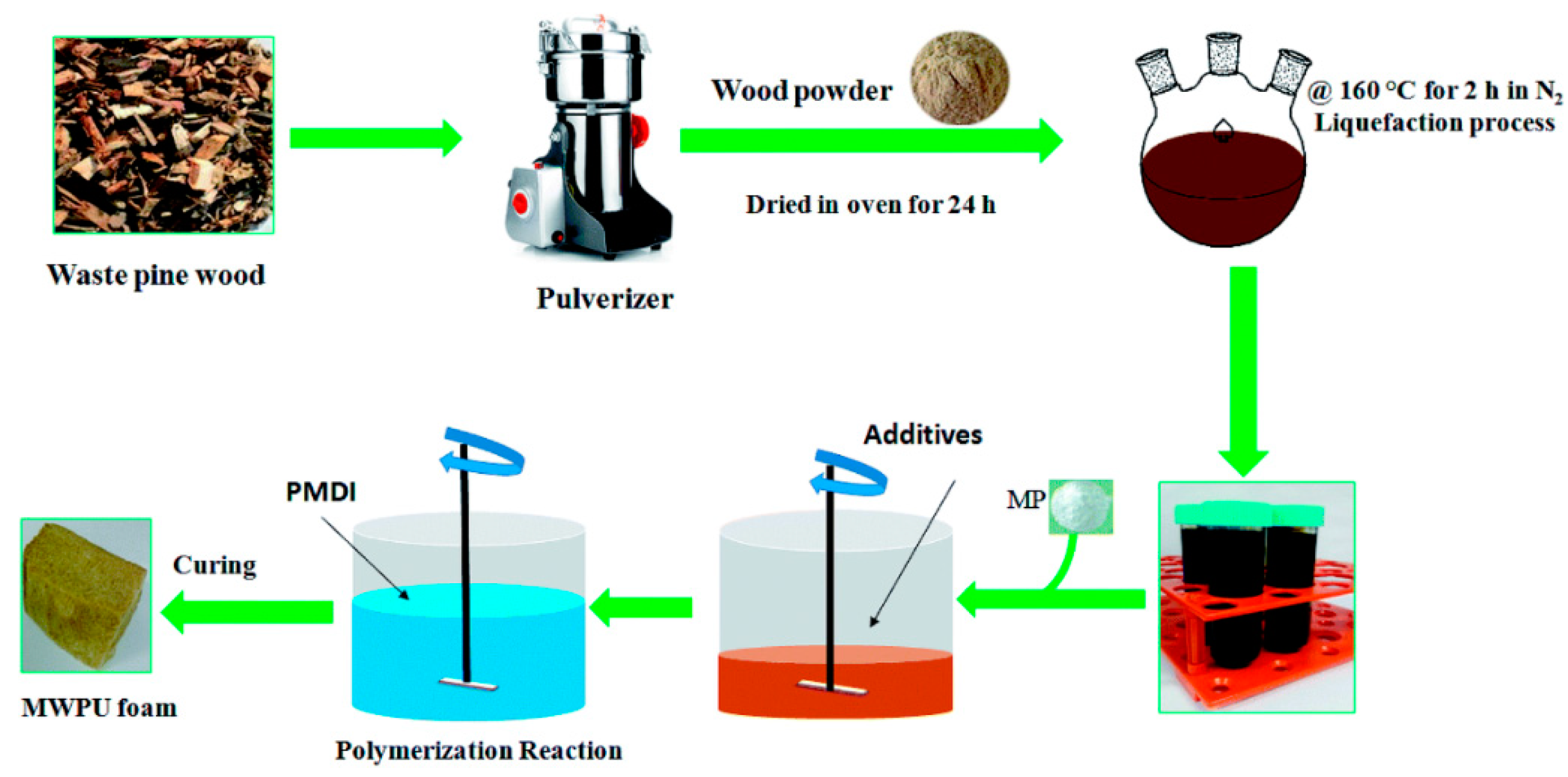
A special application consists of the synthesis of flame-retardant polyurethane foams [
100]. The stages of bio-foam fabrication are presented in
Figure 16 and the flame-retardant additive is melamine phosphate (MP).
The optimum liquefaction temperature was 160 °C which resulted in aliphatic polyols with a higher reactivity than the aromatic polyols towards the diisocyanate and a urethane bond displaying greater thermal resistance [
100]. The T
g of the foam containing 10% filler was 43.8 °C which is significantly higher than without the filler [
100]. Moreover, the MP fillers improved the storage modulus and loss modulus of the resultant foams. Thus, the foams are greener and more environmentally friendly, which is expected to pave the way toward expanding their potential applications (
Figure 17).
