
Synthesis of Novel 2,9-Disubstituted-6-morpholino Purine Derivatives Assisted by Virtual Screening and Modelling of Class I PI3K Isoforms



Abstract
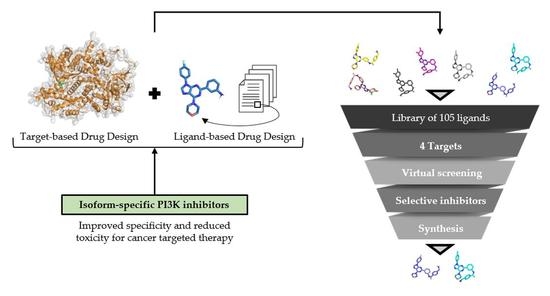
:
1. Introduction
2. Methods
2.1. In Silico
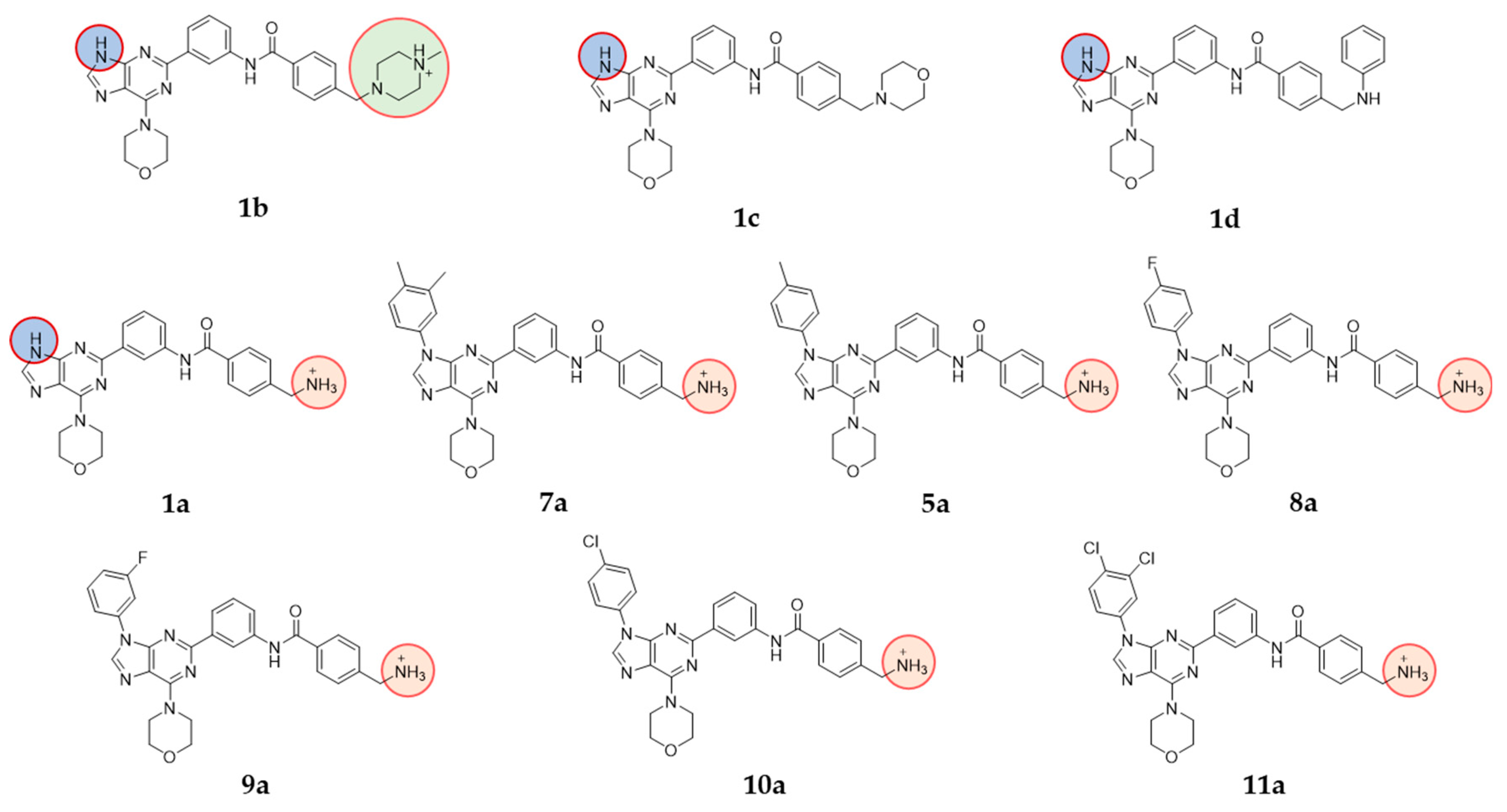
2.1.1. Class 1 Compounds
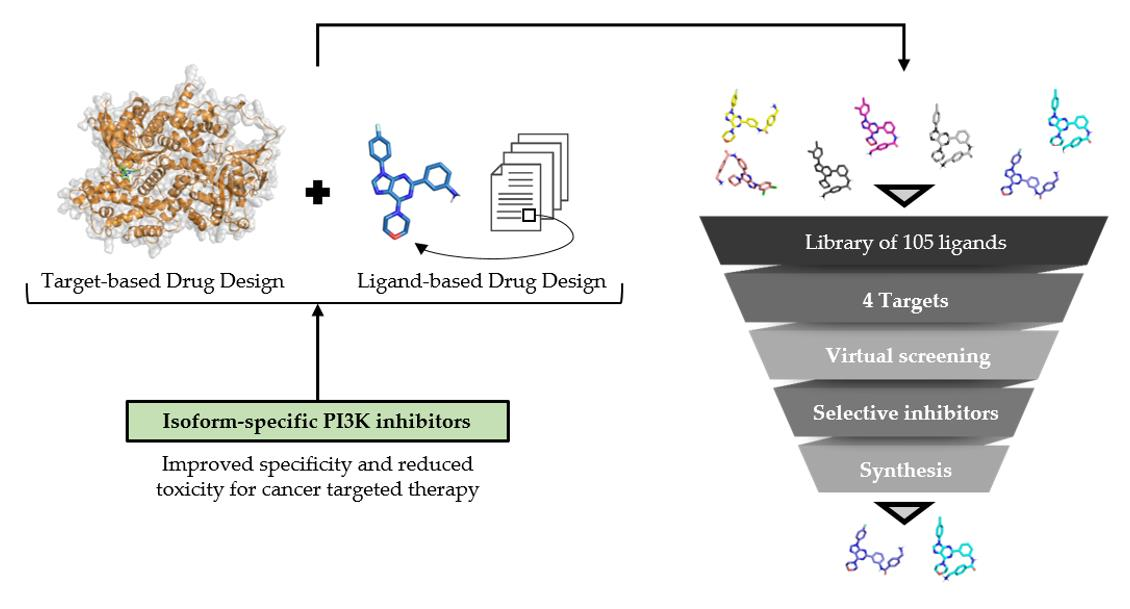
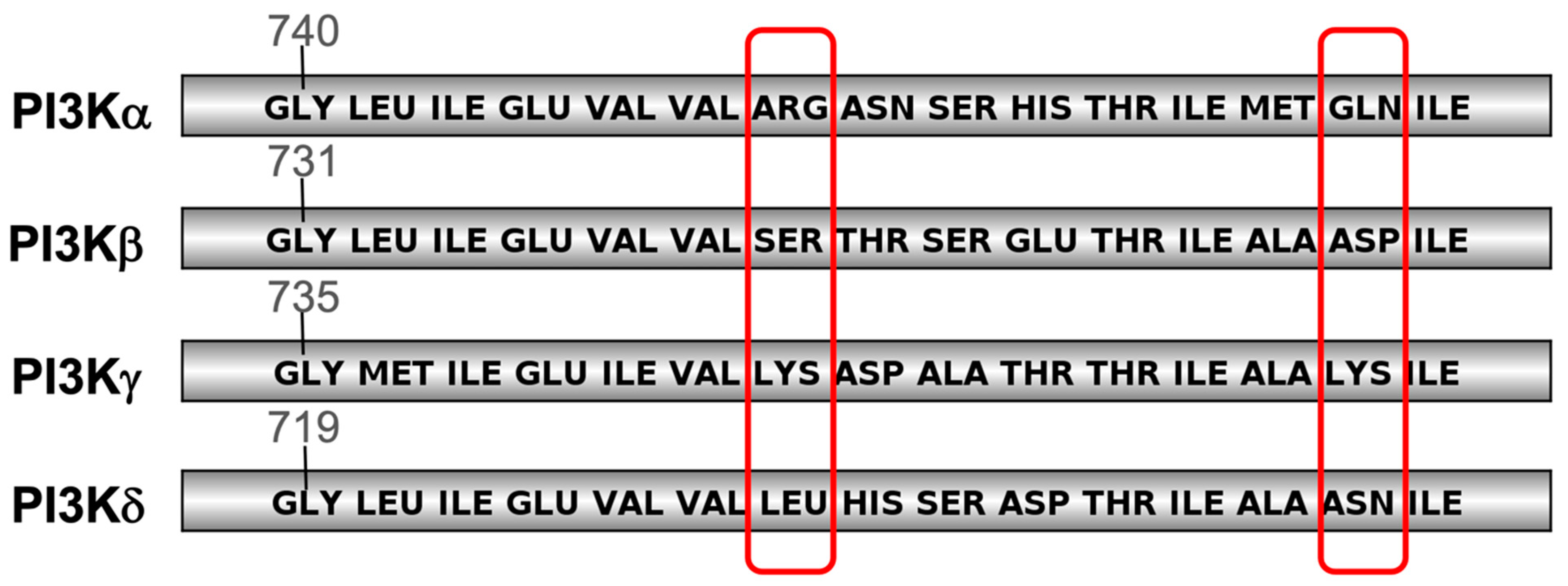
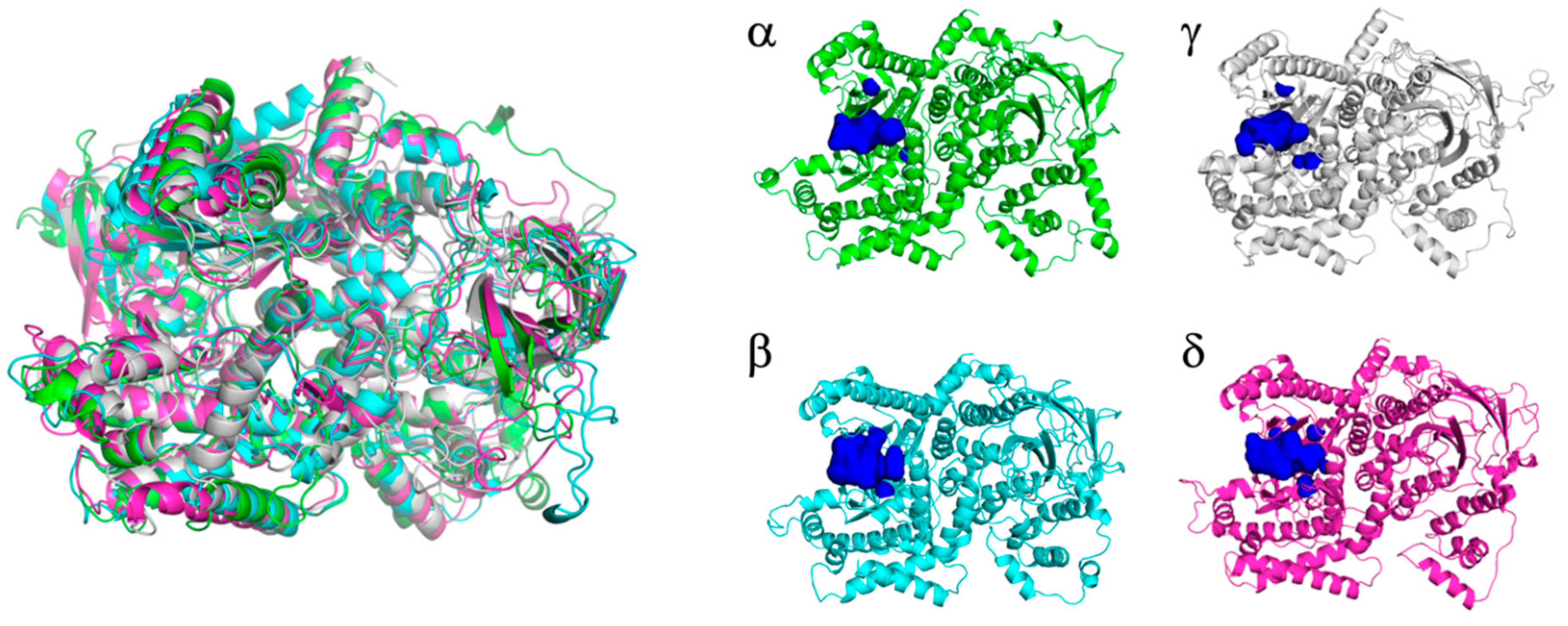
2.1.2. PI3K Isoforms
2.1.3. Virtual Screening
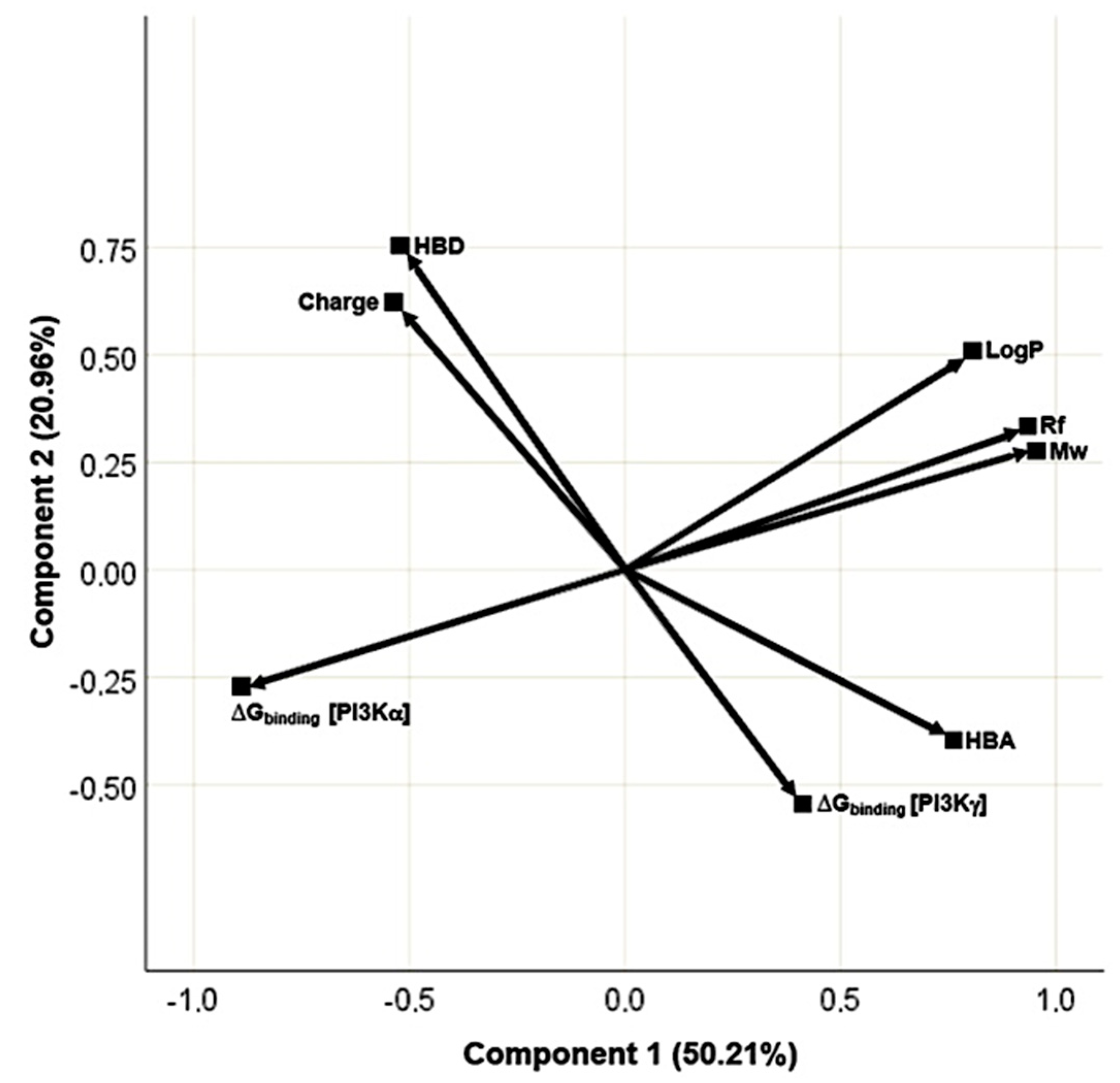
2.1.4. PCA
2.2. Chemical Synthesis
2.2.1. General Information
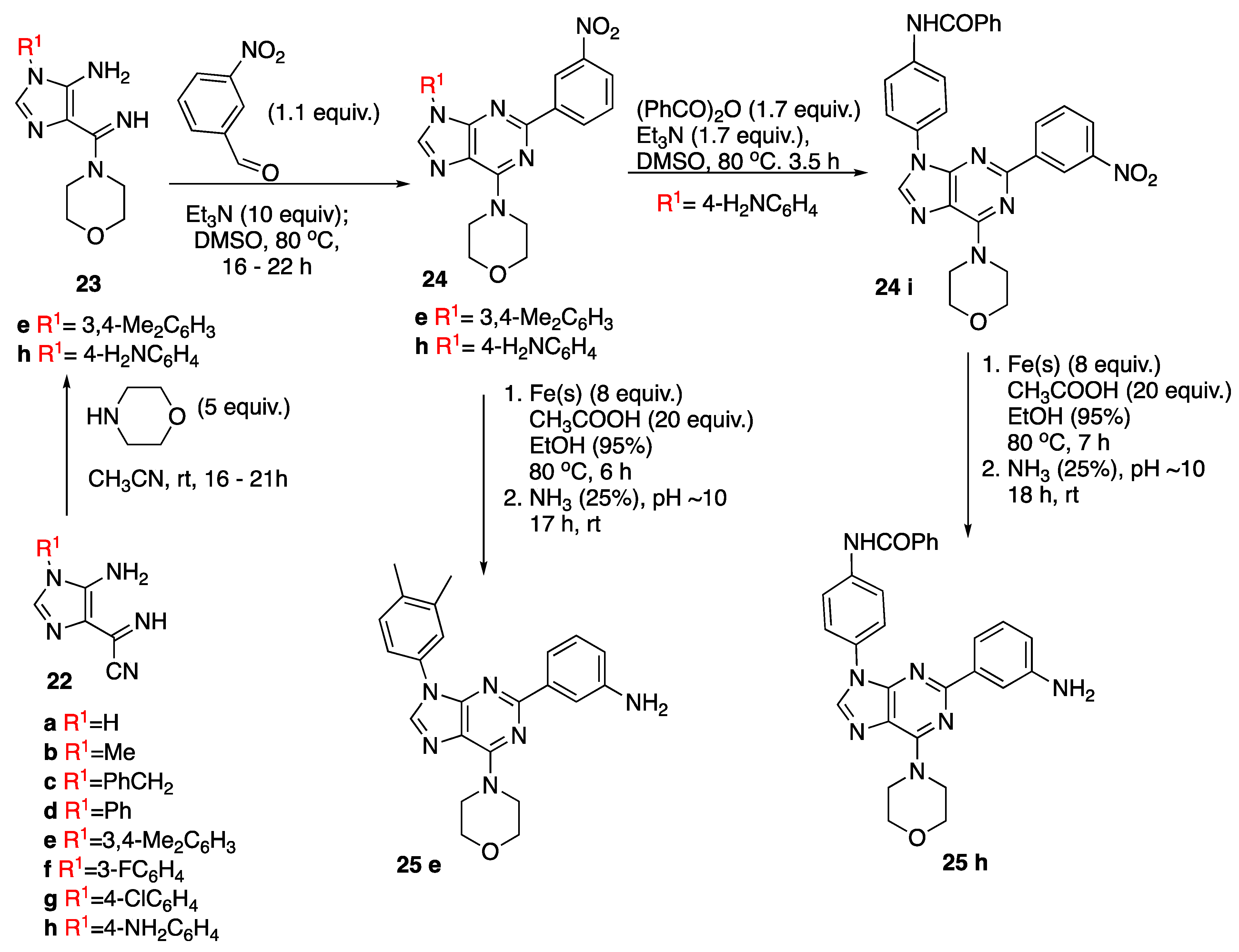
2.2.2. General Procedure for the Synthesis of Compounds 24
4-(2-(3-Nitrophenyl)-9-(3,4-dimethylphenyl)-9H-purin-6-yl)morpholine (24e)
4-(2-(3-Nitrophenyl)-9-(4-aniline)-9H-purin-6-yl)morpholine (24h)
N-(4-(6-Morpholino-2-(3-nitrophenyl)-9H-purin-9-yl)phenyl)benzamide (24i)
2.2.3. General Procedure for the Reduction of 2-(3-Nitrophenyl)-purine Derivatives (24)
3-(9-(3,4-Dimethylphenyl)-6-morpholino-9H-purin-2-yl)aniline (25e)
N-(4-(2-(3-Aminophenyl)-6-morpholino-9H-purin-9-yl)phenyl)benzamide (25h)
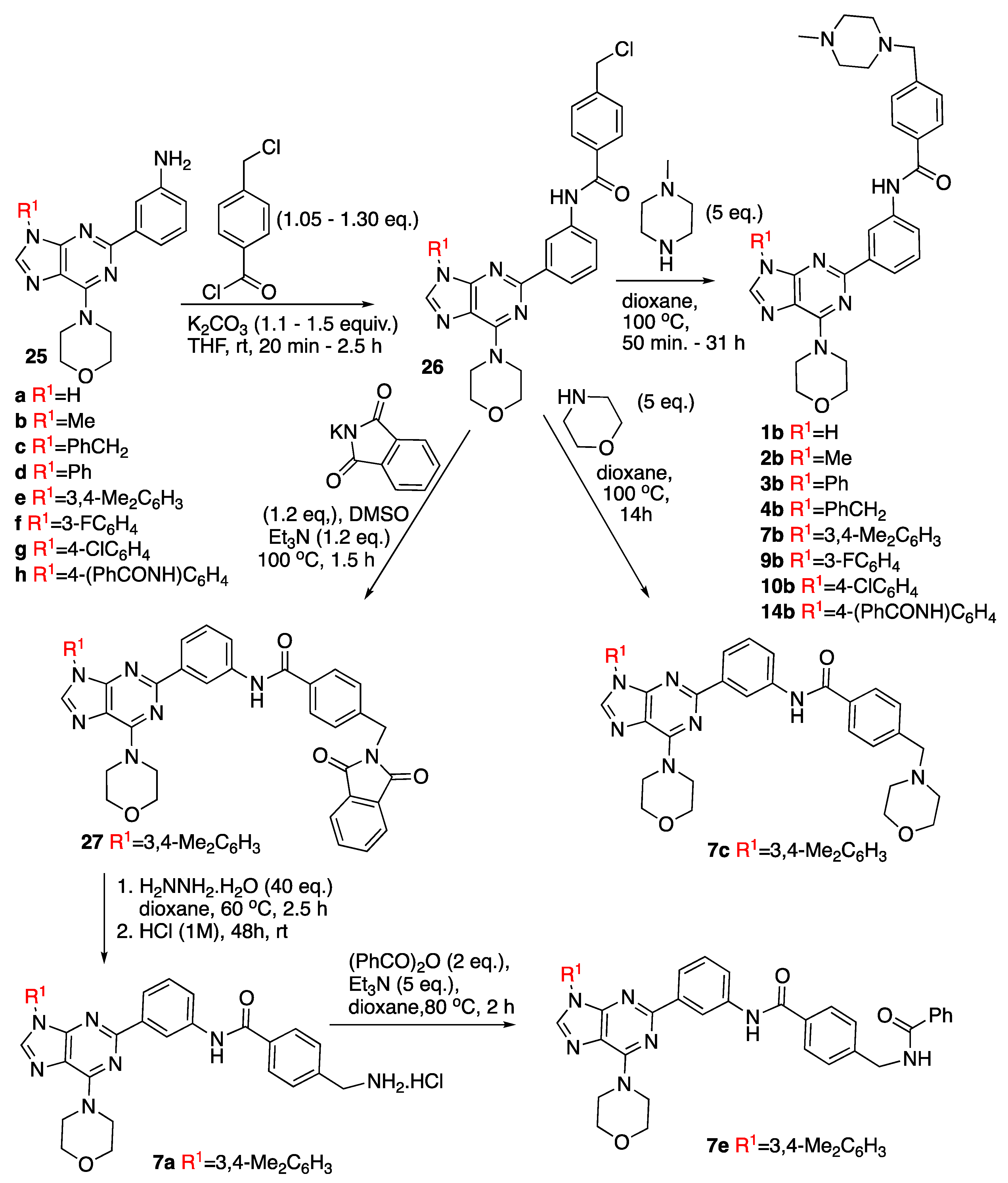
2.2.4. General Procedure for the Acylation of 2-(3-Aminophenyl)-purine Derivatives 25
Synthesis of 4-(Chloromethyl)-N-(3-(6-morpholino-9H-purin-2-yl)phenyl)benzamide (26a)
4-(Chloromethyl)-N-(3-(9-methyl-6-morpholino-9H-purin-2-yl)phenyl)benzamide (26b)
N-(3-(9-Benzyl-6-morpholino-9H-purin-2-yl)phenyl)-4-(chloromethyl)benzamide (26c)
4-(Chloromethyl)-N-(3-(6-morpholino-9-phenyl-9H-purin-2-yl)phenyl)benzamide (26d)
4-(Chloromethyl)-N-(3-(9-(3,4-dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)benzamide (26e)
4-(Chloromethyl)-N-(3-(9-(3-fluorophenyl)-6-morpholino-9H-purin-2-yl)phenyl)benzamide (26f)
4-(Chloromethyl)-N-(3-(9-(4-chlorophenyl)-6-morpholino-9H-purin-2-yl)phenyl)benzamide (26g)
N-(3-(9-(4-Benzamidophenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-(chloromethyl)benzamide (26h)
