

Sorption of CO2, CH4 and Their Mixtures in Amorphous Poly(2,6-dimethyl-1,4-phenylene)oxide (PPO)

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Theoretical Background
2.1. The NETGP-NR Model
2.2. Solution Diffusion Model of Small Molecules in Polymers
3. Materials and Methods
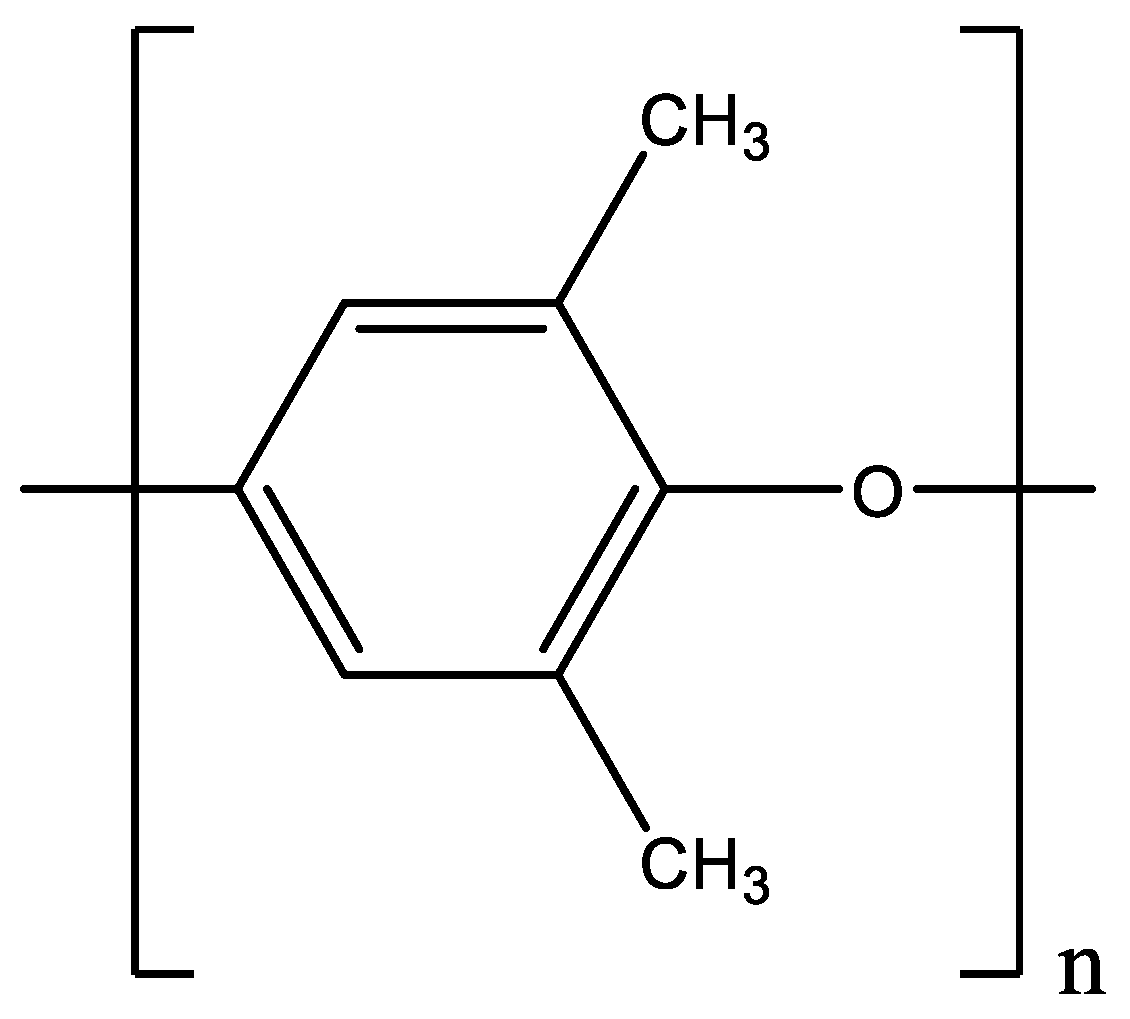
3.1. Materials
3.2. Methods
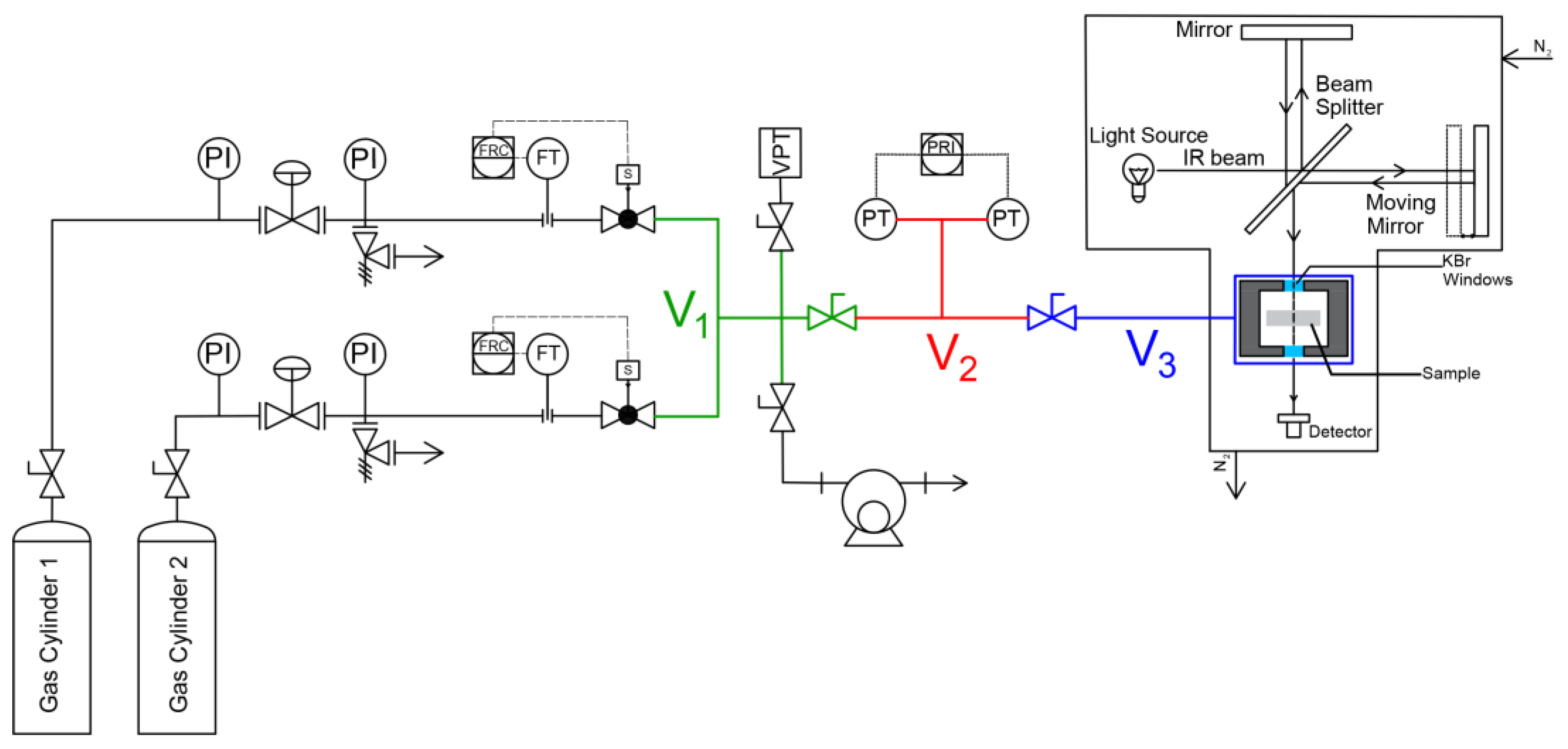
3.2.1. Closed Volume–Variable Pressure Apparatus
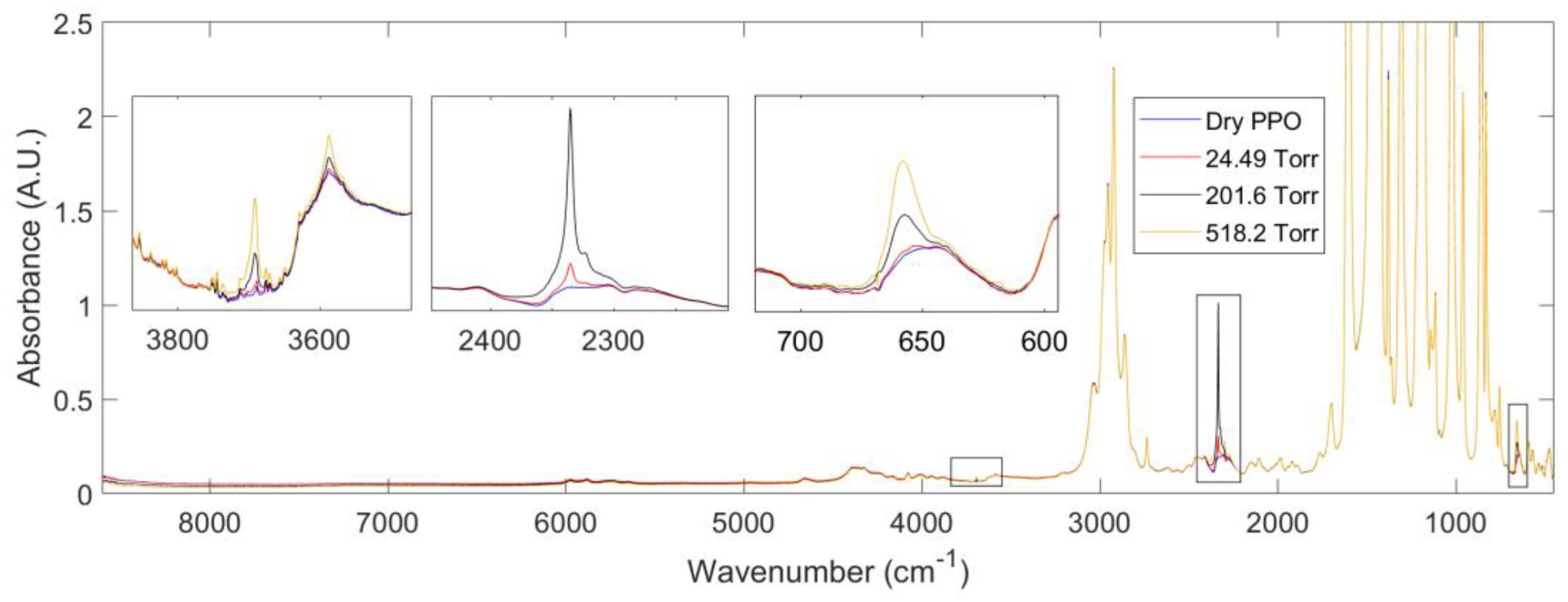
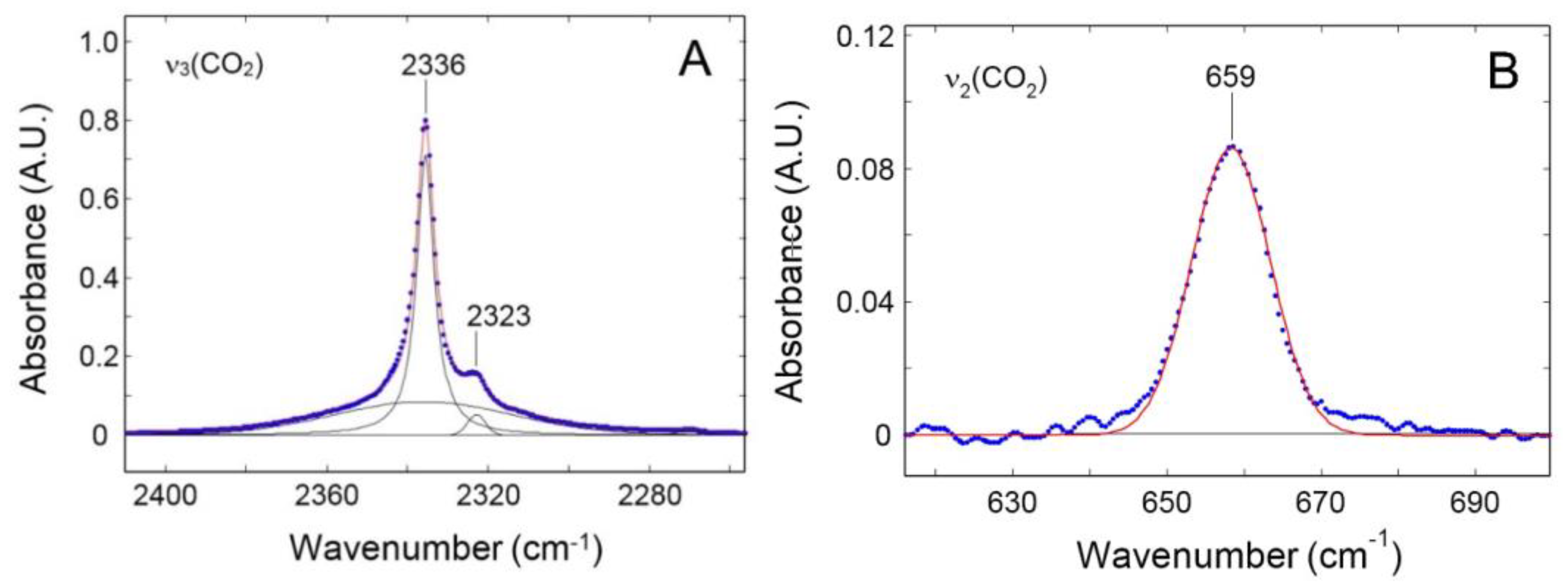
3.2.2. FTIR-Spectroscopy in the Transmission Mode
3.2.3. Performing a Sorption Experiment
4. Results and Discussion
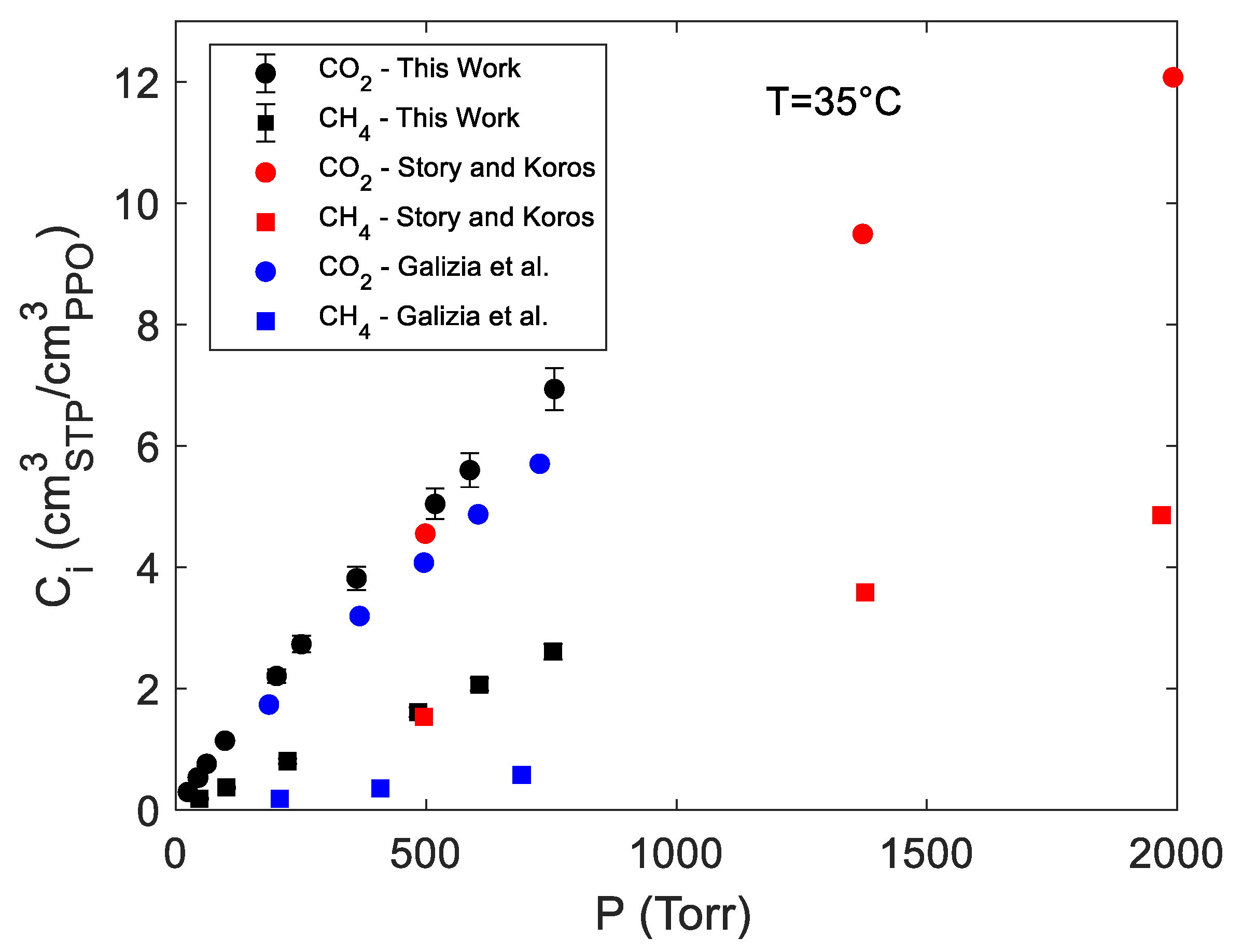
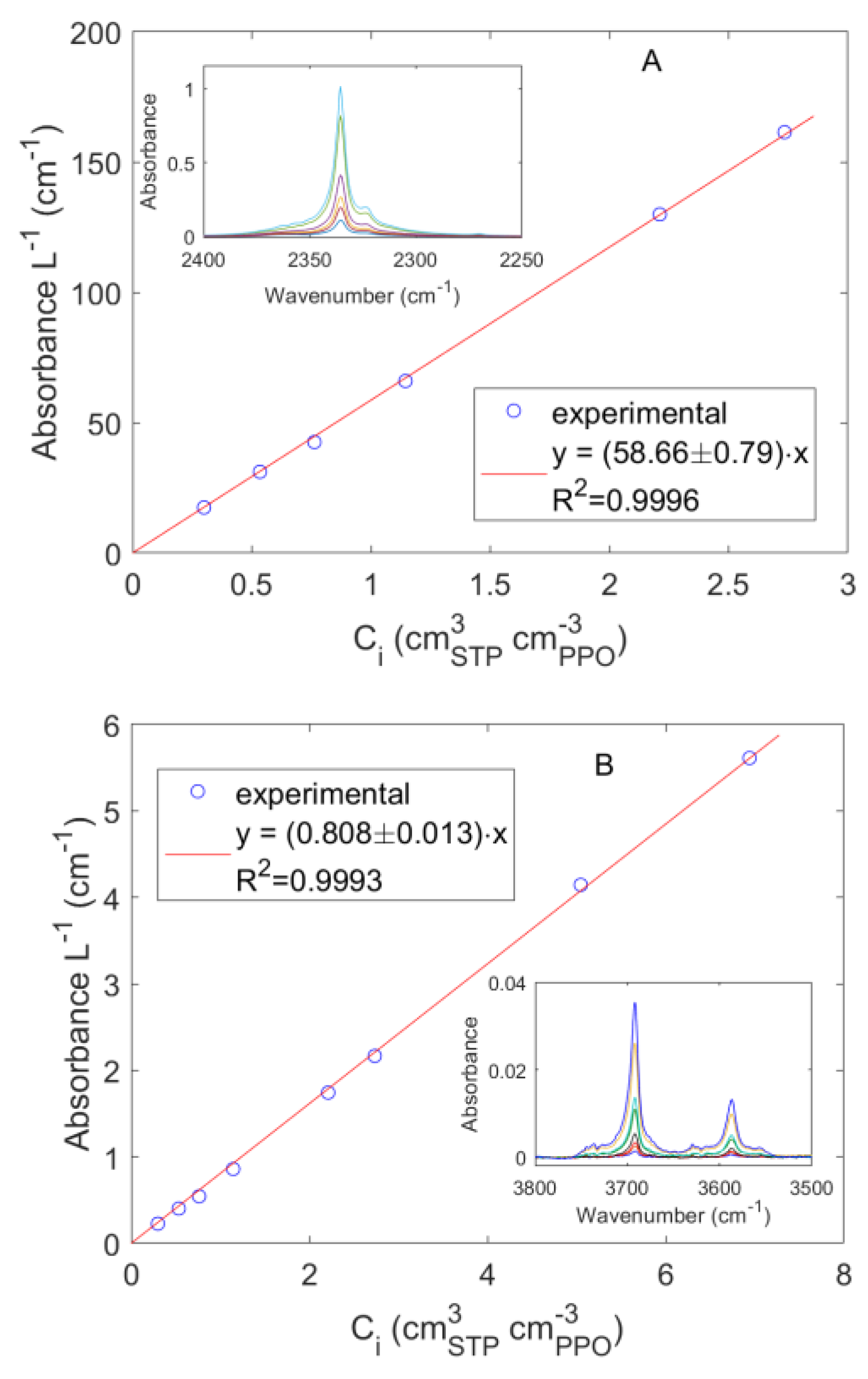
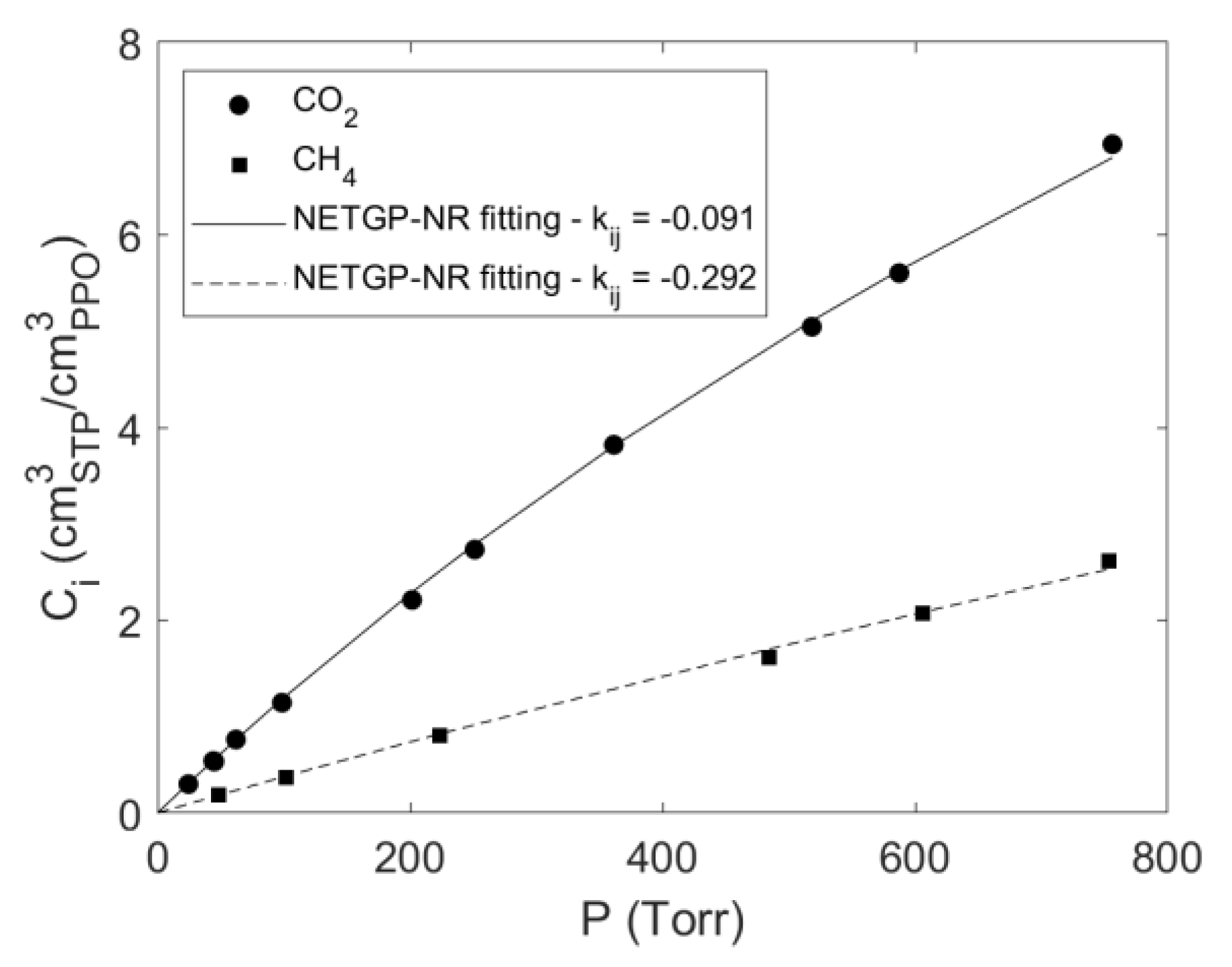
4.1. Sorption of Pure CO2 and CH4 in PPO
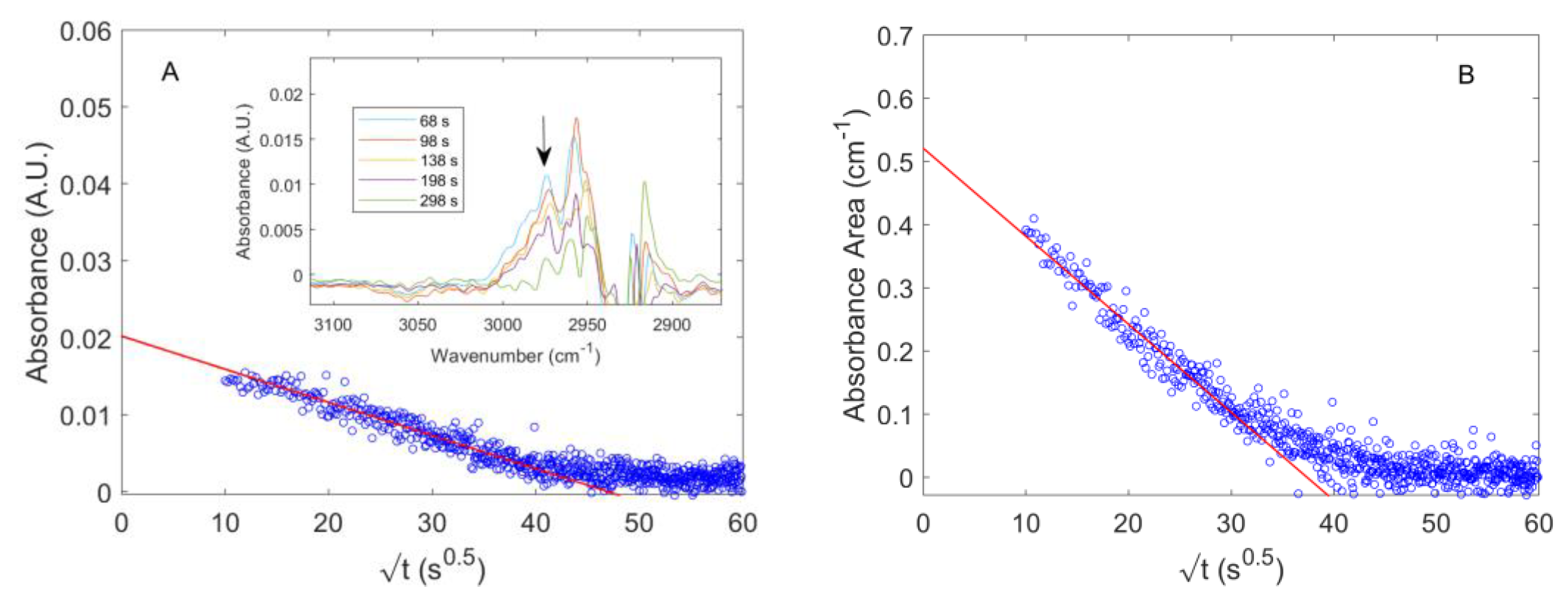
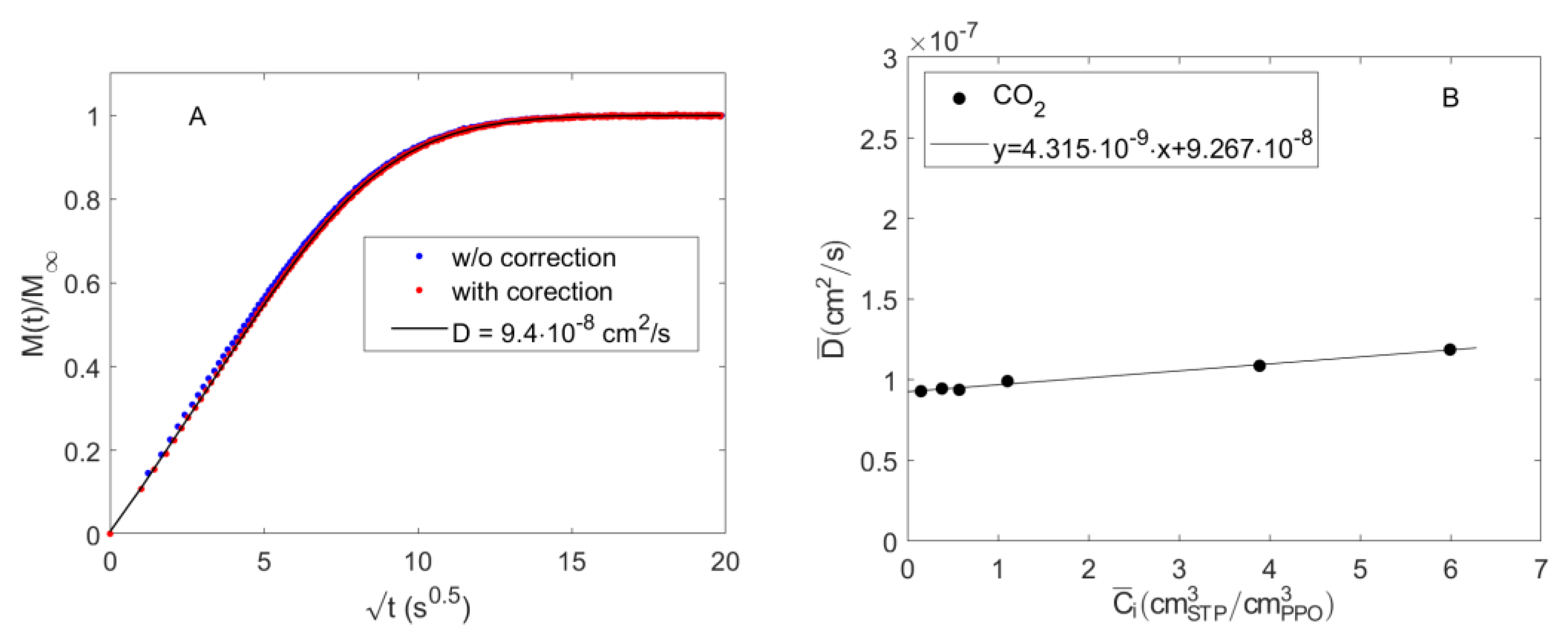
4.2. Diffusion of Pure CO2 in PPO
4.3. Modelling Sorption of Light Gases in PPO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(Jmol−1) | (Jmol−1K−1) | (cm3g−1) | Ref. | ||
|---|---|---|---|---|---|
| CO2 | 3468.4 | −4.5855 | 0.79641 | 0.909 | [38] |
| CH4 | 1956.2 | −0.9181 | 2.12519 | 0.961 | [38] |
| PPO | 5320 | 3.440 | 0.862 | 0.748 | [39] |
| CO2 | CH4 | Ref. |
|---|---|---|
| −0.091 | −0.292 | This Work |
| −0.087 | −0.278 | [16] |
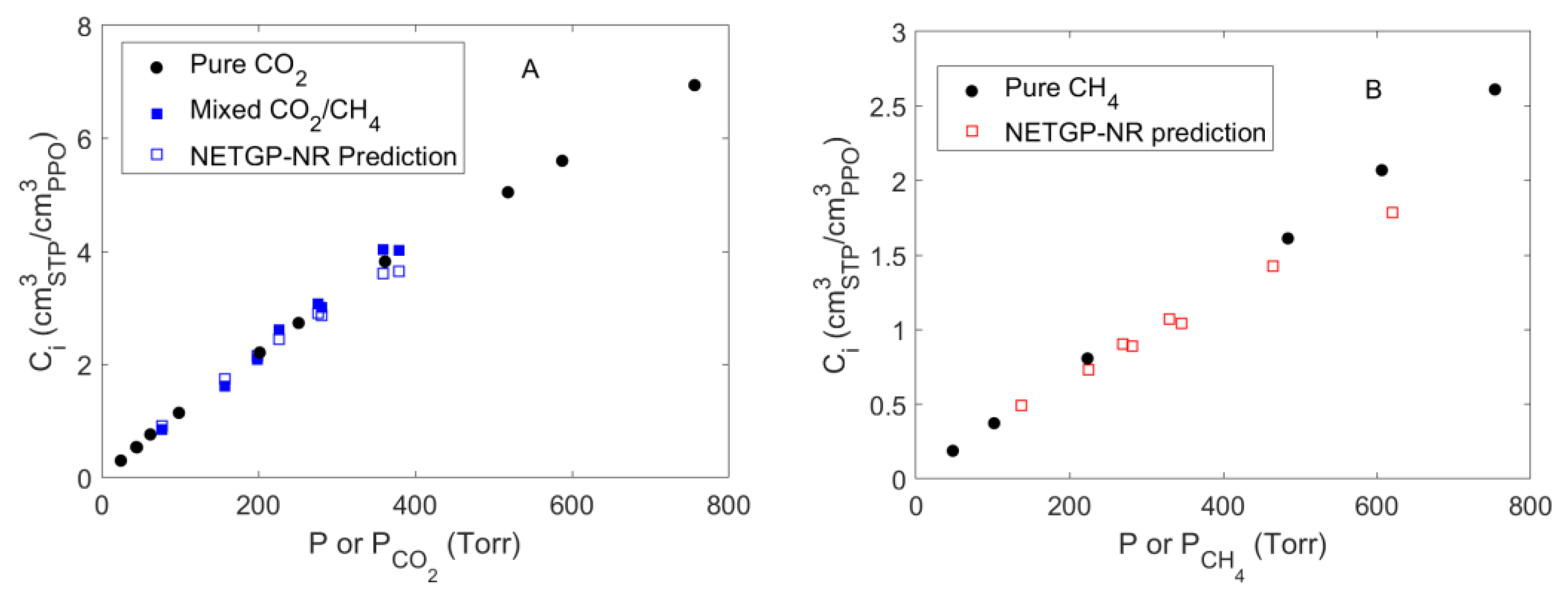
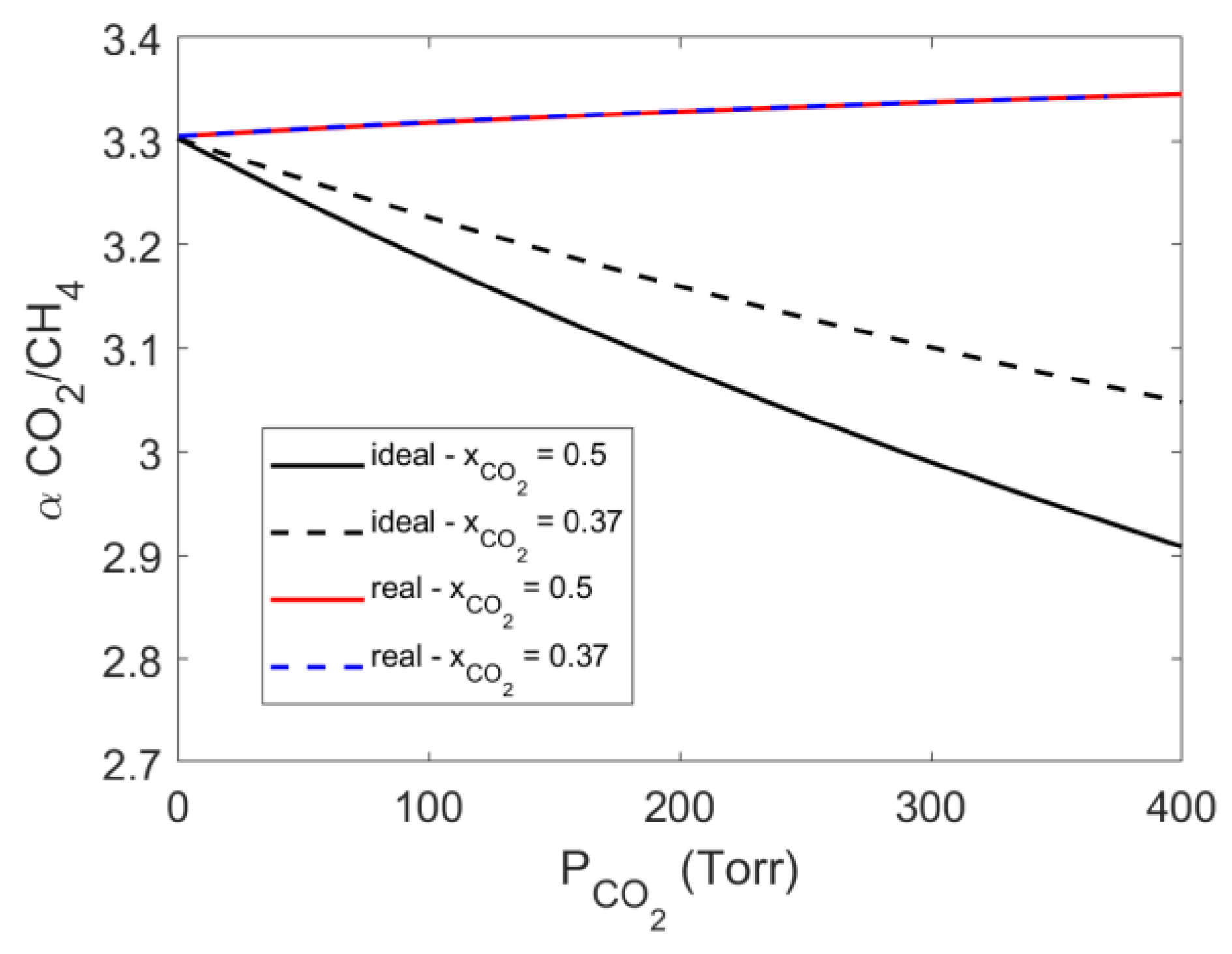
4.4. Mixed Gas Sorption in PPO
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Symbols
| gas phase contribution to the overall absorbance spectrum at time t | |
| gas phase contribution to the overall absorbance spectrum at sorption equilibrium state | |
| C | molar concentration of the pure penetrant |
| molar concentration of the pure penetrant in the polymer phase at the downstream side in a permeation experiment | |
