
Molecular Dynamic and Dissipative Particle Dynamic Simulation on the Miscibility of NR/CR Blends
 , and
, and
Abstract
:1. Introduction
2. Theory and Method
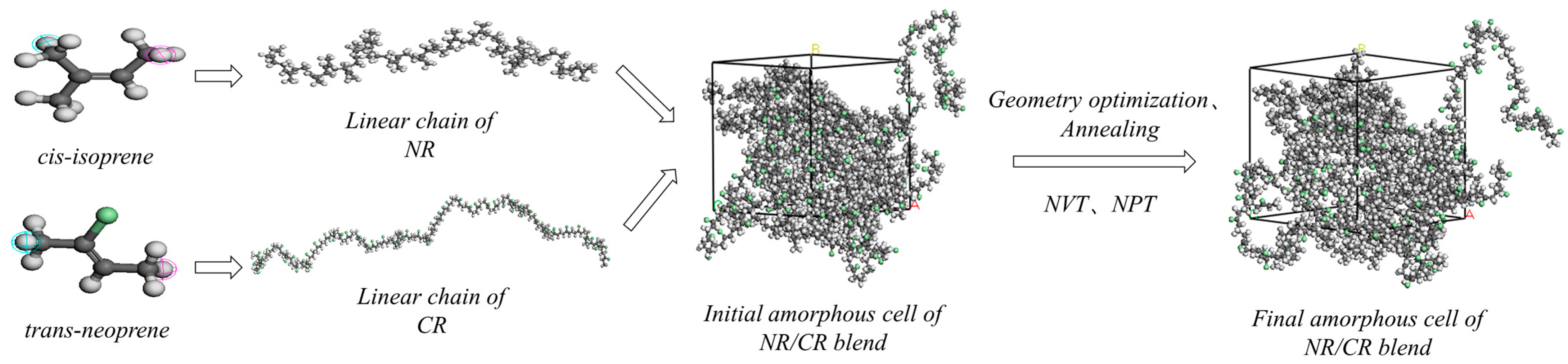
2.1. Classic Molecular Dynamics Simulation
2.2. Dissipative Particle Dynamics Simulation
2.3. Model of DPD Simulation
2.4. NR/CR Blending Experiments
3. Discussion and Results
3.1. Solubility Parameter and Flory–Huggins Parameter
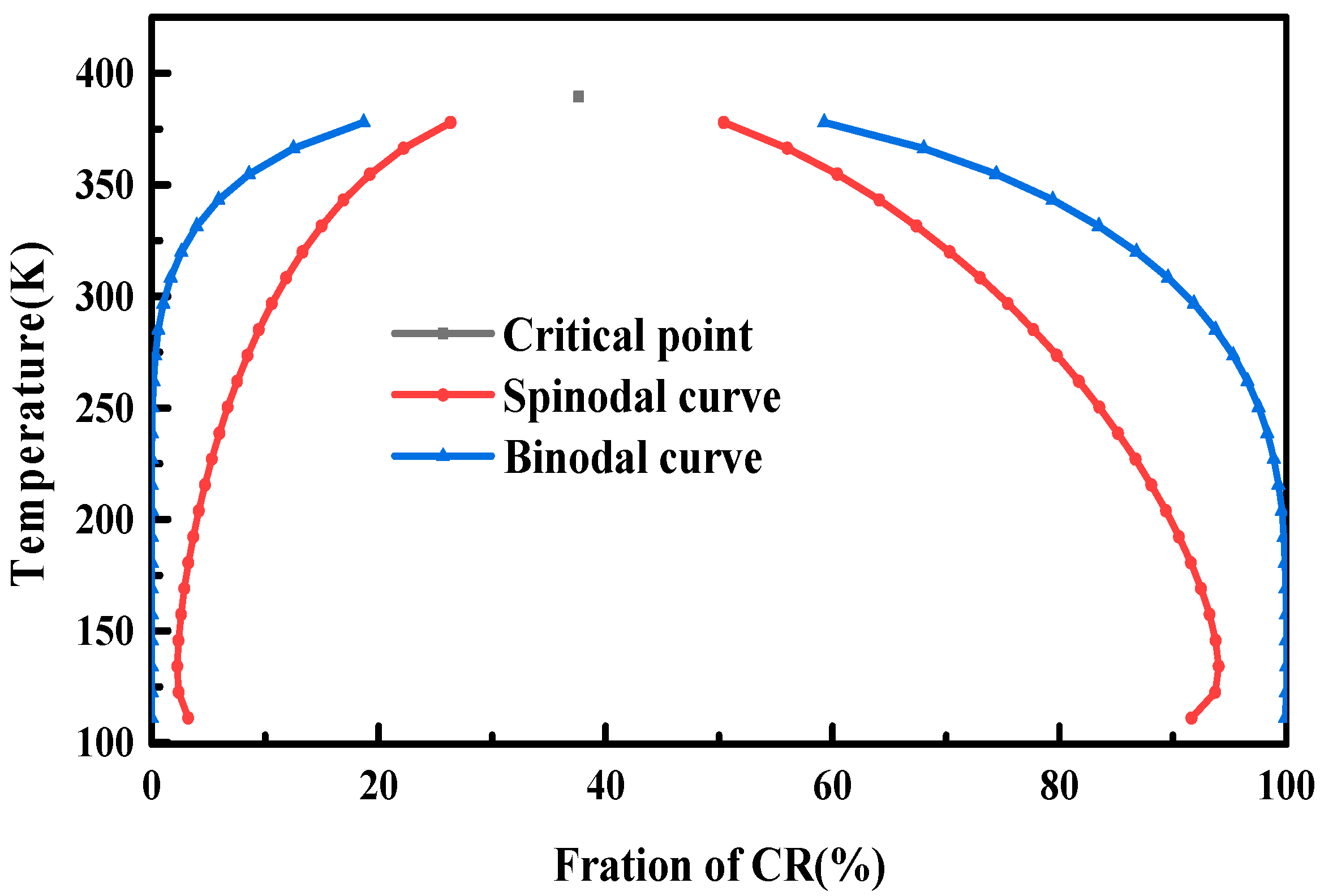
3.2. Phase Diagrams of the NR/CR Blends
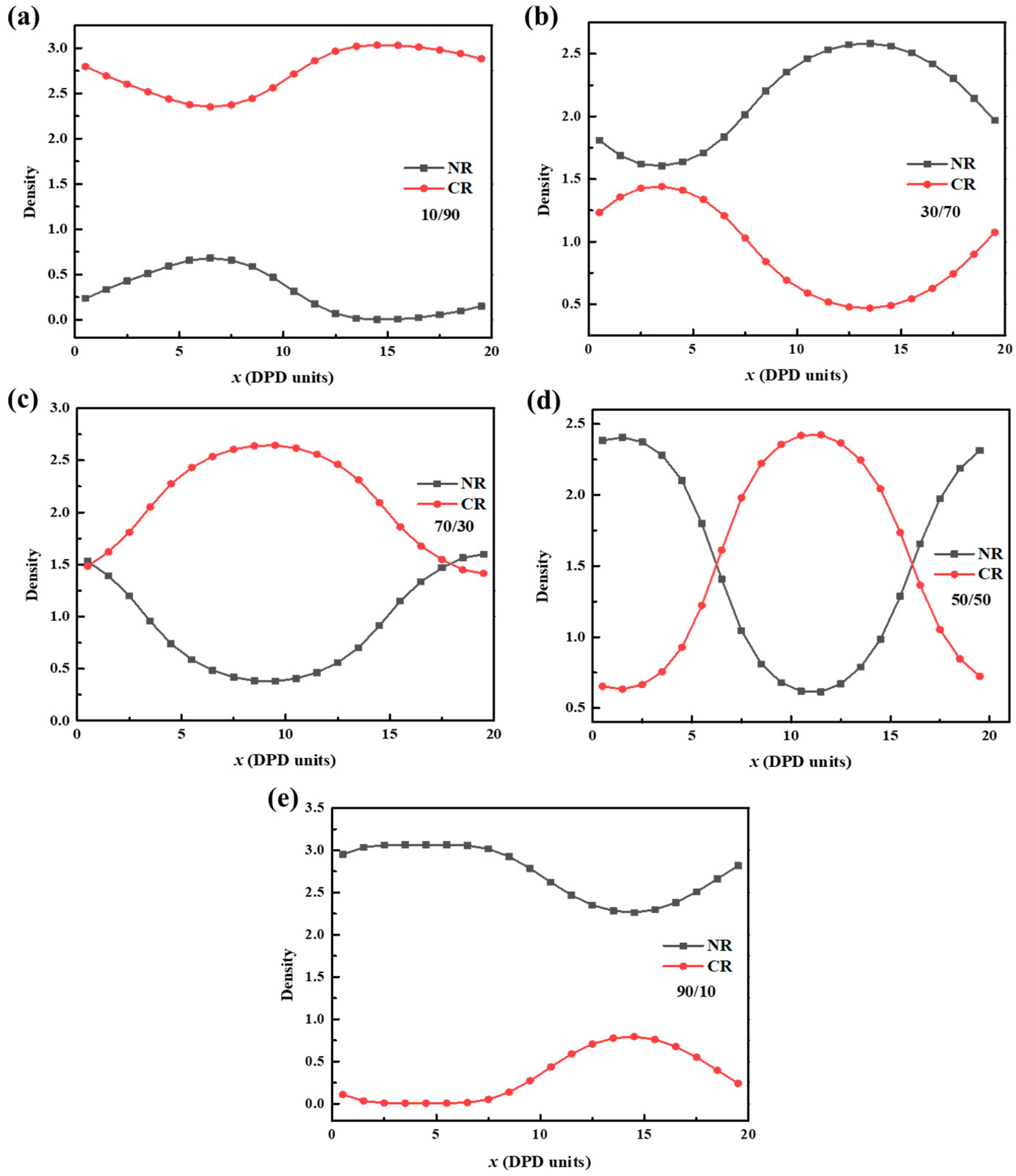
3.3. Density Variation along x-Axis of the Simulation Box
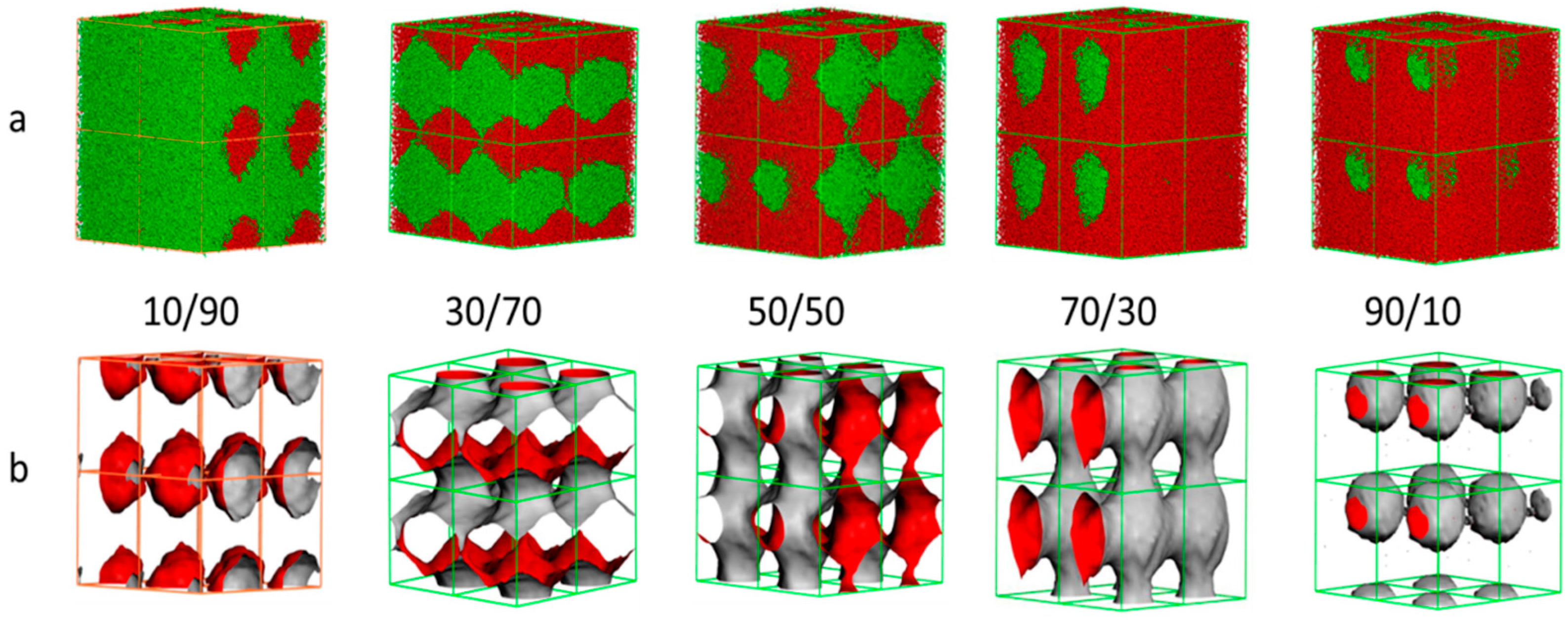
3.4. Morphology of the NR/CR Blends
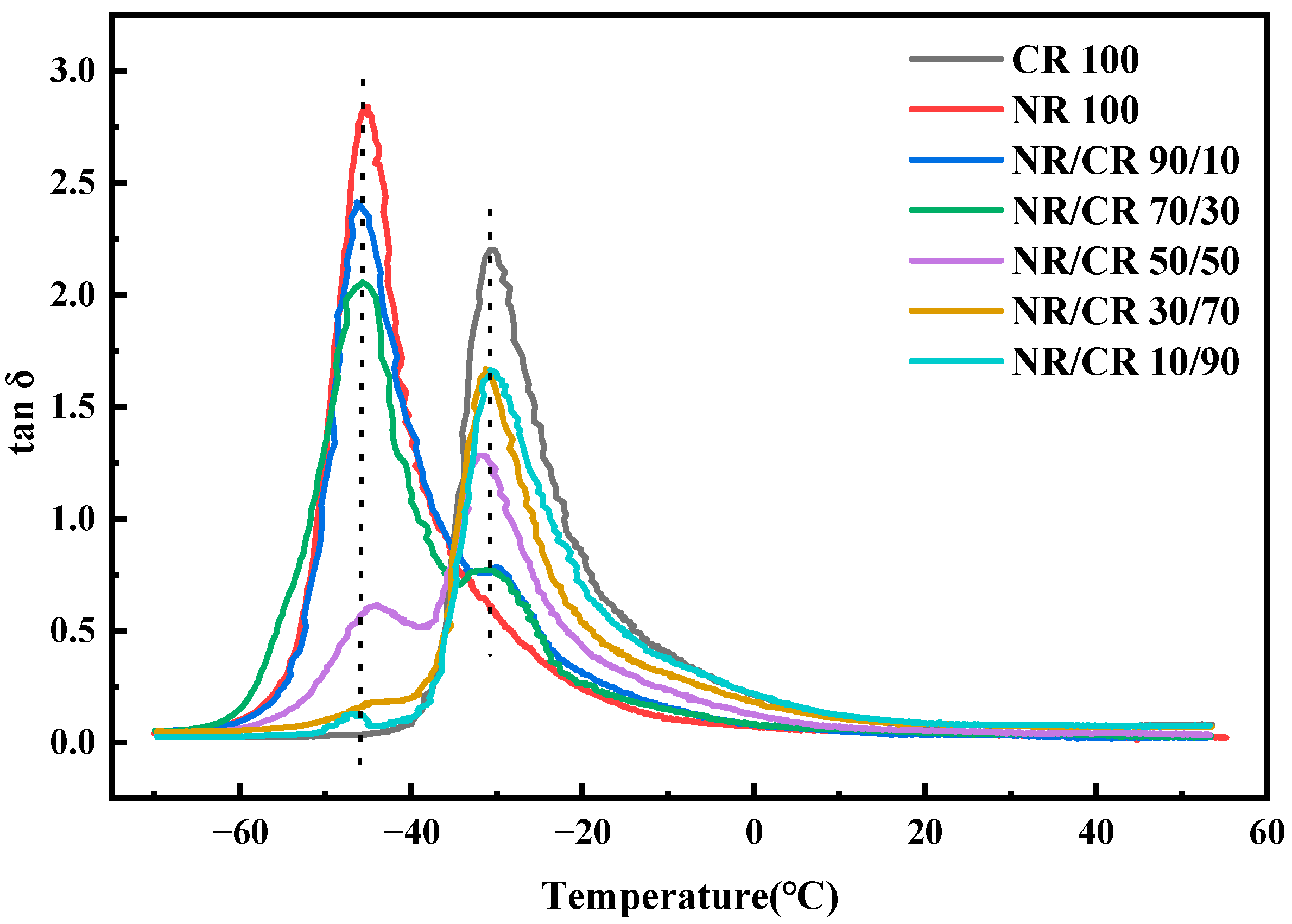
3.5. DMA Curves of the NR/CR Blends
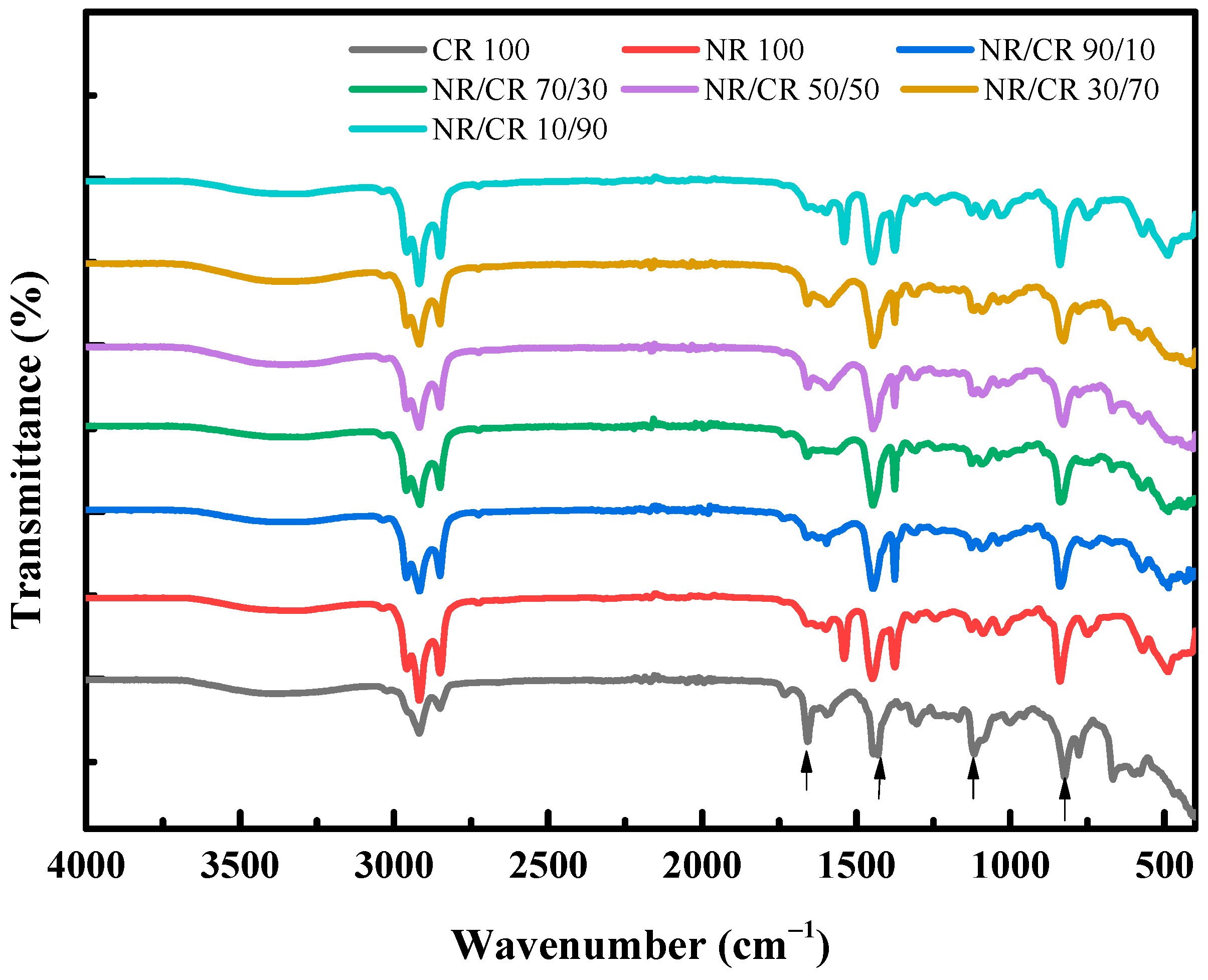
3.6. IR Curves of NR/CR Blends
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Manson, J.A. Polymer Blends and Composites; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Smitthipong, W.; Nardin, M.; Schultz, J.; Suchiva, K. Adhesion and self-adhesion of immiscible rubber blends. Int. J. Adhes. Adhes. 2009, 29, 253–258. [Google Scholar] [CrossRef]