2.2.5. Synthesis of N-(3-(9-(3,4-Dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-((1,3-dioxoisoindolin-2-yl)methyl)benzamide (27)
2.2.6. General Procedure for the Reaction of Compounds 26 with Nitrogen Nucleophiles
4-((4-Methylpiperazin-1-yl)methyl)-N-(3-(6-morpholino-9H-purin-2-yl)phenyl)benzamide (1b)
N-(3-(9-Methyl-6-morpholino-9H-purin-2-yl)phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (2b)
4-((4-Methylpiperazin-1-yl)methyl)-N-(3-(6-morpholino-9-phenyl-9H-purin-2-yl)phenyl)benzamide (3b)
N-(3-(9-Benzyl-6-morpholino-9H-purin-2-yl)phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (4b)
N-(3-(9-(3,4-Dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-((4-methylpiperazin-1-yl) methyl)benzamide (7b)
N-(3-(9-(3,4-Dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-(morpholinomethyl)benzamide (7c)
N-(3-(9-(3-Fluorophenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (9b)
N-(3-(9-(4-Chlorophenyl)-6-morpholino-9H-purin-2-yl)phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (10b)
N-(3-(9-(4-Benzamidophenyl)-6-morpholino-9H-purin-2-yl) phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (14b)
2.2.7. Synthesis of (4-((3-(9-(3,4-Dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)carbamoyl)phenyl)methanaminium Chloride (7a)
2.2.8. Synthesis of 4-(Benzamidomethyl)-N-(3-(9-(3,4-dimethylphenyl)-6-morpholino-9H-purin-2-yl)phenyl)benzamide (7e)
3. Results and Discussion
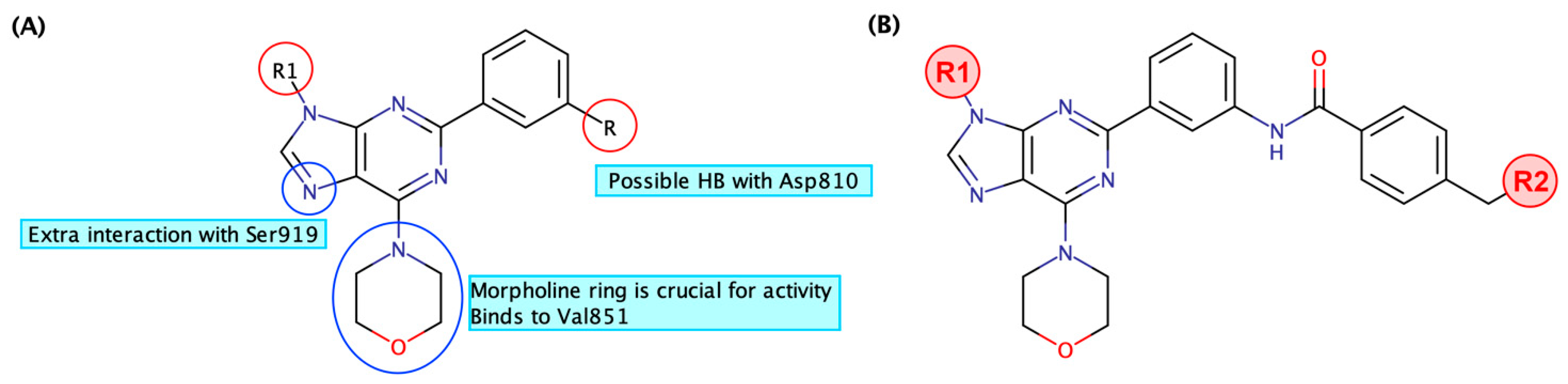
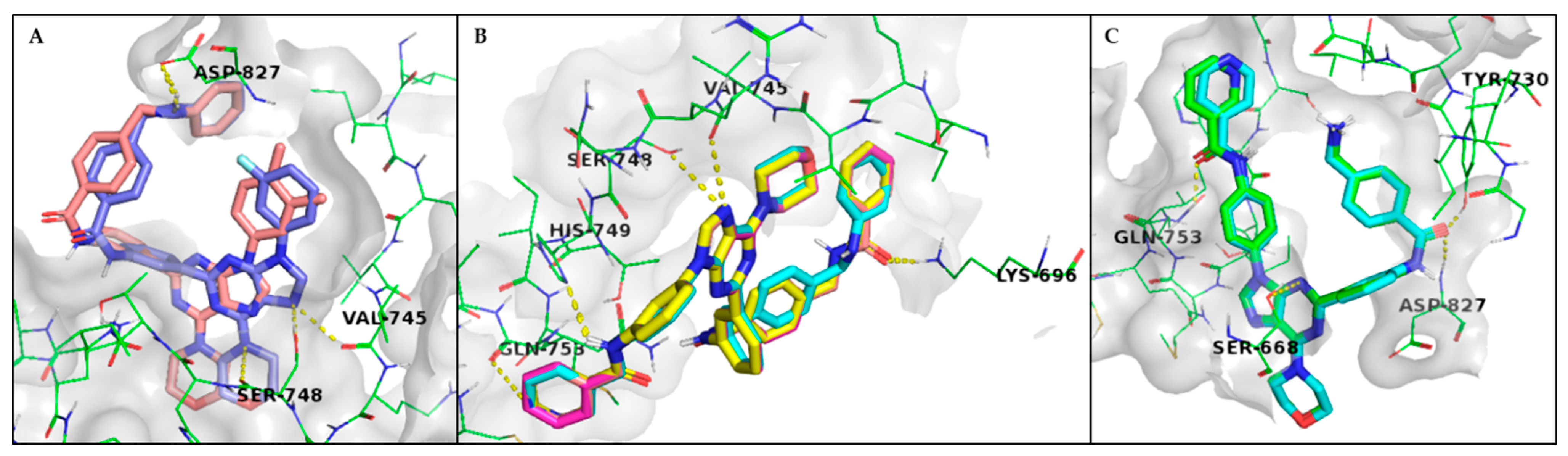
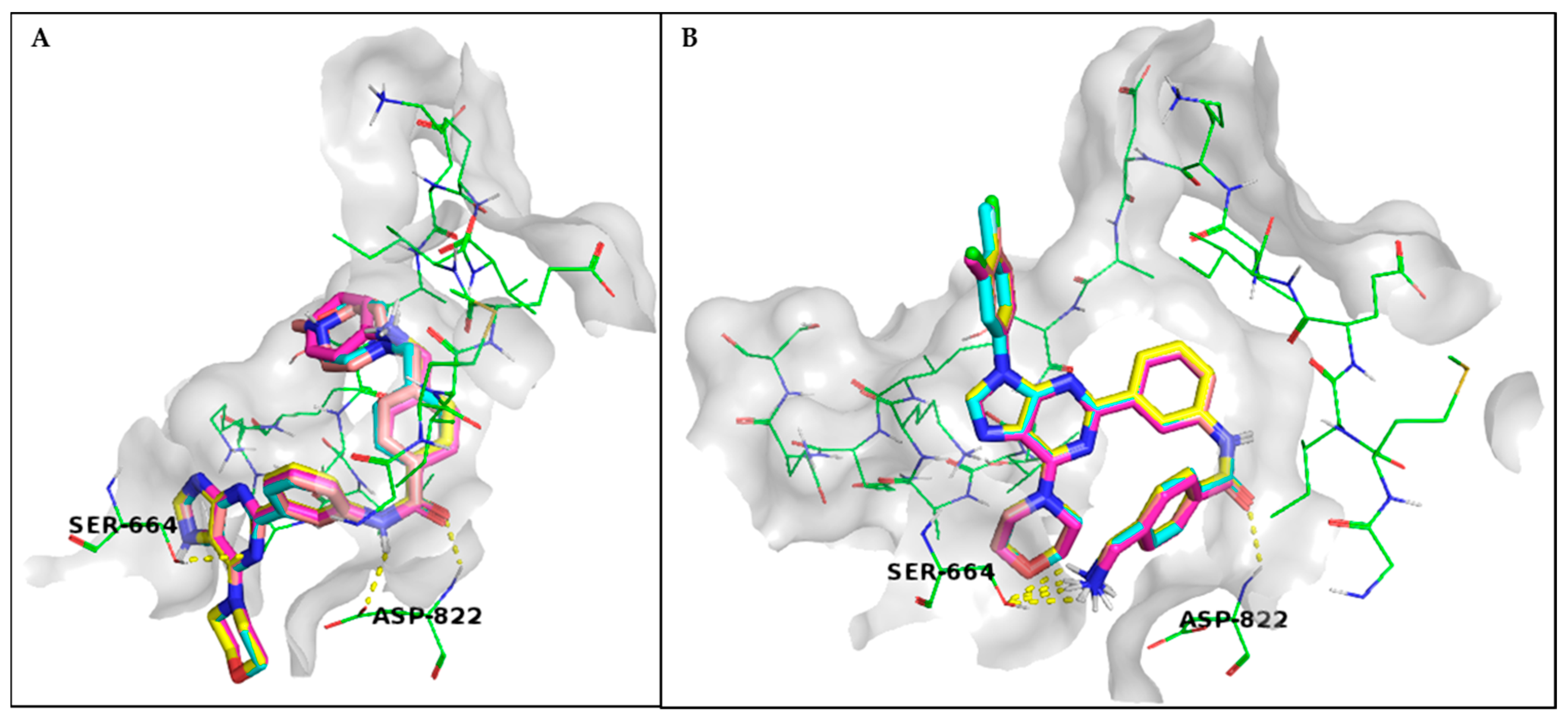
3.1. Rational Design and Modelling
3.2. Synthesis
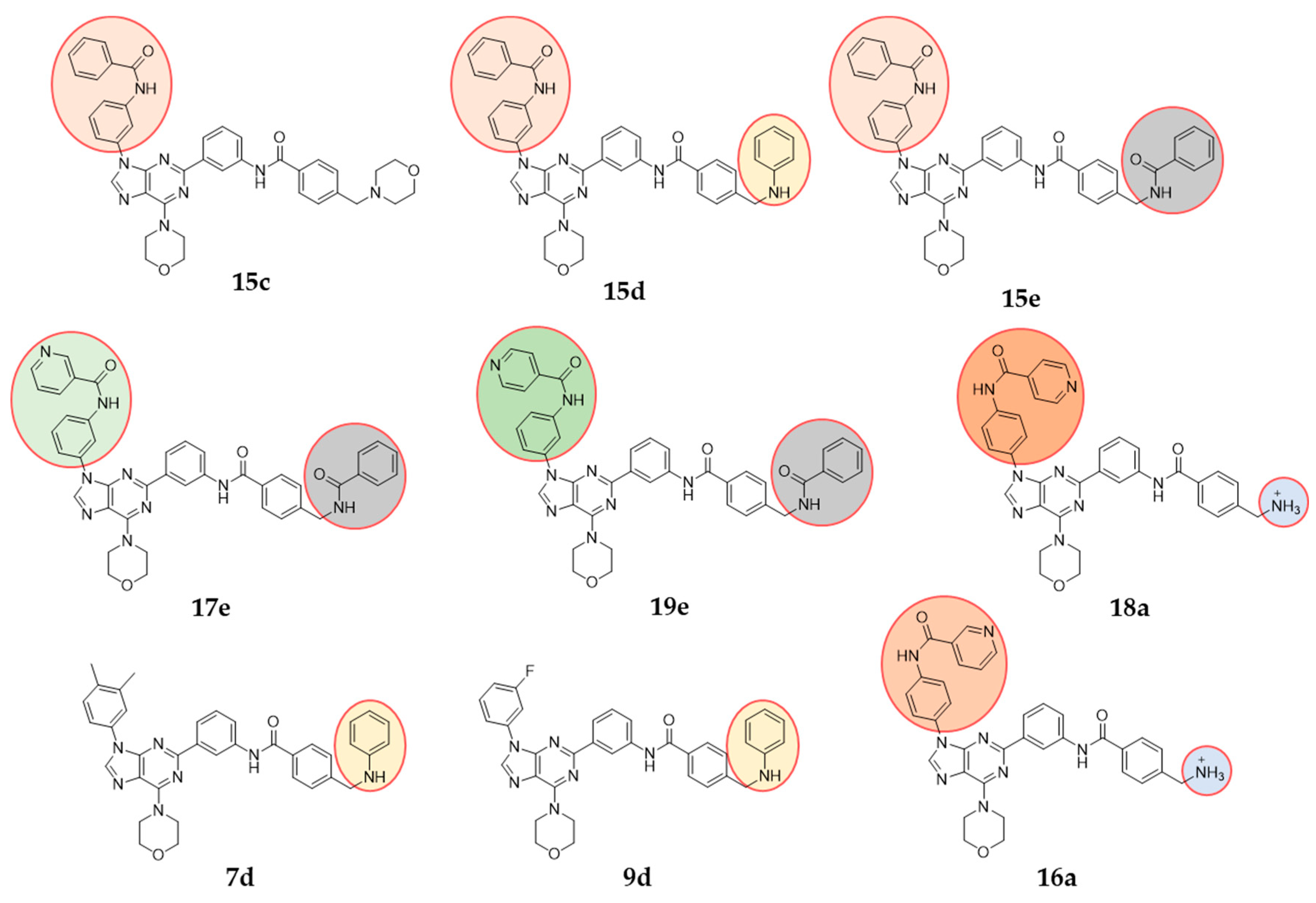
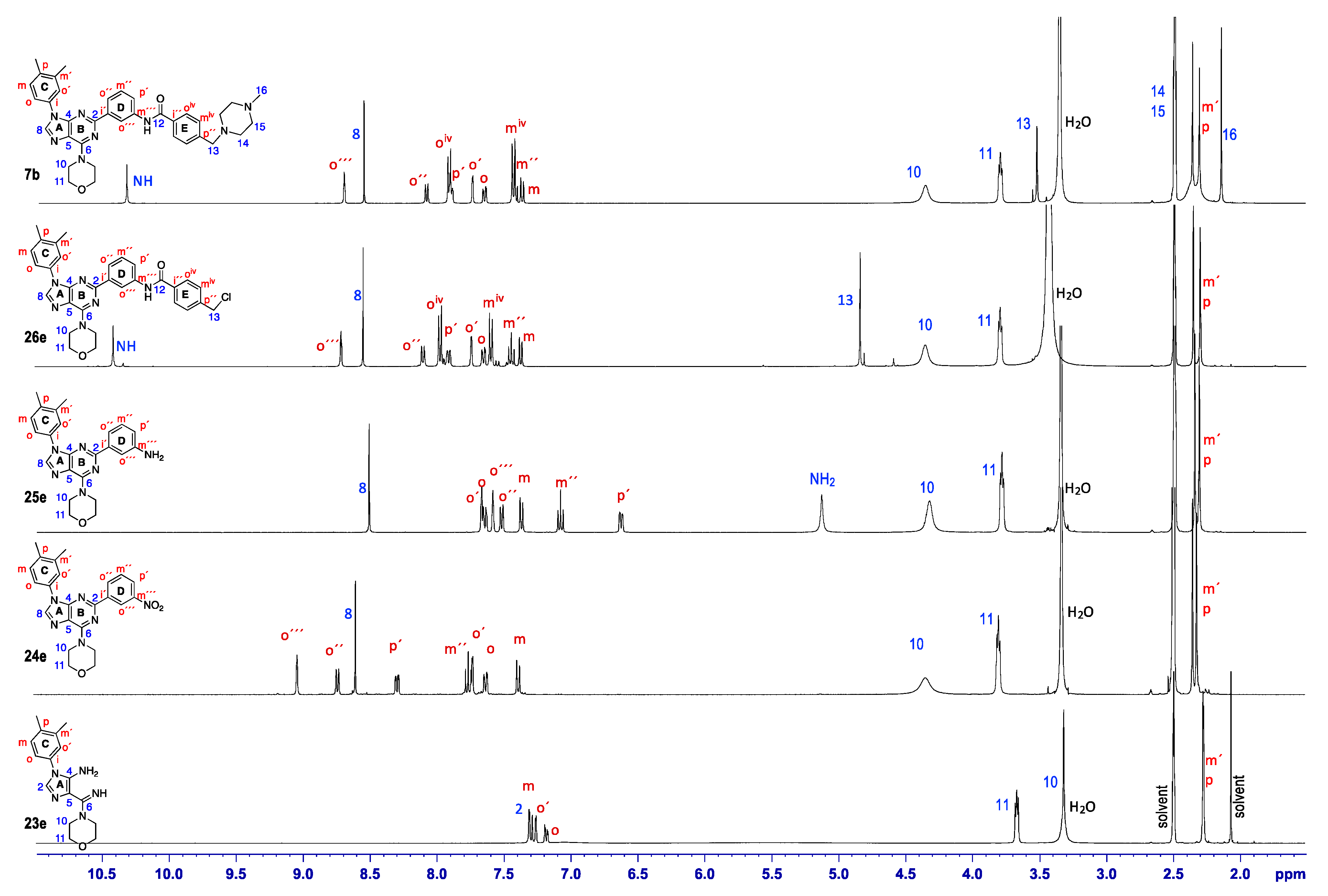
3.3. Characterisation of PI3K Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Elmenier, F.M.; Lasheen, D.S.; Abouzid, K.A.M. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur. J. Med. Chem. 2019, 183, 111718. [Google Scholar] [CrossRef]