| molar concentration of the pure penetrant in the polymer phase at the upstream side in a permeation experiment | |
| concentration of component i within the polymer phase in a sorption experiment | |
| average concentration of component i within the polymer phase in a sorption experiment | |
| initial uniform concentration of component i within the polymer phase in a sorption experiment | |
| equilibrium concentration of component i within the polymer phase in a sorption experiment | |
| molar gas phase concentration at time t | |
| molar gas phase concentration at sorption equilibrium state | |
| D | mutual diffusion coefficient |
| Di | mutual diffusion coefficient of the component i |
| effective diffusivity coefficient | |
| defined in Equation (17) and calculated from Equation (18) | |
| mean field lattice fluid binary interactional parameter between the species i and j | |
| the swelling factor associated with the polymer-penetrant couple | |
| defined as | |
| L | membrane thickness in a sorption experiment |
| m | number of components |
| mass of absorbed penetrant at time t in a sorption experiment | |
| equilibrium mass of absorbed penetrant in a sorption experiment | |
| partial pressure of the component i at the upstream side in a permeation test | |
| P | pressure |
| scaled pressure of the pure component or of the mixture | |
| mean steady-state permeability | |
| steady-state permeability of component i | |
| q | average number of lattice contacts per molecule in the mixture and it is equal to |
| qi | number of external contacts made by one molecule of species i |
| R | universal gas constant |
| r | average number of sites occupied by one molecule in the mixture and it is equal to |
| ri | number of sites occupied by one molecule of species i |
| S | apparent solubility coefficient |
| solubility coefficient of component i in the polymer–penetrant mixture | |