- Jovanović, S.; Samaržija-Jovanović, S.; Marković, G.; Jovanović, V.; Adamović, T.; Marinović-Cincović, M. Ternary NR/BR/SBR rubber blend nanocomposites. J. Thermoplast. Compos. Mater. 2018, 31, 265–287. [Google Scholar] [CrossRef]
- Cesar, M.B.; Borges, F.A.; Bilck, A.P.; Yamashita, F.; Paulino, C.G.; Herculano, R.D. Development and Characterization of Natural Rubber Latex and Polylactic Acid Membranes for Biomedical Application. J. Polym. Environ. 2019, 28, 220–230. [Google Scholar] [CrossRef]
- Lian, T.; Fu, R.; Bao, K.; Lin, J.; Zhang, J. Preparation and properties of NR/EUG composite film. Ind. Crops Prod. 2022, 187, 115423. [Google Scholar] [CrossRef]
- Kaiben, Y.U.; Tianya, L.I.; Jing, Y.; LIN, G. Effect of Curing and Reinforcing Systems on Properties of CR/NR Blends. China Rubber Ind. 2018, 65, 1002–1005. [Google Scholar]
- Das, A. Evaluation of physical and electrical properties of chloroprene rubber and natural rubber blends. Kautsch. Gummi Kunstst. 2005, 58, 230–237. [Google Scholar]
- Sae-oui, P.; Sirisinha, C.; Wantana, T.; Hatthapanit, K. Influence of silica loading on the mechanical properties and resistance to oil and thermal aging of CR/NR blends. J. Appl. Polym. Sci. 2007, 104, 3478–3483. [Google Scholar] [CrossRef]