- Helwa, A.A.; El-Dydamony, N.M.; Radwan, R.A.; Abdelraouf, S.M.; Abdelnaby, R.M. Novel antiproliferative agents bearing morpholinopyrimidine scaffold as PI3K inhibitors and apoptosis inducers; design, synthesis and molecular docking. Bioorganic Chem. 2020, 102, 104051. [Google Scholar] [CrossRef]
- Cintas, C.; Guillermet-Guibert, J. Heterogeneity of phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin activation in cancer: Is PI3k isoform specificity important? Front. Oncol. 2018, 7, 330. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Eisenreich, A.; Rauch, U. PI3K Inhibitors in Cardiovascular Disease. Cardiovasc. Ther. 2011, 29, 29–36. [Google Scholar] [CrossRef]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, e8. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Madsen, R.R.; Vanhaesebroeck, B. Cracking the context-specific PI3K signaling code. Sci. Signal. 2020, 13, eaay2940. [Google Scholar] [CrossRef] [Green Version]
- Rathinaswamy, M.K.; Burke, J.E. Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv. Biol. Regul. 2020, 75, 100657. [Google Scholar] [CrossRef]
- Hariri, S.; Rasti, B.; Mirpour, M.; Vaghar-Lahijani, G.; Attar, F.; Shiri, F. Structural insights into the origin of phosphoinositide 3-kinase inhibition. Struct. Chem. 2020, 31, 1505–1522. [Google Scholar] [CrossRef]
- Crabbe, T.; Welham, M.J.; Ward, S.G. The PI3K inhibitor arsenal: Choose your weapon! Trends Biochem. Sci. 2007, 32, 450–456. [Google Scholar] [CrossRef]
- De Santis, M.C.; Gulluni, F.; Campa, C.C.; Martini, M.; Hirsch, E. Targeting PI3K signaling in cancer: Challenges and advances. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 361–366. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.E.; Jackson, S.P. Class I phosphoinositide 3-kinases. Int. J. Biochem. Cell Biol. 2003, 35, 1028–1033. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Therville, N.; Guillermet-Guibert, J. Implication of PI3K/Akt pathway in pancreatic cancer: When PI3K isoforms matter? Adv. Biol. Regul. 2015, 59, 19–35. [Google Scholar] [CrossRef]
