
Design of New Dispersants Using Machine Learning and Visual Analytics

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset of Potential Dispersants: Data Compilation and Curation
2.2. Computational Representations of Molecules
2.3. Model Based on Structural Similarity
2.3.1. Molecular Descriptor Selection
2.3.2. Evaluation of New Molecules
2.4. Bayesian Regression Model
- The inclusion proportions of all variables in the model are calculated.
- The response vector is randomly permuted, thus breaking the relationship between the covariates and the response. The model is retrained with this new response vector, and the variable inclusion proportions are recalculated. These are considered as the existing inclusion proportions in case there is no association between the variables and the response. This step is repeated several times to create a distribution of inclusion proportions that we call the null distribution. This distribution will allow us to see whether the value of the proportion of inclusion of each observed variable is too high with respect to the expected distribution in the event that there is no association. If so, the variable in question would be considered important.
- In particular, Bleich et al. [60] described three variable inclusion rules described therein. Out of these three rules, the one with the lowest mean squared error was selected using cross-validation.
2.4.1. Evaluation of New Molecules
- Mean predictive dispersancy: the expected value of the posterior predictive distribution defined as ;
- Predictive standard deviation: the standard deviation of the posterior predictive distribution defined as ;
- The expected improvement: if we denote the best dispersancy value observed within the training set as , the expected improvement of a candidate molecule with features x is defined as . As argued in [61], expected improvement balances exploration and exploitation and is our chosen metric to decide which targets will end up being synthesized;
- Probability of improvement: the probability that a candidate molecule with features x has a dispersancy value higher than that is , where is the indicator function.
2.5. Applicability Domain
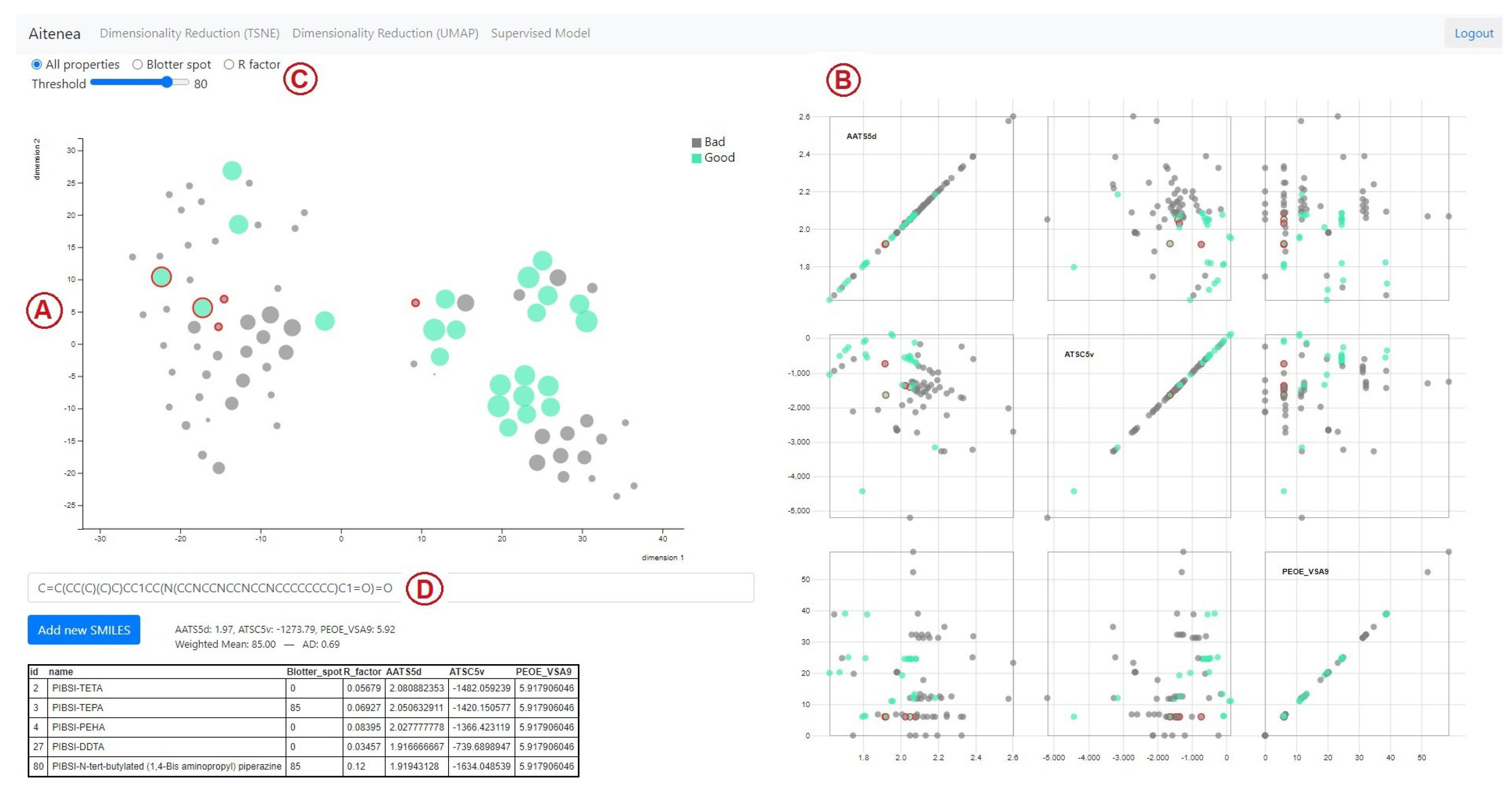
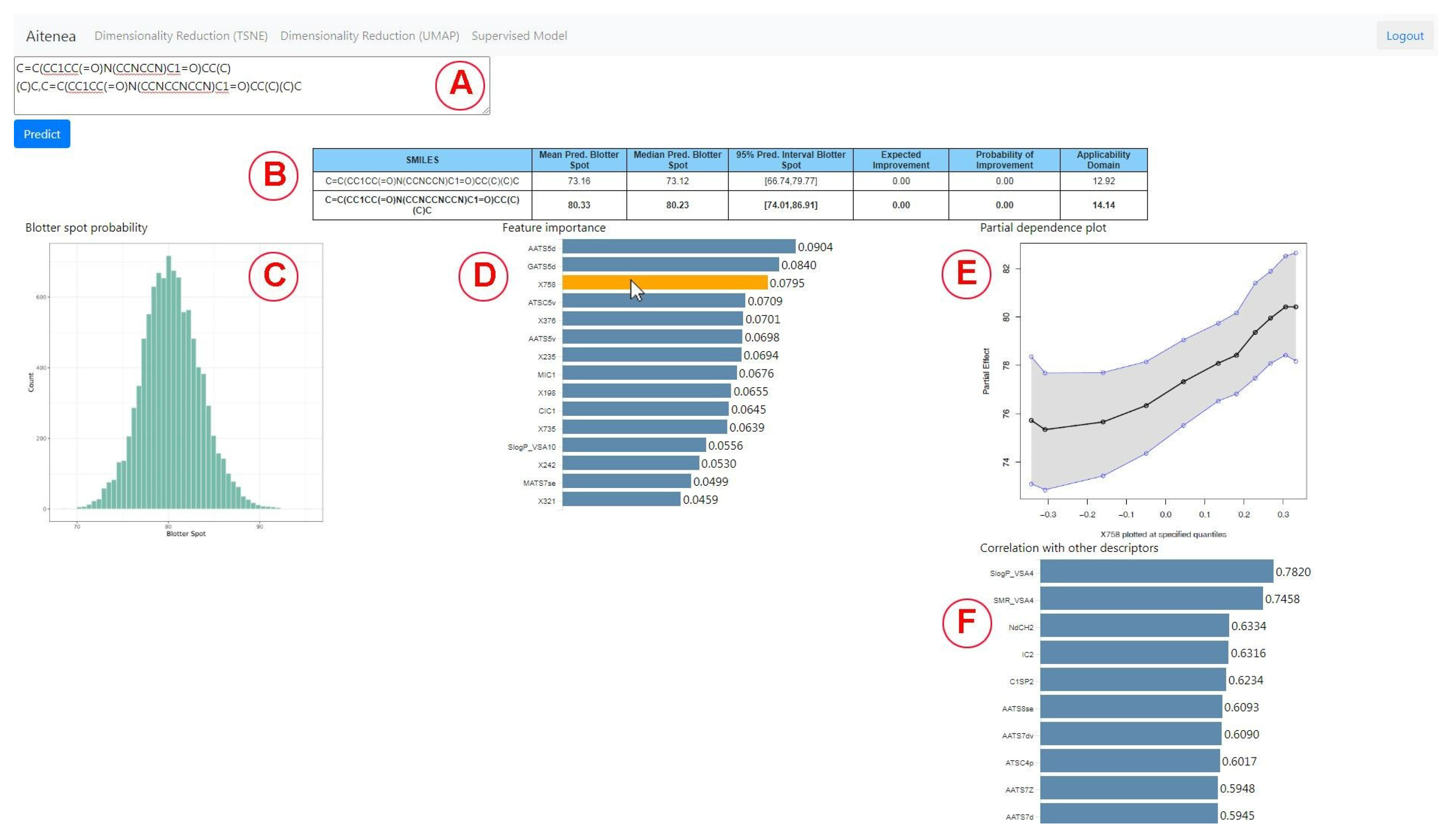
2.6. Design of the Visual Analytics Tool
- SMILES of the compound for prediction;
- Mean value of the posterior predictive distribution as described in Section 2.4.1;
- Median value of the posterior predictive distribution;
- 95% prediction interval as described in Section 2.4.1;
- Expected improvement of the predicted blotter spot value with regard to the highest blotter spot of the training dataset as described in Section 2.4.1;
- Probability of improvement with respect to the highest blotter spot of the training dataset as described in Section 2.4.1;
- Applicability domain measured as described in Section 2.5.