- Wu, W.-l.; Li, S. Preparation and properties of carbon fiber reinforced chloroprene rubber composites. China Synth. Rubber Ind. 2015, 38, 372–375. [Google Scholar]
- Maya, M.G.; George, S.C.; Jose, T.; Kailas, L.; Thomas, S. Development of a flexible and conductive elastomeric composite based on chloroprene rubber. Polym. Test. 2018, 65, 256–263. [Google Scholar] [CrossRef]
- Huang, Q.; Bai, J. Study on NR/CR blends technology. Cailiao Kaifa Yu Yingyong 2012, 27, 26–29. [Google Scholar]
- Kongvasana, N.; Kositchaiyong, A.; Wimolmala, E.; Sirisinha, C.; Sombatsompop, N. Fly ash particles and precipitated silica as fillers in NR/CR vulcanizates under thermal and thermal-oil ageing. Polym. Adv. Technol. 2011, 22, 1014–1023. [Google Scholar] [CrossRef]
- Malomo, D.; Adesigbin, M.A.; Egharevba, O.; Bello, A.; Adewuyi, S.O.; Idemudia, L. Physico-Mechanical, Solubility and Thermodynamic Studies of Natural Rubber–Neoprene Blends. J. Chem. Soc. Niger. 2019, 44, 718–725. [Google Scholar]
- Nawawi, M.A.; Har, S.L.; Han, C.C. Miscibility of polymer blends comprising poly (ethylene oxide)-epoxidized natural rubber. Int. J. Chem. Eng. Appl. 2012, 3, 410. [Google Scholar] [CrossRef]