- Yu, B.; Li, L.; Zhang, J.; Wang, X.; Zeng, Y. Single-cell Sequencing and Methylation Methods and Clinical Applications. In Single-Cell Sequencing and Methylation; Springer: Singapore, 2020; ISBN 9789811544934. [Google Scholar]
- Wang, X.; Ding, J.; Meng, L.H. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef] [Green Version]
- Esposito, A.; Viale, G.; Curigliano, G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients with Cancer: A Review. JAMA Oncol. 2019, 5, 1347–1354. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Venkatesan, A.M.; Dehnhardt, C.M.; Chen, Z.; Santos, E.D.; Dos Santos, O.; Bursavich, M.; Gilbert, A.M.; Ellingboe, J.W.; Ayral-Kaloustian, S.; Khafizova, G.; et al. Novel imidazolopyrimidines as dual PI3-Kinase/mTOR inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 653–656. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Nishiyama, J.; Ellison, E.C.; Mizuno, G.R.; Chipault, J.R. Micro-determinations of alpha-tocopherol in tissue lipids. J. Nutr. Sci. Vitaminol. 1975, 21, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 720–723. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Fradera, X.; Methot, J.L.; Achab, A.; Christopher, M.; Altman, M.D.; Zhou, H.; McGowan, M.A.; Kattar, S.D.; Wilson, K.; Garcia, Y.; et al. Design of selective PI3Kδ inhibitors using an iterative scaffold-hopping workflow. Bioorganic Med. Chem. Lett. 2019, 29, 2575–2580. [Google Scholar] [CrossRef]
- Certal, V.; Carry, J.C.; Halley, F.; Virone-Oddos, A.; Thompson, F.; Filoche-Rommé, B.; El-Ahmad, Y.; Karlsson, A.; Charrier, V.; Delorme, C.; et al. Discovery and optimization of pyrimidone indoline amide PI3Kβ inhibitors for the treatment of phosphatase and tensin homologue (PTEN)-deficient cancers. J. Med. Chem. 2014, 57, 903–920. [Google Scholar] [CrossRef]