3. Results and Discussion
3.1. Evaluation of the Models
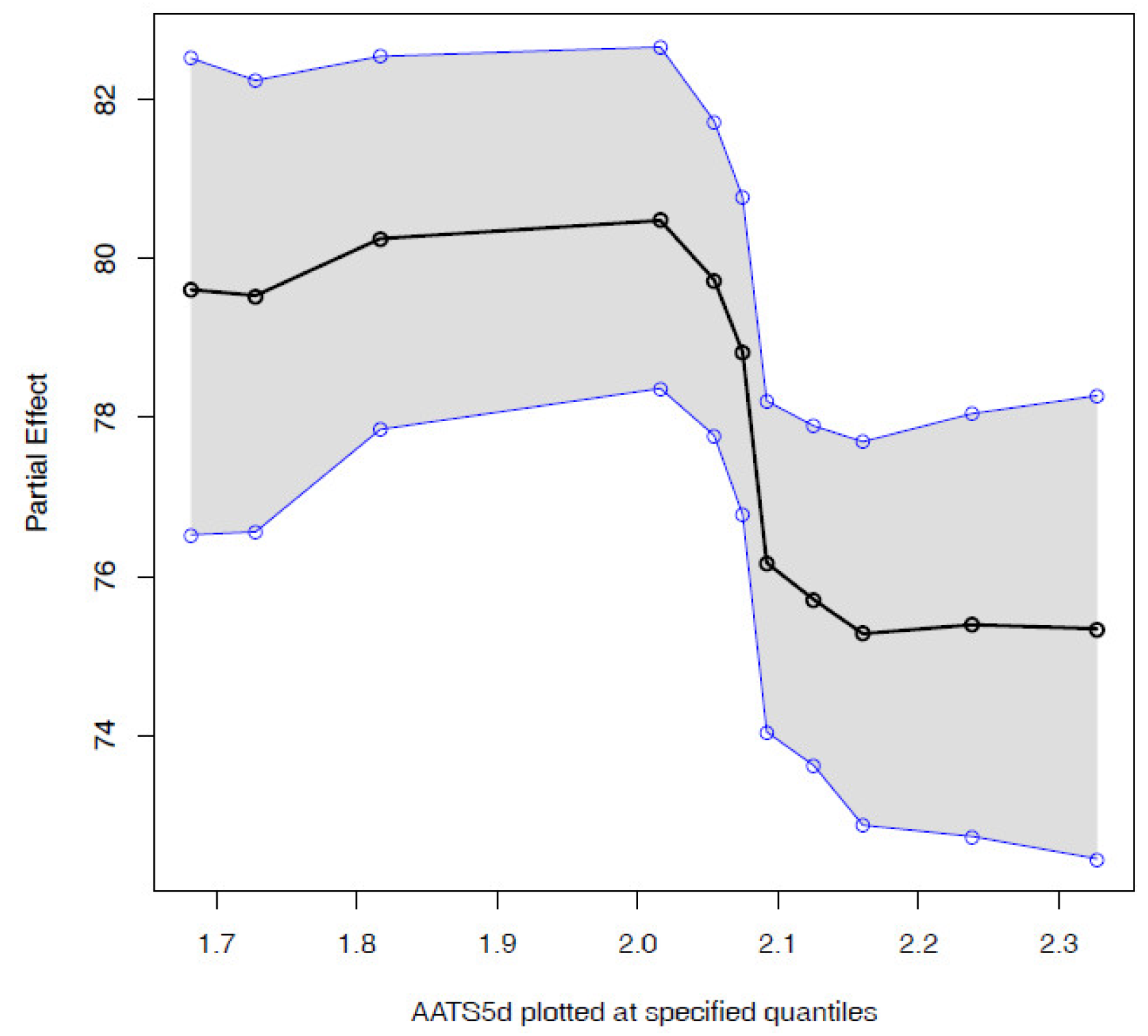
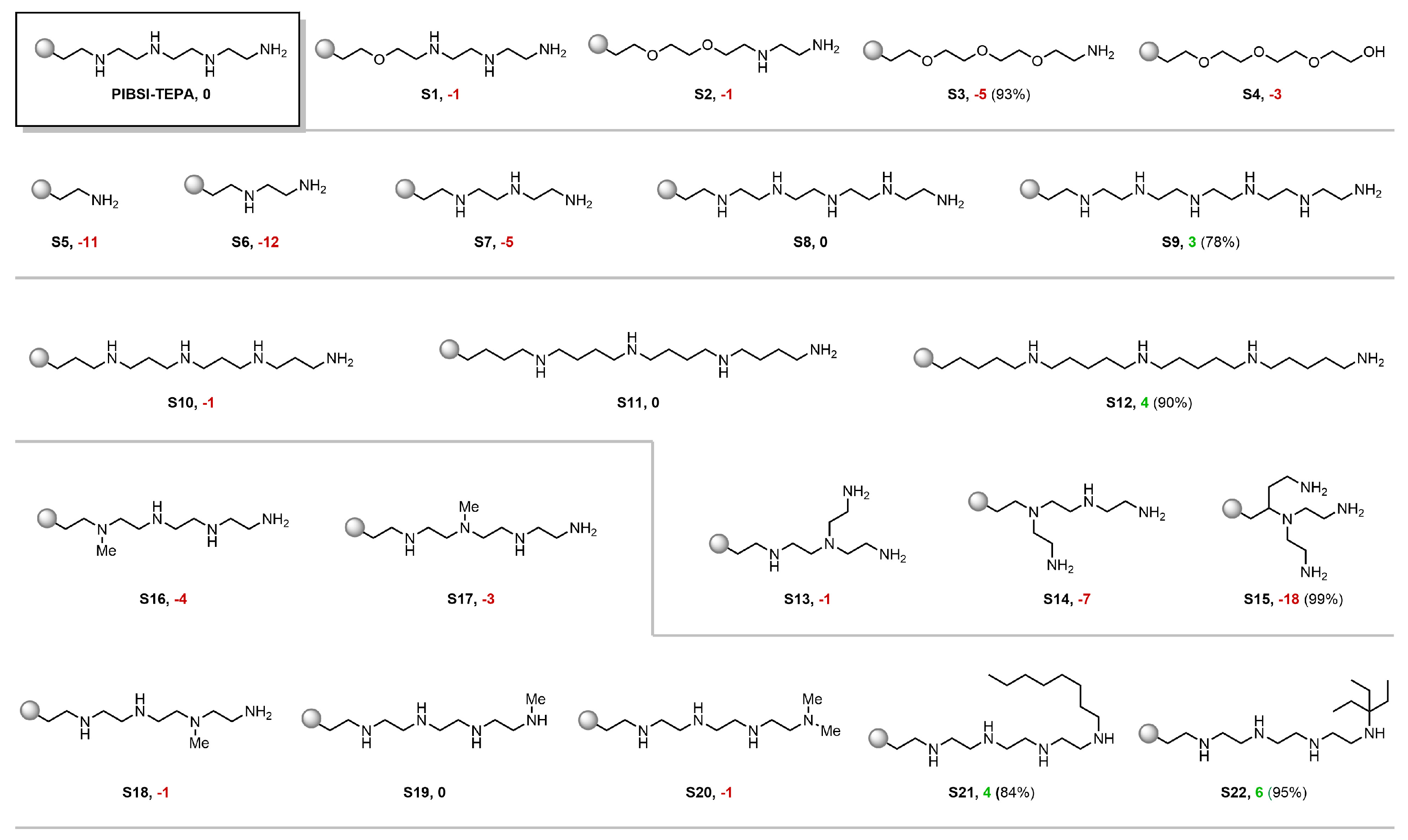
3.2. Virtual Screening Using the Visual Analytics Tool
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, S.; Josephson, T.R.; Athaley, A.; Chen, Q.P.; Norton, A.; Ierapetritou, M.; Siepmann, J.I.; Saha, B.; Vlachos, D.G. Renewable lubricants with tailored molecular architecture. Sci. Adv. 2019, 5, eaav5487. [Google Scholar] [CrossRef] [PubMed]
- Childs, C.M.; Washburn, N.R. Embedding domain knowledge for machine learning of complex material systems. MRS Commun. 2019, 9, 806–820. [Google Scholar] [CrossRef]
- Patra, T.K. Data-Driven Methods for Accelerating Polymer Design. ACS Polym. Au 2022, 2, 8–26. [Google Scholar] [CrossRef]
- Deutch, J. Is net zero carbon 2050 possible? Joule 2020, 4, 2237–2240. [Google Scholar] [CrossRef]
- Seto, K.C.; Churkina, G.; Hsu, A.; Keller, M.; Newman, P.W.; Qin, B.; Ramaswami, A. From low-to net-zero carbon cities: The next global agenda. Annu. Rev. Environ. Resour. 2021, 46, 377–415. [Google Scholar] [CrossRef]
- Bouckaert, S.; Pales, A.F.; McGlade, C.; Remme, U.; Wanner, B.; Varro, L.; D’Ambrosio, D.; Spencer, T. Net Zero by 2050: A Roadmap for the Global Energy Sector, 4th rev. ed.; The National Academies of Sciences, Engineering, and Medicine: Washington, DC, USA, 2021; Available online: www.iea.org/corrections (accessed on 10 February 2023).