- Ganesan, V.; Jayaraman, A. Theory and simulation studies of effective interactions, phase behavior and morphology in polymer nanocomposites. Soft Matter 2014, 10, 13–38. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Yang, J.; Yang, B.; Li, Y.; Wang, S. Study on Compatibility and low temperature Resistance of butadiene rubber/Butadiene Rubber Blends by Molecular Dynamics Simulation. Synth. Rubber Ind. 2020, 43, 291–295. [Google Scholar]
- Jiang, H.; Yue, H.; Zhao, J.Y.; Sha, Q.E. Molecular dynamics simulation of the mechanical properties of NR/TPI. Adv. Mater. Res. 2012, 560–561 Pt 2, 1114–1118. [Google Scholar]
- Ryu, M.S.; Kim, H.G.; Kim, H.Y.; Min, K.S.; Kim, H.J.; Lee, H.M. Prediction of the glass transition temperature and design of phase diagrams of butadiene rubber and styrene-butadiene rubber via molecular dynamics simulations. Phys. Chem. Chem. Phys. 2017, 19, 16498–16506. [Google Scholar] [CrossRef]
- Fan, Z.; Guo, R.; Yang, Z.; Yang, Y.; Liu, X. The Effect of the Co−Blending Process on the Sensing Characteristics of Conductive Chloroprene Rubber/Natural Rubber Composites. Polymers 2022, 14, 3326. Available online: https://0-doi-org.brum.beds.ac.uk/10.3390/polym14163326 (accessed on 7 January 