- Safina, B.S.; Elliott, R.L.; Forrest, A.K.; Heald, R.A.; Murray, J.M.; Nonomiya, J.; Pang, J.; Salphati, L.; Seward, E.M.; Staben, S.T.; et al. Design of Selective Benzoxazepin PI3Kδ Inhibitors Through Control of Dihedral Angles. ACS Med. Chem. Lett. 2017, 8, 936–940. [Google Scholar] [CrossRef]
- Henley, Z.A.; Amour, A.; Barton, N.; Bantscheff, M.; Bergamini, G.; Bertrand, S.M.; Convery, M.; Down, K.; Dümpelfeld, B.; Edwards, C.D.; et al. Optimization of Orally Bioavailable PI3KδInhibitors and Identification of Vps34 as a Key Selectivity Target. J. Med. Chem. 2020, 63, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, 162–173. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: http://www.pymol.org/pymol (accessed on 15 December 2021).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- IBM Corp. IBM SPSS Statistics for Windows; Version 26.0; IBM Corp.: Armonk, NY, USA, 2019. [Google Scholar]
- Jollife, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 7391–7401. [Google Scholar] [CrossRef] [Green Version]
- Alves, M.J.; Booth, B.L.; Kl-Duaj, O.; Eastwood, P.; Nezhat, L.; Proença, M.F.; Ramos, A.S. No Title. J. Chem. Res. (S) 1993, 402, 2701. [Google Scholar]
- Alves, M.J.; Carvalho, M.A.; Carvalho, S.; Dias, A.M.; Fernandes, F.H.; Proença, M.F. A new approach to the synthesis of N,N-dialkyladenine derivatives. Eur. J. Org. Chem. 2007, 2007, 4881–4887. [Google Scholar] [CrossRef]
- Correia, C.; Carvalho, M.A.; Proença, M.F. Synthesis and in vitro activity of 6-amino-2,9-diarylpurines for Mycobacterium tuberculosis. Tetrahedron 2009, 65, 6903–6911. [Google Scholar] [CrossRef]
- Correia, C.; Carvalho, M.A.; Rocha, A.; Proença, M.F. General synthetic approach to 2-phenolic adenine derivatives. Synlett 2012, 23, 1923–1926. [Google Scholar] [CrossRef] [Green Version]