- Dubois-Clochard, M.C.; Durand, J.P.; Delfort, B.; Gateau, P.; Barré, L.; Blanchard, I.; Chevalier, Y.; Gallo, R. Adsorption of Polyisobutenylsuccinimide Derivatives at a Solid- Hydrocarbon Interface. Langmuir 2001, 17, 5901–5910. [Google Scholar] [CrossRef]
- Shen, Y.; Duhamel, J. Micellization and adsorption of a series of succinimide dispersants. Langmuir 2008, 24, 10665–10673. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.J.; Matsunaga, T.; Fowkes, F.M. The dispersibility and stability of carbon black in media of low dielectric constant. 1. Electrostatic and steric contributions to colloidal stability. Colloids Surfaces 1983, 7, 183–207. [Google Scholar] [CrossRef]
- Growney, D.J.; Mykhaylyk, O.O.; Derouineau, T.; Fielding, L.A.; Smith, A.J.; Aragrag, N.; Lamb, G.D.; Armes, S.P. Star diblock copolymer concentration dictates the degree of dispersion of carbon black particles in nonpolar media: Bridging flocculation versus steric stabilization. Macromolecules 2015, 48, 3691–3704. [Google Scholar] [CrossRef]
- Wang, Q.; Chung, Y. Encyclopedia of Tribology; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2013. [Google Scholar]
- Myers, D. Surfactant Science and Technology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2020. [Google Scholar]
- Le Suer, W.M.; Norman, G.R. Reaction Product of High Molecular Weight Succinic Acids and Succinic Anhydrides with an Ethylene Polyamine. U.S. Patent 3172892A, 30 March 1959. [Google Scholar]
- Stuart, F.A.; Anderson, R.G.; Drummond, A.Y. Alkenyl Succinimides of Tetraethylene Pentamine. U.S. Patent 3202678A, 24 August 1965. [Google Scholar]
- Jablonka, K.M.; Jothiappan, G.M.; Wang, S.; Smit, B.; Yoo, B. Bias free multiobjective active learning for materials design and discovery. Nat. Commun. 2021, 12, 2312. [Google Scholar] [CrossRef]
- Jia, D.; Duan, H.; Zhan, S.; Jin, Y.; Cheng, B.; Li, J. Design and development of lubricating material database and research on performance prediction method of machine learning. Sci. Rep. 2019, 9, 20277. [Google Scholar] [CrossRef]
- Marian, M.; Tremmel, S. Current trends and applications of machine learning in tribology—A review. Lubricants 2021, 9, 86. [Google Scholar] [CrossRef]
- Rosenkranz, A.; Marian, M.; Profito, F.J.; Aragon, N.; Shah, R. The use of artificial intelligence in tribology—A perspective. Lubricants 2021, 9, 2. [Google Scholar] [CrossRef]
- Zhou, L.; Song, Y.; Ji, W.; Wei, H. Machine learning for combustion. Energy AI 2022, 7, 100128. [Google Scholar] [CrossRef]
- Aghbashlo, M.; Peng, W.; Tabatabaei, M.; Kalogirou, S.A.; Soltanian, S.; Hosseinzadeh-Bandbafha, H.; Mahian, O.; Lam, S.S. Machine learning technology in biodiesel research: A review. Prog. Energy Combust. Sci. 2021, 85, 100904. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Rasulev, B.; Casanola-Martin, G. QSAR/QSPR in polymers: Recent developments in property modeling. Int. J. Quant. Struct.-Prop. Relationships (IJQSPR) 2020, 5, 80–88. [Google Scholar] [CrossRef]
- Menon, A.; Gupta, C.; Perkins, K.M.; DeCost, B.L.; Budwal, N.; Rios, R.T.; Zhang, K.; Póczos, B.; Washburn, N.R. Elucidating multi-physics interactions in suspensions for the design of polymeric dispersants: A hierarchical machine learning approach. Mol. Syst. Des. Eng. 2017, 2, 263–273. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.; Hyeon, D.H.; Han, J.S.; Chun, B.H.; Jeong, B.H.; Kim, S.H. Development of PIBSI type dispersants for carbon deposit from thermal oxidative decomposition of Jet A-1. Fuel 2015, 158, 91–97. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, R.K. A search for ecofriendly detergent/dispersant additives for vegetable-oil based lubricants. J. Surfactants Deterg. 2012, 15, 399–409. [Google Scholar] [CrossRef]