| t | time |
| T | temperature |
| Tg | glass-to-rubber transition temperature |
| scaled temperature of the pure component or of the mixture | |
| scaled temperature of the pure component i in the mixture | |
| the temperature-independent contribution to the close packed specific volume of the pure component i | |
| close packed specific volume of the pure component i | |
| scaled lattice fluid volume of the pure components or of the mixture and it is equal to | |
| the ratio of molar surface to molar volume of component i | |
| xi | molar fraction of species i |
| molar fraction of species i at Pseudo-Equilibrium of phase condition | |
| z | lattice coordination number |
| Greek letters | |
| solubility selectivity defined by Equation (15) | |
| αid | ideal selectivity |
| solubility contribution to the ideal selectivity | |
| diffusivity contribution to the ideal selectivity | |
| non-random factor | |
| exchanged interaction energy determined by breaking one i-ì and one j-j interaction for the formation of two i–j interactions in the lattice | |
| flexibility factor of species i | |
| mean field interaction energy within the mixture | |
| mean field interaction energy per molar segment | |
| mean interaction energy per molar segment, enthalpic contribution | |
| mean interaction energy per molar segment, entropic contribution | |
| surface fraction of component i, | |
| molar chemical potential of the species i | |
| equilibrium molar chemical potential of the species i | |
| non-equilibrium molar chemical potential of the species i within the glassy polymer–penetrant phase | |
| non-equilibrium polymer mass density within the mixture | |
| closed-packed density of the polymer–penetrant mixture | |
| polymer mass density induced by elastic instantaneous swelling | |
