



Physical Aging Behavior of the Side Chain of a Conjugated Polymer PBTTT

 , , ,
, , ,
Abstract
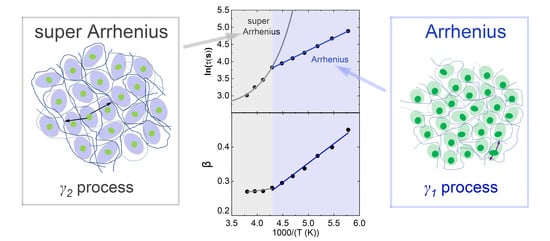
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, X.; Baumgarten, M.; Müllen, K. Designing π-conjugated polymers for organic electronics. Prog. Polym. Sci. 2013, 38, 1832–1908. [Google Scholar] [CrossRef]
- Müller, C.D.; Falcou, A.; Reckefuss, N.; Rojahn, M.; Wiederhirn, V.; Rudati, P.; Frohne, H.; Nuyken, O.; Becker, H.; Meerholz, K. Multi-colour organic light-emitting displays by solution processing. Nature 2003, 421, 829–833. [Google Scholar] [CrossRef]
- Endrődi, B.; Mellár, J.; Gingl, Z.; Visy, C.; Janáky, C. Molecular and Supramolecular Parameters Dictating the Thermoelectric Performance of Conducting Polymers: A Case Study Using Poly(3-alkylthiophene)s. J. Phys. Chem. C 2015, 119, 8472–8479. [Google Scholar] [CrossRef]
- Caire da Silva, L.; Rojas, G.; Schulz, M.D.; Wagener, K.B. Acyclic diene metathesis polymerization: History, methods and applications. Prog. Polym. Sci. 2017, 69, 79–107. [Google Scholar] [CrossRef]
- Ma, W.; Shi, K.; Wu, Y.; Lu, Z.-Y.; Liu, H.-Y.; Wang, J.-Y.; Pei, J. Enhanced Molecular Packing of a Conjugated Polymer with High Organic Thermoelectric Power Factor. Acs Appl. Mater. Inter. 2016, 8, 24737–24743. [Google Scholar] [CrossRef] [PubMed]
- Kajiya, D.; Koganezawa, T.; Saitow, K.-I. Enhancement of Out-of-Plane Mobilities of Three Poly(3-alkylthiophene)s and Associated Mechanism. J. Phys. Chem. C 2016, 120, 23351–23357. [Google Scholar] [CrossRef]
- Stevens, D.M.; Speros, J.C.; Hillmyer, M.A.; Frisbie, C.D. Relationship between Diode Saturation Current and Open Circuit Voltage in Poly(3-alkylthiophene) Solar Cells as a Function of Device Architecture, Processing Conditions, and Alkyl Side Chain Length. J. Phys. Chem. C 2011, 115, 20806–20816. [Google Scholar] [CrossRef]
- Schuettfort, T.; Watts, B.; Thomsen, L.; Lee, M.; Sirringhaus, H.; McNeill, C.R. Microstructure of Polycrystalline PBTTT Films: Domain Mapping and Structure Formation. ACS Nano 2012, 6, 1849–1864. [Google Scholar] [CrossRef]
- Liu, C.; Yang, W.; Jiang, H.; Zhang, B.; Liu, G.; Chen, E.; Boué, F.; Wang, D. Chain Conformation and Liquid-Crystalline Structures of a Poly(thieno)thiophene. Macromolecules 2022, 55, 2892–2903. [Google Scholar] [CrossRef]
- Li, Y.; Sonar, P.; Singh, S.P.; Soh, M.S.; van Meurs, M.; Tan, J. Annealing-Free High-Mobility Diketopyrrolopyrrole−Quaterthiophene Copolymer for Solution-Processed Organic Thin Film Transistors. J. Am. Chem. Soc. 2011, 133, 2198–2204. [Google Scholar] [CrossRef]
- Otep, S.; Lin, Y.-C.; Matsumoto, H.; Mori, T.; Wei, K.-H.; Michinobu, T. Diketopyrrolopyrrole–thiophene–methoxythiophene based random copolymers for organic field effect transistor applications. Org. Electron. 2020, 87, 105986. [Google Scholar] [CrossRef]