- Pirouz, S.; Wang, Y.; Chong, J.M.; Duhamel, J. Chemical modification of polyisobutylene succinimide dispersants and characterization of their associative properties. J. Phys. Chem. 2015, 119, 12202–12211. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, T.P.; Masson, G.M.; Storey, R.F. Synthesis, characterization, and evaluation of polyisobutylene-based imido-amine-type dispersants containing exclusively non-nucleophilic nitrogen. J. Polym. Sci. Part Polym. Chem. 2018, 56, 1657–1675. [Google Scholar] [CrossRef]
- Mekewi, M.A. Synthesis and characterization of antioxidants and detergent dispersant based on some polyisobutylene copolymers. Mater. Res. Innov. 2002, 6, 214–217. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Nassar, A.M.; Abdel-Azim, A.A.A. Synthesis and evaluation of some detergent/dispersant additives for lube oil. Int. J. Polym. Mater. 2007, 57, 114–124. [Google Scholar] [CrossRef]
- Amal, M.; Nehal, S.; Khalid, I.; Ahmed, F.; Abdel-Azim, A. Synthesis and evaluation of detergent/dispersant additives from polyisobutylene succinimides. Int. J. Polym. Mater. 2006, 55, 703–713. [Google Scholar]
- Holbrook, T.P.; Masson, G.M.; Storey, R.F. Synthesis of comb-like dispersants and a study on the effect of dispersant architecture and carbon black dispersion. J. Polym. Sci. Part Polym. Chem. 2019, 57, 1682–1696. [Google Scholar] [CrossRef]
- Kozaka, D.; Moreton, D.; Vincent, B. The adsorption of nonionic surfactants on carbon black particles in hydrocarbon media. Colloids Surf. A Physicochem. Eng. Asp. 2009, 347, 245–250. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Nassar, A.M.; Kabel, K.I.; Abdel Azim, A.A.A.; El-Kafrawy, A.F. Deposit Control Agents for Lubricating Oil. Pet. Coal 2017, 59. [Google Scholar]
- Ahmed, N.S.; Nassar, A.M. Lubricating oil additives based on polyalkylpolyamines. Int. J. Polym. Mater. 2009, 58, 178–190. [Google Scholar] [CrossRef]
- Singh, R.K.; Pandey, S.; Saxena, R.C.; Thakre, G.D.; Atray, N.; Ray, S.S. Derivatizing L-histidine to develop a novel additive for a polyol-based biolubricant. New J. Chem. 2015, 39, 5354–5359. [Google Scholar] [CrossRef]
- Sammaiah, A.; Padmaja, K.V.; Kaki, S.S.; Prasad, R.B. Multifunctional lubricant additives derived from natural amino acids and methyl oleate. RSC Adv. 2015, 5, 77538–77544. [Google Scholar] [CrossRef]
- Kontham, V.; Ansari, K.R.; Padmaja, K.V. Tribological properties of 10-Undecenoic acid-derived Schiff Base lubricant additives. Arab. J. Sci. Eng. 2021, 46, 5593–5603. [Google Scholar] [CrossRef]
- Nassar, A.M.; Ahmed, N.S.; Abdel-Hameed, H.S.; El-Kafrawy, A.F. Synthesis and utilization of non-metallic detergent/dispersant and antioxidant additives for lubricating engine oil. Tribol. Int. 2016, 93, 297–305. [Google Scholar]
- Kitchin, R.; Lauriault, T.P. Small data in the era of big data. GeoJournal 2015, 80, 463–475. [Google Scholar] [CrossRef]
- Qi, G.J.; Luo, J. Small data challenges in big data era: A survey of recent progress on unsupervised and semi-supervised methods. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 44, 2168–2187. [Google Scholar] [CrossRef] [PubMed]
- Ameur, H.; Njah, H.; Jamoussi, S. Merits of Bayesian networks in overcoming small data challenges: A meta-model for handling missing data. Int. J. Mach. Learn. Cybern. 2023, 14, 229–251. [Google Scholar] [CrossRef]
- Li, H.; Fang, S.; Mukhopadhyay, S.; Saykin, A.J.; Shen, L. Interactive machine learning by visualization: A small data solution. In Proceedings of the 2018 IEEE International Conference on Big Data (Big Data), Seattle, WA, USA, 10–13 December 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 3513–3521. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminfor. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Infor. 2018, 37, 1700111. [Google Scholar] [CrossRef]