| the value of the unpenetrated polymer mass density right before the start of the sorption process | |
| scaled lattice fluid density of a pure component or of the mixture ( | |
| “close-packed” volumetric fraction of component i | |
| polymer mass fraction | |
References
- Eurostat. Available online: https://Ec.Europa.Eu/Eurostat/ (accessed on 13 October 2022).
- Short-Term Energy Outlook. 2022. Available online: https://www.Eia.Gov/Todayinenergy/Detail.Php?Id=53839 (accessed on 26 October 2022).
- Baker, R.W.; Lokhandwala, A. Natural Gas Processing with Membranes: An Overview. Ind. Eng. Chem. 2008, 47, 2109–2121. [Google Scholar] [CrossRef]
- White, L.S. Effect of Operating Environment on Membrane Performance. Curr. Opin. Chem. Eng. 2020, 28, 105–111. [Google Scholar] [CrossRef]
- Genduso, G.; Ghanem, B.S.; Pinnau, I. Experimental Mixed-Gas Permeability, Sorption and Diffusion of CO2-CH4 Mixtures in 6FDA-MPDA Polyimide Membrane: Unveiling the Effect of Competitive Sorption on Permeability Selectivity. Membranes 2019, 9, 10. [Google Scholar] [CrossRef]
- Pourafshari Chenar, M.; Soltanieh, M.; Matsuura, T.; Tabe-Mohammadi, A.; Khulbe, K.C. The Effect of Water Vapor on the Performance of Commercial Polyphenylene Oxide and Cardo-Type Polyimide Hollow Fiber Membranes in CO2/CH4 Separation Applications. J. Membr. Sci. 2006, 285, 265–271. [Google Scholar] [CrossRef]
- Story, B.J.; Koros, W.J. Sorption of CO2/CH4 Mixtures in Poly(Phenylene Oxide) and a Carboxylated Derivative. J. Appl. Polym. Sci. 1991, 42, 2613–2626. [Google Scholar] [CrossRef]
- Sanders, E.S.; Koros, W.J.; Hopfenberg, H.B.; Stannett, V.T. Mixed Gas Sorption in Glassy Polymers: Equipment Design Considerations and Preliminary Results. J. Membr. Sci. 1983, 13, 161–174. [Google Scholar] [CrossRef]