2023.). [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Ndoro, T.V.; Böhm, M.C.; Müller-Plathe, F. Interface and interphase dynamics of polystyrene chains near grafted and ungrafted silica nanoparticles. Macromolecules 2012, 45, 171–179. [Google Scholar] [CrossRef]
- Voyiatzis, E.; Rahimi, M.; Müller-Plathe, F.; Böhm, M.C. How thick is the polymer interphase in nanocomposites? Probing it by local stress anisotropy and gas solubility. Macromolecules 2014, 47, 7878–7889. [Google Scholar] [CrossRef]
- Brown, D.; Mele, P.; Marceau, S.; Albérola, N.D. A molecular dynamics study of a model nanoparticle embedded in a polymer matrix. Macromolecules 2003, 36, 1395–1406. [Google Scholar] [CrossRef]
- Eslami, H.; Rahimi, M.; Müller-Plathe, F. Molecular dynamics simulation of a silica nanoparticle in oligomeric poly (methyl methacrylate): A model system for studying the interphase thickness in a polymer–nanocomposite via different properties. Macromolecules 2013, 46, 8680–8692. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applications—Overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; Van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Koelman, J.M.V.A.; Hoogerbrugge, P.J. Dynamic simulations of hard-sphere suspensions under steady shear. Europhys. Lett. 1993, 21, 363. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19, 155. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Espanol, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995, 30, 191. [Google Scholar] [CrossRef]
- Pryamitsyn, V.; Ganesan, V. Mechanisms of steady-shear rheology in polymer-nanoparticle composites. J. Rheol. 2006, 50, 655–683. [Google Scholar] [CrossRef]
- Pryamitsyn, V.; Ganesan, V. Origins of Linear Viscoelastic Behavior of Polymer−Nanoparticle Composites. Macromolecules 2006, 39, 844–856. [Google Scholar] [CrossRef]
- Espanol, P.; Warren, P.B. Perspective: Dissipative particle dynamics. J. Chem. Phys. 2017, 146, 150901. [Google Scholar] [CrossRef]
- Li, Z.; Bian, X.; Tang, Y.H.; Karniadakis, G.E. A dissipative particle dynamics method for arbitrarily complex geometries. J. Comput. Phys. 2018, 355, 534–547. [Google Scholar] [CrossRef]
- Shiling, Y.; Heng, Z.; Dongju, Z. Molecular Simulation: Theory and Experiment; Chemical Industry Press: Beijing, China, 2016; pp. 155–166. [Google Scholar]
- Saad, A.L.; El-Sabbagh, S. Compatibility studies on some polymer blend systems by electrical and mechanical techniques. J. Appl. Polym. Sci. 2001, 79, 60–71. [Google Scholar] [CrossRef]