- Sabando, M.V.; Ponzoni, I.; Milios, E.E.; Soto, A.J. Using molecular embeddings in QSAR modeling: Does it make a difference? Briefings Bioinform. 2022, 23, bbab365. [Google Scholar] [CrossRef]
- Hugging Face Model Page. Available online: https://huggingface.co/mrm8488/chEMBL26_smiles_v2 (accessed on 20 August 2022).
- Mosqueira-Rey, E.; Hernández-Pereira, E.; Alonso-Ríos, D.; Bobes-Bascarán, J.; Fernández-Leal, Á. Human-in-the-loop machine learning: A state of the art. Artif. Intell. Rev. 2022, 1–50. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Maimon, O.Z.; Rokach, L. Data Mining with Decision Trees: Theory and Applications; World Scientific: Singapore, 2014; Volume 81. [Google Scholar]
- Martínez, M.J.; Ponzoni, I.; Díaz, M.F.; Vazquez, G.E.; Soto, A.J. Visual analytics in cheminformatics: User-supervised descriptor selection for QSAR methods. J. Cheminfor. 2015, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Kramer, O.; Kramer, O. K-nearest neighbors. In Dimensionality Reduction With Unsupervised Nearest Neighbors; Springer: Berlin/Heidelberg, Germany, 2013; pp. 13–23. [Google Scholar]
- Khan, K.; Rehman, S.U.; Aziz, K.; Fong, S.; Sarasvady, S. DBSCAN: Past, present and future. In Proceedings of the Fifth International Conference on the Applications of Digital Information and Web Technologies (ICADIWT 2014), Chennai, India, 17–19 February 2014; IEEE: Piscataway, NJ, USA, 2014; pp. 232–238. [Google Scholar]
- Scikit Learn KMeans Page. Available online: https://scikit-learn.org/stable/modules/generated/sklearn.cluster.KMeans.html (accessed on 20 August 2022).
- Scikit Learn DBSCAN Page. Available online: https://scikit-learn.org/stable/modules/generated/sklearn.cluster.DBSCAN.html. (accessed on 20 August 2022).
- Chipman, H.A.; George, E.I.; McCulloch, R.E. BART: Bayesian additive regression trees. Ann. Appl. Stat. 2010, 4, 266–298. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Polson, N.G.; Scott, J.G. Handling sparsity via the horseshoe. In Proceedings of the Artificial Intelligence and Statistics, Shanghai, China, 7–8 November 2009; pp. 73–80. [Google Scholar]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef]
- Bleich, J.; Kapelner, A.; Jensen, S.; George, E. Variable selection inference for bayesian additive regression trees. arXiv 2013, arXiv:1310.4887. [Google Scholar]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical bayesian optimization of machine learning algorithms. In Advances in Neural Information Processing Systems 25; Curran Associates, Incorporated: Red Hook, NY, USA, 2012. [Google Scholar]
- Weaver, S.; Gleeson, M.P. The importance of the domain of applicability in QSAR modeling. J. Mol. Graph. Model. 2008, 26, 1315–1326. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability domain: A step toward confident predictions and decidability for QSAR modeling. In Computational Toxicology; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2018; pp. 141–169. [Google Scholar]
- Rakhimbekova, A.; Madzhidov, T.I.; Nugmanov, R.I.; Gimadiev, T.R.; Baskin, I.I.; Varnek, A. Comprehensive analysis of applicability domains of QSPR models for chemical reactions. Int. J. Mol. Sci. 2020, 21, 5542. [Google Scholar] [CrossRef]
- Keim, D.; Kohlhammer, J.; Ellis, G.; Mansmann, F. Mastering the Information Age: Solving Problems with Visual Analytics; Eurographics Association: Goslar, Germany, 2010. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Scikit Learn manifold TSNE Page. Available online: https://scikit-learn.org/stable/modules/generated/sklearn.manifold.TSNE.html (accessed on 20 August 2022).