- Ricci, E.; Benedetti, F.M.; Noto, A.; Merkel, T.C.; Jin, J.; De Angelis, M.G. Enabling Experimental Characterization and Prediction of Ternary Mixed-Gas Sorption in Polymers: C2H6/CO2/CH4 in PIM-1. Chem. Eng. J. 2021, 426, 130715. [Google Scholar] [CrossRef]
- Genduso, G.; Litwiller, E.; Ma, X.; Zampini, S.; Pinnau, I. Mixed-Gas Sorption in Polymers via a New Barometric Test System: Sorption and Diffusion of CO2-CH4 Mixtures in Polydimethylsiloxane (PDMS). J. Membr. Sci. 2019, 577, 195–204. [Google Scholar] [CrossRef]
- Loianno, V.; Mensitieri, G.; Baldanza, A.; Scherillo, G.; Musto, P. Combining FTIR Spectroscopy and Pressure-Decay Techniques to Analyze Sorption Isotherms and Sorption Kinetics of Pure Gases and Their Mixtures in Polymers: The Case of CO2 and CH4 Sorption in Polydimethylsiloxane. J. Membr. Sci. 2022, 652, 120445. [Google Scholar] [CrossRef]
- Hong, S.U.; Barbari, T.A.; Sloan, J.M. Multicomponent Diffusion of Methyl Ethyl Ketone and Toluene in Polyisobutylene from Vapor Sorption FTIR-ATR Spectroscopy. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 337–344. [Google Scholar] [CrossRef]
- Shade, D.; Bout, B.W.S.; Sholl, D.S.; Walton, K.S. Opening the Toolbox: 18 Experimental Techniques for Measurement of Mixed Gas Adsorption. Ind. Eng. Chem. Res. 2022, 61, 2367–2391. [Google Scholar] [CrossRef]
- Minelli, M.; Campagnoli, S.; De Angelis, M.G.; Doghieri, F.; Sarti, G.C. Predictive Model for the Solubility of Fluid Mixtures in Glassy Polymers. Macromolecules 2011, 44, 4852–4862. [Google Scholar] [CrossRef]
- Minelli, M.; Sarti, G.C. Modeling Mass Transport in Dense Polymer Membranes: Cooperative Synergy among Multiple Scale Approaches. Curr. Opin. Chem. Eng. 2020, 28, 43–50. [Google Scholar] [CrossRef]
- Baldanza, A.; Loianno, V.; Mensitieri, G.; Scherillo, G. Predictive Approach for the Solubility and Permeability of Binary Gas Mixtures in Glassy Polymers Based on an NETGP-NRHB Model. Ind. Eng. Chem. Res. 2022, 61, 3439–3456. [Google Scholar] [CrossRef]