- Ashly Rocha, M.F.; Proença, M.A.C. Synthesis of 2-(Aminophenyl)adenine Derivatives: A Simple Protocol using the Classical Iron Powder/Acetic Acid Reduction Methodology. Can. J. Chem. 2020, 98, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Correcting the impact of docking pose generation error on binding affinity prediction. BMC Bioinform. 2016, 17, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Cortés Franco, K.-D.; Brakmann, I.C.; Feoktistova, M.; Panayotova-Dimitrova, D.; Gründer, S.; Tian, Y. Aggressive migration in acidic pH of a glioblastoma cancer stem cell line in vitro is independent of ASIC and KCa3.1 ion channels, but involves phosphoinositide 3-kinase. Pflugers Arch. Eur. J. Physiol. 2023, 475, 405–416. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
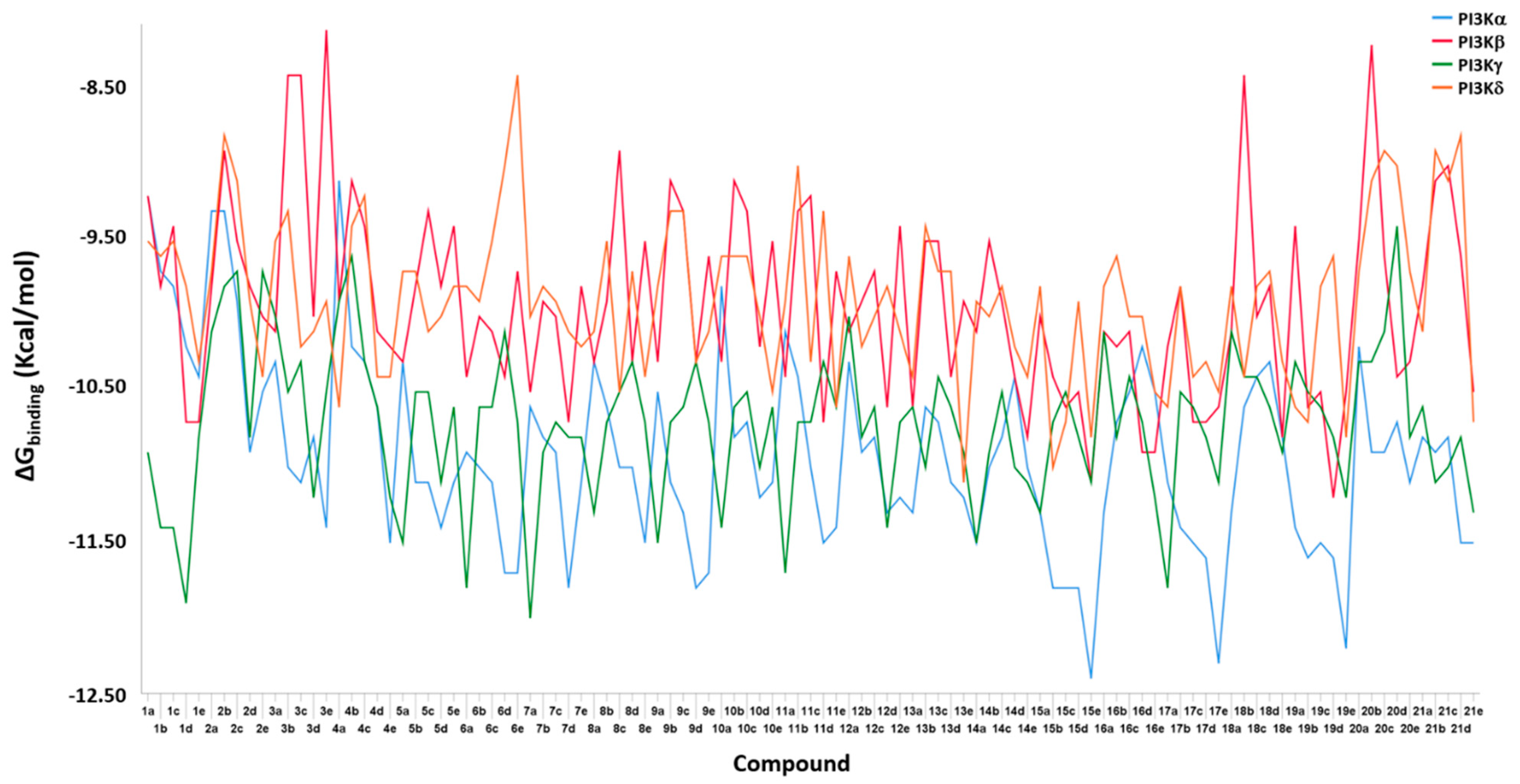
 |  | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Physicochemical and Structural Properties | ΔGbinding (kcal/mol) | ||||||||
| Charge | HBA | HBD | LogP | Mw | Rf | PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | ||
 1 | a | +1 | 8 | 5 | 2.33 | 429.48 | 134.82 | −9.2 | −9.2 | −10.9 | −9.5 |
| b | +1 | 9 | 3 | 3.40 | 512.62 | 160.94 | −9.7 | −9.8 | −11.4 | −9.6 | |
| c | 0 | 11 | 2 | 3.28 | 499.58 | 153.96 | −9.8 | −9.4 | −11.4 | −9.5 | |
| d | 0 | 9 | 3 | 4.93 | 505.58 | 161.15 | −10.2 | −10.7 | −11.9 | −9.8 | |
| e | 0 | 10 | 3 | 4.38 | 533.59 | 164.93 | −10.4 | −10.7 | −10.8 | −10.3 | |
 2 | a | +1 | 8 | 4 | 3.01 | 443.51 | 139.72 | −9.3 | −9.8 | −10.1 | −9.7 |
| b | +1 | 9 | 2 | 3.64 | 526.65 | 165.84 | −9.3 | −8.9 | −9.8 | −8.8 | |
| c | 0 | 11 | 1 | 3.52 | 513.60 | 158.86 | −9.9 | −9.5 | −9.7 | −9.1 | |
| d | 0 | 9 | 2 | 5.16 | 519.61 | 166.05 | −10.9 | −9.8 | −10.8 | −9.9 | |
| e | 0 | 10 | 2 | 4.62 | 547.62 | 169.83 | −10.5 | −10.0 | −9.7 | −10.4 | |
 3 | a | +1 | 8 | 4 | 4.66 | 505.58 | 169.81 | −10.3 | −10.1 | −10.0 | −9.5 |
| b | +1 | 9 | 2 | 5.29 | 588.72 | 195.93 | −11.0 | −8.4 | −10.5 | −9.3 | |
| c | 0 | 11 | 1 | 5.17 | 575.67 | 188.96 | −11.1 | −8.4 | −10.3 | −10.2 | |
| d | 0 | 9 | 2 | 6.81 | 581.68 | 196.15 | −10.8 | −10.0 | −11.2 | −10.1 | |