- Son, S.Y.; Park, T.; You, W. Understanding of Face-On Crystallites Transitioning to Edge-On Crystallites in Thiophene-Based Conjugated Polymers. Chem. Mater. 2021, 33, 4541–4550. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, Y.; Li, J.; Ong, B.S.; Shuai, Z.; Xu, J.; Zhao, N. Spectroscopic Study of Electron and Hole Polarons in a High-Mobility Donor–Acceptor Conjugated Copolymer. J. Phys. Chem. C 2013, 117, 6835–6841. [Google Scholar] [CrossRef]
- Rodriquez, D.; Savagatrup, S.; Valle, E.; Proctor, C.M.; McDowell, C.; Bazan, G.C.; Nguyen, T.-Q.; Lipomi, D.J. Mechanical Properties of Solution-Processed Small-Molecule Semiconductor Films. Acs Appl. Mater. Inter. 2016, 8, 11649–11657. [Google Scholar] [CrossRef] [PubMed]
- Root, S.E.; Alkhadra, M.A.; Rodriquez, D.; Printz, A.D.; Lipomi, D.J. Measuring the Glass Transition Temperature of Conjugated Polymer Films with Ultraviolet–Visible Spectroscopy. Chem. Mater. 2017, 29, 2646–2654. [Google Scholar] [CrossRef]
- Luo, N.; Ren, P.; Feng, Y.; Shao, X.; Zhang, H.-L.; Liu, Z. Side-Chain Engineering of Conjugated Polymers for High-Performance Organic Field-Effect Transistors. J. Phys. Chem. Lett. 2022, 13, 1131–1146. [Google Scholar] [CrossRef]
- Kline, R.J.; DeLongchamp, D.M.; Fischer, D.A.; Lin, E.K.; Richter, L.J.; Chabinyc, M.L.; Toney, M.F.; Heeney, M.; McCulloch, I. Critical Role of Side-Chain Attachment Density on the Order and Device Performance of Polythiophenes. Macromolecules 2007, 40, 7960–7965. [Google Scholar] [CrossRef]
- Giri, G.; DeLongchamp, D.M.; Reinspach, J.; Fischer, D.A.; Richter, L.J.; Xu, J.; Benight, S.; Ayzner, A.; He, M.; Fang, L.; et al. Effect of Solution Shearing Method on Packing and Disorder of Organic Semiconductor Polymers. Chem. Mater. 2015, 27, 2350–2359. [Google Scholar] [CrossRef]
- McCulloch, I.; Heeney, M.; Bailey, C.; Genevicius, K.; MacDonald, I.; Shkunov, M.; Sparrowe, D.; Tierney, S.; Wagner, R.; Zhang, W.; et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat. Mater. 2006, 5, 328–333. [Google Scholar] [CrossRef]
- Biniek, L.; Leclerc, N.; Heiser, T.; Bechara, R.; Brinkmann, M. Large Scale Alignment and Charge Transport Anisotropy of pBTTT Films Oriented by High Temperature Rubbing. Macromolecules 2013, 46, 4014–4023. [Google Scholar] [CrossRef]
- Gao, Y.; Bai, J.; Sui, Y.; Han, Y.; Deng, Y.; Tian, H.; Geng, Y.; Wang, F. High Mobility Ambipolar Diketopyrrolopyrrole-Based Conjugated Polymers Synthesized via Direct Arylation Polycondensation: Influence of Thiophene Moieties and Side Chains. Macromolecules 2018, 51, 8752–8760. [Google Scholar] [CrossRef]
- Balar, N.; Siddika, S.; Kashani, S.; Peng, Z.; Rech, J.J.; Ye, L.; You, W.; Ade, H.; O’Connor, B.T. Role of Secondary Thermal Relaxations in Conjugated Polymer Film Toughness. Chem. Mater. 2020, 32, 6540–6549. [Google Scholar] [CrossRef]
- Tang, L.; McNeill, C.R. Capturing the Phase Transformation and Thermal Behavior of P(NDI2OD-T2) with In Situ Grazing Incidence WAXS. Macromolecules 2022, 55, 7273–7283. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Sugiyama, S.; Shimada, S.; Kaneko, T.; Takagi, S.; Kinefuchi, I. Molecular Insights into the Mechanical Properties of Polymer–Fullerene Bulk Heterojunctions for Organic Photovoltaic Applications. Macromolecules 2021, 54, 958–969. [Google Scholar] [CrossRef]