6. Discussion and Limitations
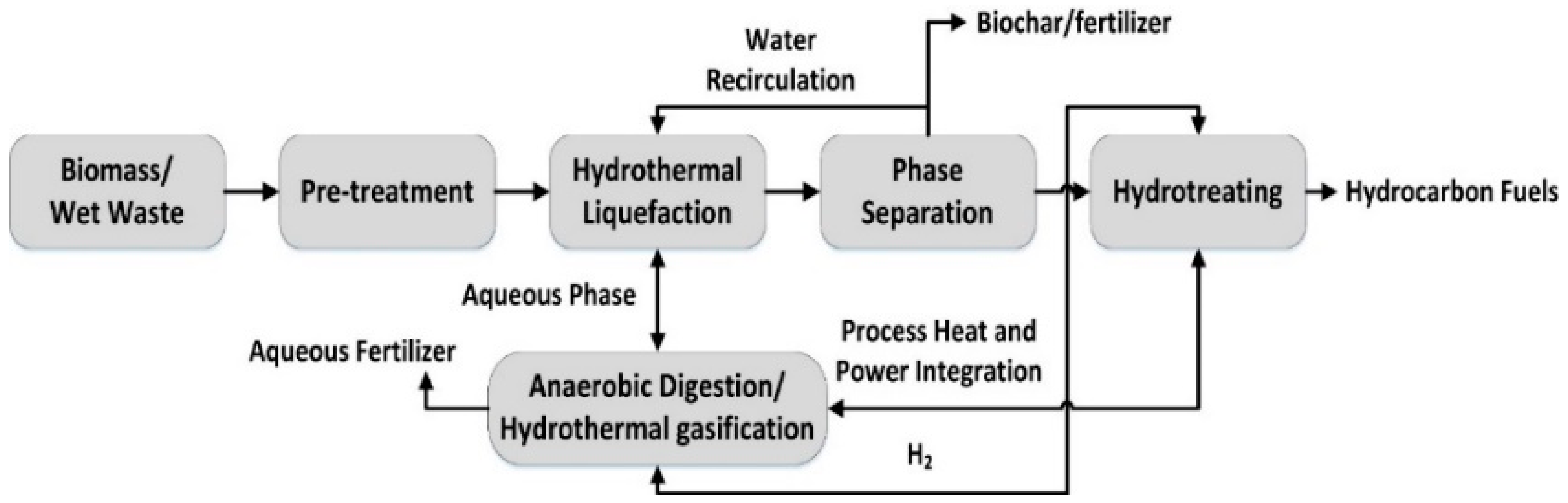
The HTL process has been developed to convert biomass into bio-oil/bio-crude fuels and organic derivatives. This method has limitations that make the scale-up difficult to be implemented due to the high reaction temperatures (200–400 °C) and pressure range (5–20 MPa). However, the quantity of residue is very small, almost inexistent and there is the possibility for straightforward obtained final products such as coatings and adhesives. HTL can be performed in the absence of a catalyst in terms of downstream processing, which constitutes an advantage, in addition to water serving as the solvent. MACL has the advantage of a lower cost due to a lower energy requirement, which can also be improved using ultrasounds and/or microwaves in the presence of an acidic catalyst. Nevertheless, the acidity of the medium requires a corrosion resistant installation both for the reaction and separation stage, since the solvolysis process doesn’t assure a complete conversion. However, MACL has the advantage of a lower energy requirement due to the use of a catalyst and the use of a solvent different from water can facilitate control over the properties of the final products. Both the nature of the employed solvent and catalyst influences the lignocellulose conversion process. In the case of MACL, often used as solvents for the production of biopolyols are glycerol, diethylene glycol, and polyethylene glycol oligomers, while the use of phenol as a solvent allows access to phenolic derivatives. If ethylene or propylene carbonate is used as a solvent, the release of toxic gas takes place. More recently, the literature presents the utilization of ionic liquids as a green solvent alternative; however, for the moment, the price prohibits large-scale implementation.
Comparing the processes, from the reaction products point of view, in the case of HTL bio-oil for energy applications is obtained. In contrast, MACL employing polyols permits the production of biopolyols for coating, adhesives, and foams, while the use of phenols as solvent leads to phenolic derivatives products suitable for applications in adhesive or active carbon fibers. A major drawback in the case of polyols-based solvolysis is the obtaining of reaction mixtures with a high molecular weight, and high viscosity for the reduction of which a higher quantity of solvent is required. In the case of MACL utilizing phenol, a high quantity of phenol is required, which has to be recycled.
Wood is still the largest biomass energy resource today. Other sources include food crops, grassy and woody plants, residues from agriculture or forestry, oil-rich algae, and the organic component of municipal and industrial wastes.
In conclusion, all lignocellulose biomass conversion methods presented have their respective advantages and limitations. However, through the optimization of the process, value-added products presenting the desired properties can be obtained.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}