| e | 0 | 10 | 2 | 6.27 | 609.69 | 199.93 | −11.4 | −8.1 | −10.5 | −9.9 | |
 4 | a | +1 | 8 | 4 | 4.74 | 519.61 | 164.33 | −9.1 | −9.9 | −9.9 | −10.6 |
| b | +1 | 9 | 2 | 5.37 | 602.74 | 190.45 | −10.2 | −9.1 | −9.6 | −9.4 | |
| c | 0 | 11 | 1 | 5.25 | 589.70 | 183.47 | −10.3 | −9.4 | −10.3 | −9.2 | |
| d | 0 | 9 | 2 | 6.89 | 595.71 | 190.66 | −10.6 | −10.1 | −10.6 | −10.4 | |
| e | 0 | 10 | 2 | 6.35 | 623.72 | 194.44 | −11.5 | −10.2 | −11.2 | −10.4 | |
 5 | a | +1 | 8 | 4 | 5.16 | 519.61 | 174.85 | −10.3 | −10.3 | −11.5 | −9.7 |
| b | +1 | 9 | 2 | 5.79 | 602.74 | 200.97 | −11.1 | −9.8 | −10.5 | −9.7 | |
| c | 0 | 11 | 1 | 5.67 | 589.70 | 194.00 | −11.1 | −9.3 | −10.5 | −10.1 | |
| d | 0 | 9 | 2 | 7.31 | 595.71 | 201.19 | −11.4 | −9.8 | −11.1 | −10.0 | |
| e | 0 | 10 | 2 | 6.77 | 623.72 | 204.97 | −11.1 | −9.4 | −10.6 | −9.8 | |
 6 | a | +1 | 8 | 4 | 5.16 | 519.61 | 174.85 | −10.9 | −10.4 | −11.8 | −9.8 |
| b | +1 | 9 | 2 | 5.79 | 602.74 | 200.97 | −11.0 | −10.0 | −10.6 | −9.9 | |
| c | 0 | 11 | 1 | 5.67 | 589.70 | 194.00 | −11.1 | −10.1 | −10.6 | −9.5 | |
| d | 0 | 9 | 2 | 7.31 | 595.71 | 201.19 | −11.7 | −10.4 | −10.1 | −9.0 | |
| e | 0 | 10 | 2 | 6.77 | 623.72 | 204.97 | −11.7 | −9.7 | −10.7 | −8.4 | |
 7 | a | +1 | 8 | 4 | 5.66 | 533.64 | 179.90 | −10.6 | −10.5 | −12.0 | −10.0 |
| b | +1 | 9 | 2 | 6.29 | 616.77 | 206.02 | −10.8 | −9.9 | −10.9 | −9.8 | |
| c | 0 | 11 | 1 | 6.17 | 603.73 | 199.04 | −10.9 | −10.0 | −10.7 | −9.9 | |
| d | 0 | 9 | 2 | 7.81 | 609.73 | 206.23 | −11.8 | −10.7 | −10.8 | −10.1 | |
| e | 0 | 10 | 2 | 7.27 | 637.74 | 210.01 | −11.1 | −9.8 | −10.8 | −10.2 | |
 8 | a | +1 | 8 | 4 | 4.81 | 523.57 | 170.03 | −10.3 | −10.3 | −11.3 | −10.1 |
| b | +1 | 9 | 2 | 5.44 | 606.71 | 196.15 | −10.6 | −9.9 | −10.7 | −9.5 | |
| c | 0 | 11 | 1 | 5.32 | 593.66 | 189.17 | −11.0 | −8.9 | −10.5 | −10.5 | |
| d | 0 | 9 | 2 | 6.96 | 599.67 | 196.36 | −11.0 | −10.3 | −10.3 | −9.7 | |
| e | 0 | 10 | 2 | 6.42 | 627.68 | 200.14 | −11.5 | −9.5 | −10.7 | −10.4 | |