- Panayiotou, C.; Pantoula, M.; Stefanis, E.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 1. Pure Fluids. Ind. Eng. Chem. Res. 2004, 43, 6592–6606. [Google Scholar] [CrossRef]
- Panayiotou, C.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 2. Multicomponent Mixtures. Ind. Eng. Chem. Res. 2007, 46, 2628–2636. [Google Scholar] [CrossRef]
- Mensitieri, G.; Scherillo, G.; Panayiotou, C.; Musto, P. Towards a Predictive Thermodynamic Description of Sorption Processes in Polymers: The Synergy between Theoretical EoS Models and Vibrational Spectroscopy. Mater. Sci. Eng. R Rep. 2020, 140, 100525. [Google Scholar] [CrossRef]
- Neau, E. A consistent method for phase equilibrium calculation using the Sanchez–Lacombe lattice–fluid equation-of-state. Fluid Phase Equilibria 2002, 203, 133–140. [Google Scholar] [CrossRef]
- Von Konigslow, K.; Park, C.B.; Thompson, R.B. Polymeric Foaming Predictions from the Sanchez-Lacombe Equation of State: Application to Polypropylene-Carbon Dioxide MixturesPhys. Rev. Appl. 2017, 8, 044009. [Google Scholar] [CrossRef]
- Von Konigslow, K.; Park, C.B.; Thompson, R.B. Application of a Constant Hole Volume Sanchez–Lacombe Equation of State to Mixtures Relevant to Polymeric Foaming. Soft Matter 2018, 14, 4603–4614. [Google Scholar] [CrossRef]
- Baldanza, A.; Loianno, V.; Mensitieri, G.; Panayiotou, C.; Scherillo, G. On the Thermodynamic Consistency of Non-Random Hydrogen Bonding Lattice-Fluid Model for Multicomponent Mixtures. Fluid Phase Equilibria 2022, 553, 113302. [Google Scholar] [CrossRef]
- Doghieri, F.; Sarti, G.C. Nonequilibrium Lattice Fluids: A Predictive Model for the Solubility in Glassy Polymers. Macromolecules 1996, 29, 7885–7896. [Google Scholar] [CrossRef]
- Sarti, G.C.; Doghieri, F. Predictions of the Solubility of Gases in Glassy Polymers Based on the NELF Model. Chem. Eng. Sci. 1998, 53, 3435–3447. [Google Scholar] [CrossRef]