- Siddika, S.; Balar, N.; Booth, R.E.; O’Connor, B.T. Dynamic Mechanical Analysis of Polymer Thin Films Using a Kirigami-Inspired Support. ACS Macro Lett. 2021, 10, 1107–1112. [Google Scholar] [CrossRef]
- Bruner, C.; Dauskardt, R. Role of Molecular Weight on the Mechanical Device Properties of Organic Polymer Solar Cells. Macromolecules 2014, 47, 1117–1121. [Google Scholar] [CrossRef]
- Root, S.E.; Savagatrup, S.; Printz, A.D.; Rodriquez, D.; Lipomi, D.J. Mechanical Properties of Organic Semiconductors for Stretchable, Highly Flexible, and Mechanically Robust Electronics. Chem. Rev. 2017, 117, 6467–6499. [Google Scholar] [CrossRef]
- Lu, W.; Wang, R.; Li, R.; Wang, Y.; Wang, Q.; Qin, Y.; Chen, Y.; Lai, W.; Zhang, X. Stable Ultrathin Ag Electrodes by Tailoring the Surface of Plastic Substrates for Flexible Organic Light-Emitting Devices. Acs Appl. Mater. Inter. 2022, 14, 55905–55914. [Google Scholar] [CrossRef]
- Dimov, I.B.; Moser, M.; Malliaras, G.G.; McCulloch, I. Semiconducting Polymers for Neural Applications. Chem. Rev. 2022, 122, 4356–4396. [Google Scholar] [CrossRef]
- Balar, N.; Rech, J.J.; Siddika, S.; Song, R.; Schrickx, H.M.; Sheikh, N.; Ye, L.; Bonilla, A.M.; Awartani, O.; Ade, H. Resolving the Molecular Origin of Mechanical Relaxations in Donor–Acceptor Polymer Semiconductors. Adv. Funct. Mater. 2022, 13, 1131–1146. [Google Scholar] [CrossRef]
- Zhao, B.; Awartani, O.; O’Connor, B.; Zikry, M.A. A direct correlation of x-ray diffraction orientation distributions to the in-plane stiffness of semi-crystalline organic semiconducting films. Appl. Phys. Lett. 2016, 108, 181902. [Google Scholar] [CrossRef]
- Luo, S.; Li, N.; Zhang, S.; Zhang, C.; Qu, T.; Ocheje, M.U.; Xue, G.; Gu, X.; Rondeau-Gagné, S.; Hu, W.; et al. Observation of Stepwise Ultrafast Crystallization Kinetics of Donor–Acceptor Conjugated Polymers and Correlation with Field Effect Mobility. Chem. Mater. 2021, 33, 1637–1647. [Google Scholar] [CrossRef]
- Luo, S.; Kui, X.; Xing, E.; Wang, X.; Xue, G.; Schick, C.; Hu, W.; Zhuravlev, E.; Zhou, D. Interplay between Free Surface and Solid Interface Nucleation on Two-Step Crystallization of Poly(ethylene terephthalate) Thin Films Studied by Fast Scanning Calorimetry. Macromolecules 2018, 51, 5209–5218. [Google Scholar] [CrossRef]
- Gibbs, J.H.; Dimarzio, E.A. Nature of the Glass Transition and the Glassy State. J. Chem. Phys. 1958, 28, 373–383. [Google Scholar] [CrossRef]
- Calderwood, H.J. Dielectric Spectroscopy of Polymers. Phys. Bull. 1977, 27, 572. [Google Scholar] [CrossRef]
- Cowie, J.; Ferguson, R. Aging of Poly(vinyl methyl ether) as determined from enthalpy relaxation measurements. Polym. Commun. Guildf. 1986, 27, 258–260. [Google Scholar]
- Alegría, A.; Goitiandia, L.; Colmenero, J. Interpretation of the TSDC fractional polarization experiments on the α-relaxation of polymers. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 2105–2113. [Google Scholar] [CrossRef]