- Ahn, W.S.; Park, D.R. Effects of CR Contents on Rubber Vulcanization and Mechanical Properties of NR/CR Blends. Elastomers Compos. 2011, 46, 152–157. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System Number | NR/CR (Mass Ratio) | Number of Chain | Density (g/cm3) | Box Length (Å) |
|---|---|---|---|---|
| 1 | 90/10 | 32NR/1CR | 0.961 | 44.62 |
| 2 | 70/30 | 8NR/1CR | 1.203 | 30.07 |
| 3 | 50/50 | 7NR/2BR | 1.085 | 31.46 |
| 4 | 30/70 | 3NR/2BR | 1.147 | 27.45 |
| 5 | 90/10 | 3NR/8NR | 1.209 | 39.20 |
| Sample | Relative Molecular Weight | Limiting Characteristic Ratio | Particle Number |
|---|---|---|---|
| NR | 300,000 | 5.45 | 810 |
| CR | 120,000 | 5.44 | 263 |
| Sample Reagent | NR/CR 0/100 | NR/CR 10/90 | NR/CR 30/70 | NR/CR 50/50 | NR/CR 30/70 | NR/CR 100/0 |
|---|---|---|---|---|---|---|
| NR | 0 | 10 | 30 | 50 | 30 | 100 |
| CR | 100 | 90 | 70 | 50 | 70 | 0 |
| Stearic Acid | 2 | 2 | 2 | 2 | 2 | 2 |
| Zinc Oxide | 5 | 5 | 5 | 5 | 5 | 5 |
| Promoter CZ | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Magnesium Oxide | 5 | 4 | 3 | 2 | 1 | 0.5 |
| Sulfur | 0 | 0.5 | 1 | 1.5 | 2 | 2.5 |
| NR/CR | 10/90 | 30/70 | 50/50 | 70/30 | 90/10 |
|---|---|---|---|---|---|