- Fredenslund, A.; Jones, R.L.; Prausnitz, J.M. Group-Contribution Estimation of Activity Coefficients in Nonideal Liquid Mixtures. AIChE J. 1975, 21, 1086–1099. [Google Scholar] [CrossRef]
- Graham, T.L.V. On the Absorption and Dialytic Separation of Gases by Colloid Septa. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1866, 32, 401–420. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Loianno, V.; Mensitieri, G. A Novel Dynamic Method for the Storage of Calibration Gas Mixtures Based on Thermal Mass Flow Controllers. Meas. Sci. Technol. 2022, 33, 065017. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, SRD 69. Available online: https://Webbook.Nist.Gov/Chemistry/Fluid/ (accessed on 21 July 2022).
- Galizia, M.; Daniel, C.; Fasano, G.; Guerra, G.; Mensitieri, G. Gas Sorption and Diffusion in Amorphous and Semicrystalline Nanoporous Poly(2,6-Dimethyl-1,4-Phenylene)Oxide. Macromolecules 2012, 45, 3604–3615. [Google Scholar] [CrossRef]
- Story, B.J.; Koros, W.J. Sorption and Transport of CO2 and CH4 in Chemically Modified Poly(Phenylene Oxide). J. Membr. Sci. 1992, 67, 191–210. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Vincent, M.F.; Bright, F.V.; Liotta, C.L.; Eckert, C.A. Specific Intermolecular Interaction of Carbon Dioxide with Polymers. J. Am. Chem. Soc. 1996, 118, 1729–1736. [Google Scholar] [CrossRef]
- Turner, J.J. Bandwidths. In Handbook of Vibrational Spectroscopy; Chalmers, J., Griffiths, P., Eds.; John Wiley & Son: Chichester, UK, 2002; Volume 1, pp. 101–127. [Google Scholar]
- Clarke, J.H.R. Band Shapes and Molecular Dynamics in Liquids. In Advances in Infrared and Raman Spectroscopy; Clarke, J.H.R., Hester, R.E., Eds.; Hey Den: London, UK, 1978; Volume 4, pp. 109–193. [Google Scholar]