- Williams, G.; Watts, D.C. Non-symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Trans. Faraday Soc. 1970, 66, 80. [Google Scholar] [CrossRef]
- Dixon, P.K.; Lei, W.; Nagel, S.R.; Williams, B.D.; Carini, J.P. Scaling in the relaxation of supercooled liquids. Phys. Rev. Lett. 1990, 65, 1108–1111. [Google Scholar] [CrossRef]
- Cardona, M.; Chamberlin, R.V.; Marx, W. Comment on the history of the stretched exponential function. arXiv 2007, arXiv:0710.4446. [Google Scholar]
- Androsch, R.; Zhuravlev, E.; Schmelzer, J.W.P.; Schick, C. Relaxation and crystal nucleation in polymer glasses. Eur. Polym. J. 2018, 102, 195–208. [Google Scholar] [CrossRef]
- Hodge, I.M. Enthalpy relaxation and recovery in amorphous materials. J. Non. Cryst. Solids 1994, 169, 211–266. [Google Scholar] [CrossRef]
- Boucher, V.M.; Cangialosi, D.; Alegría, A.; Colmenero, J. Enthalpy Recovery of Glassy Polymers: Dramatic Deviations from the Extrapolated Liquidlike Behavior. Macromolecules 2011, 4, 8333–8342. [Google Scholar] [CrossRef]
- Grassia, L.; Koh, Y.P.; Rosa, M.; Simon, S.L. Complete Set of Enthalpy Recovery Data Using Flash DSC: Experiment and Modeling. Macromolecules 2018, 51, 1549–1558. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Dranca, I. Physical Stability and Relaxation of Amorphous Indomethacin. J. Phys. Chem. B 2005, 10, 18637–18644. [Google Scholar] [CrossRef]
- Hodge, I.M. Effects of annealing and prior history on enthalpy relaxation in glassy polymers. 4. Comparison of five polymers. Macromolecules 1983, 16, 898–902. [Google Scholar] [CrossRef]
- Bendler, J.T.; Ngai, K.L. Microscopic approach to volume recovery of polymers. Macromolecules 1984, 17, 1174–1177. [Google Scholar] [CrossRef]
- Yang, A.C.M.; Wang, R.C.; Lin, J.H. Ductile-brittle transition induced by aging in poly(phenylene oxide) thin films. Polymer 1996, 37, 5751–5754. [Google Scholar] [CrossRef]
- Zhao, G.; Gomes, F.; Marway, H.; Thompson, M.R.; Zhu, Z. Physical Aging as the Driving Force for Brittle–Ductile Transition of Polylactic Acid. Macromol. Chem. Phys. 2020, 30, 5327–5338. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, T.; Meng, F.; Li, L.; Zhang, C.; Wang, X.; Chen, W.; Xue, G.; Zhuravlev, E.; Luo, S.; Zhou, D. Physical Aging Behavior of the Side Chain of a Conjugated Polymer PBTTT. Polymers 2023, 15, 794. https://doi.org/10.3390/polym15040794
Qu T, Meng F, Li L, Zhang C, Wang X, Chen W, Xue G, Zhuravlev E, Luo S, Zhou D. Physical Aging Behavior of the Side Chain of a Conjugated Polymer PBTTT. Polymers. 2023; 15(4):794. https://doi.org/10.3390/polym15040794
Chicago/Turabian StyleQu, Tengfei, Fanzhang Meng, Linling Li, Chen Zhang, Xiaoliang Wang, Wei Chen, Gi Xue, Evgeny Zhuravlev, Shaochuan Luo, and Dongshan Zhou. 2023. "Physical Aging Behavior of the Side Chain of a Conjugated Polymer PBTTT" Polymers 15, no. 4: 794. https://doi.org/10.3390/polym15040794
APA StyleQu, T., Meng, F., Li, L., Zhang, C., Wang, X., Chen, W., Xue, G., Zhuravlev, E., Luo, S., & Zhou, D. (2023). Physical Aging Behavior of the Side Chain of a Conjugated Polymer PBTTT. Polymers, 15(4), 794. https://doi.org/10.3390/polym15040794