| (%) | 12.39 | 36.22 | 56.98 | 75.31 | 92.37 |
| (%) | 87.61 | 63.78 | 43.02 | 24.69 | 7.63 |
| CEDNR (×108 J.m−3) | 2.68 | 2.68 | 2.68 | 2.68 | 2.68 |
| CEDCR (×108 J.m−3) | 3.50 | 3.50 | 3.50 | 3.50 | 3.50 |
| CEDmix (×108 J.m−3) | 3.36 | 3.07 | 2.95 | 2.80 | 2.63 |
| (×106 J.m−3) | 4.19 | 13.9 | 8.62 | 9.20 | 11.7 |
| V(Å3) | 60,239.85 | 20,697.66 | 31,153.17 | 27,181.1 | 88,840.9 |
| Vmono (×10−5 m3/mol) | 7.26 | 7.33 | 7.50 | 7.61 | 7.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Yuan, X.; Jiang, R.; Liao, J.; Yu, R.; Chen, Y.; Liao, L. Molecular Dynamic and Dissipative Particle Dynamic Simulation on the Miscibility of NR/CR Blends. Polymers 2023, 15, 856. https://doi.org/10.3390/polym15040856
Ma Y, Yuan X, Jiang R, Liao J, Yu R, Chen Y, Liao L. Molecular Dynamic and Dissipative Particle Dynamic Simulation on the Miscibility of NR/CR Blends. Polymers. 2023; 15(4):856. https://doi.org/10.3390/polym15040856
Chicago/Turabian StyleMa, Yanbin, Xiaoqin Yuan, Ruifeng Jiang, Jianhe Liao, Rentong Yu, Yongping Chen, and Lusheng Liao. 2023. "Molecular Dynamic and Dissipative Particle Dynamic Simulation on the Miscibility of NR/CR Blends" Polymers 15, no. 4: 856. https://doi.org/10.3390/polym15040856