- Li, C.; Xin, Q. FT-IR Spectroscopic Investigation of Methane Adsorption on Cerium Oxide. J. Phys. Chem. 1992, 96, 7714–7718. [Google Scholar] [CrossRef]
- Yoshida, H.; Yamazaki, T.; Ozawa, S. IR Spectra of CH4 Physisorbed on an Active Carbon at Low Temperature. J. Colloid Interface Sci. 2000, 224, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Tsivintzelis, I.; Spyriouni, T.; Economou, I.G. Modeling of Fluid Phase Equilibria with Two Thermodynamic Theories: Non-Random Hydrogen Bondin g (NRHB) and Statistical Associating Fluid Theory (SAFT). Fluid Phase Equilibria 2007, 253, 19–28. [Google Scholar] [CrossRef]
- Musto, P.; Loianno, V.; Scherillo, G.; La Manna, P.; Galizia, M.; Guerra, G.; Mensitieri, G. Benzene-Induced Crystallization of PPO: A Combined Thermodynamic and Vibrational Spectroscopy Study. Ind. Eng. Chem. Res. 2020, 59, 5402–5411. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loianno, V.; Baldanza, A.; Scherillo, G.; Musto, P.; Mensitieri, G. Sorption of CO2, CH4 and Their Mixtures in Amorphous Poly(2,6-dimethyl-1,4-phenylene)oxide (PPO). Polymers 2023, 15, 1144. https://doi.org/10.3390/polym15051144
Loianno V, Baldanza A, Scherillo G, Musto P, Mensitieri G. Sorption of CO2, CH4 and Their Mixtures in Amorphous Poly(2,6-dimethyl-1,4-phenylene)oxide (PPO). Polymers. 2023; 15(5):1144. https://doi.org/10.3390/polym15051144
Chicago/Turabian StyleLoianno, Valerio, Antonio Baldanza, Giuseppe Scherillo, Pellegrino Musto, and Giuseppe Mensitieri. 2023. "Sorption of CO2, CH4 and Their Mixtures in Amorphous Poly(2,6-dimethyl-1,4-phenylene)oxide (PPO)" Polymers 15, no. 5: 1144. https://doi.org/10.3390/polym15051144
APA StyleLoianno, V., Baldanza, A., Scherillo, G., Musto, P., & Mensitieri, G. (2023). Sorption of CO2, CH4 and Their Mixtures in Amorphous Poly(2,6-dimethyl-1,4-phenylene)oxide (PPO). Polymers, 15(5), 1144. https://doi.org/10.3390/polym15051144