- Umap Learn Home Page. Available online: https://umap-learn.readthedocs.io/en/latest/ (accessed on 20 August 2022).
- Probst, D.; Reymond, J.L. SmilesDrawer: Parsing and drawing SMILES-encoded molecular structures using client-side JavaScript. J. Chem. Inf. Model. 2018, 58, 1–7. [Google Scholar] [CrossRef]
- Ware, C. Information Visualization: Perception for Design; Morgan Kaufmann: Burlington, MA, USA, 2019. [Google Scholar]
- Scherr, M. Multiple and coordinated views in information visualization. Trends Inf. Vis. 2008, 38, 1–33. [Google Scholar]
- Fushiki, T. Estimation of prediction error by using K-fold cross-validation. Stat. Comput. 2011, 21, 137–146. [Google Scholar] [CrossRef]
- Wong, T.T.; Yeh, P.Y. Reliable Accuracy Estimates from k-Fold Cross Validation. IEEE Trans. Knowl. Data Eng. 2020, 32, 1586–1594. [Google Scholar] [CrossRef]
- Ghosh, P.; Hoque, M. Multifunctional lube oil additives based on maleic anhydride. Pet. Sci. Technol. 2016, 34, 1761–1767. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subset | Feature Selection Method | Pre-Selected Molecular Descriptor |
|---|---|---|
| 1 | Random Forest | AATS5d, AATS5Z, SIC0, ATSC3d, ATSC5v, GATS5d, |
| AATS5v, GATS3d | ||
| 2 | Decision Tree | AATS5d, ATSC5v, ATSC6Z, AATS8p, AATSC7d, |
| MINssCH2, PEOE_VSA9, GATS8i | ||
| 3 | Decision Tree | AATS5d, ATSC5v, GATS8p, GATS8v, GATS1se, |
| MINssCH2, AATS1Z, GATS8i | ||
| 4 | Expert Criterion | nBase, nN, MATS1se, AATSC0p, TopoPSA(NO) |
| Data Sampling | MAE | RMSE |
|---|---|---|
| KMeans | ||
| DBSCAN |
| Algorithm | MAE | RMSE |
|---|---|---|
| Bayesian Regression + horseshoe prior | ||
| BART | ||
| BART + Variable Selection | ||
| Random Forest | ||
| Gradient Boosting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, M.J.; Naveiro, R.; Soto, A.J.; Talavante, P.; Kim Lee, S.-H.; Gómez Arrayas, R.; Franco, M.; Mauleón, P.; Lozano Ordóñez, H.; Revilla López, G.; et al. Design of New Dispersants Using Machine Learning and Visual Analytics. Polymers 2023, 15, 1324. https://doi.org/10.3390/polym15051324
Martínez MJ, Naveiro R, Soto AJ, Talavante P, Kim Lee S-H, Gómez Arrayas R, Franco M, Mauleón P, Lozano Ordóñez H, Revilla López G, et al. Design of New Dispersants Using Machine Learning and Visual Analytics. Polymers. 2023; 15(5):1324. https://doi.org/10.3390/polym15051324
Chicago/Turabian StyleMartínez, María Jimena, Roi Naveiro, Axel J. Soto, Pablo Talavante, Shin-Ho Kim Lee, Ramón Gómez Arrayas, Mario Franco, Pablo Mauleón, Héctor Lozano Ordóñez, Guillermo Revilla López, and et al. 2023. "Design of New Dispersants Using Machine Learning and Visual Analytics" Polymers 15, no. 5: 1324. https://doi.org/10.3390/polym15051324
APA StyleMartínez, M. J., Naveiro, R., Soto, A. J., Talavante, P., Kim Lee, S.-H., Gómez Arrayas, R., Franco, M., Mauleón, P., Lozano Ordóñez, H., Revilla López, G., Bernabei, M., Campillo, N. E., & Ponzoni, I. (2023). Design of New Dispersants Using Machine Learning and Visual Analytics. Polymers, 15(5), 1324. https://doi.org/10.3390/polym15051324