 9 | a | +1 | 8 | 4 | 4.81 | 523.57 | 170.03 | −10.5 | −10.3 | −11.5 | −9.8 |
| b | +1 | 9 | 2 | 5.44 | 606.71 | 196.15 | −11.1 | −9.1 | −10.7 | −9.3 | |
| c | 0 | 11 | 1 | 5.32 | 593.66 | 189.17 | −11.3 | −9.3 | −10.6 | −9.3 | |
| d | 0 | 9 | 2 | 6.96 | 599.67 | 196.36 | −11.8 | −10.3 | −10.3 | −10.3 | |
| e | 0 | 10 | 2 | 6.42 | 627.68 | 200.14 | −11.7 | −9.6 | −10.7 | −10.1 | |
 10 | a | +1 | 8 | 4 | 5.23 | 540.02 | 174.62 | −9.8 | −10.3 | −11.4 | −9.6 |
| b | +1 | 9 | 2 | 5.86 | 623.16 | 200.74 | −10.8 | −9.1 | −10.6 | −9.6 | |
| c | 0 | 11 | 1 | 5.74 | 610.12 | 193.76 | −10.7 | −9.3 | −10.5 | −9.6 | |
| d | 0 | 9 | 2 | 7.38 | 616.12 | 200.95 | −11.2 | −10.2 | −11.0 | −10.0 | |
| e | 0 | 10 | 2 | 6.83 | 644.13 | 204.73 | −11.1 | −9.5 | −10.6 | −10.5 | |
 11 | a | +1 | 8 | 4 | 5.79 | 574.47 | 179.42 | −10.1 | −10.4 | −11.7 | −9.9 |
| b | +1 | 9 | 2 | 6.42 | 657.60 | 205.54 | −10.4 | −9.3 | −10.7 | −9.0 | |
| c | 0 | 11 | 1 | 6.30 | 644.56 | 198.57 | −11.0 | −9.2 | −10.7 | −10.3 | |
| d | 0 | 9 | 2 | 7.94 | 650.56 | 205.76 | −11.5 | −10.7 | −10.3 | −9.3 | |
| e | 0 | 10 | 2 | 7.40 | 678.57 | 209.54 | −11.4 | −9.7 | −10.6 | −10.6 | |
 12 | a | +1 | 10 | 5 | 3.99 | 562.63 | 184.68 | −10.3 | −10.1 | −10.0 | −9.6 |
| b | +1 | 11 | 3 | 4.62 | 645.77 | 210.80 | −10.9 | −9.9 | −10.8 | −10.2 | |
| c | 0 | 13 | 2 | 4.50 | 632.73 | 203.82 | −10.8 | −9.7 | −10.6 | −10.0 | |
| d | 0 | 11 | 3 | 6.14 | 638.73 | 211.01 | −11.3 | −10.6 | −11.4 | −9.8 | |
| e | 0 | 12 | 3 | 5.60 | 666.74 | 214.79 | −11.2 | −9.4 | −10.7 | −10.1 | |
 13 | a | +1 | 10 | 5 | 3.99 | 562.63 | 184.68 | −11.3 | −10.6 | −10.6 | −10.4 |
| b | +1 | 11 | 3 | 4.62 | 645.77 | 210.80 | −10.6 | −9.5 | −11.0 | −9.4 | |
| c | 0 | 13 | 2 | 4.50 | 632.73 | 203.82 | −10.7 | −9.5 | −10.4 | −9.7 | |
| d | 0 | 11 | 3 | 6.14 | 638.73 | 211.01 | −11.1 | −10.4 | −10.6 | −9.7 | |
| e | 0 | 12 | 3 | 5.60 | 666.74 | 214.79 | −11.2 | −9.9 | −10.9 | −11.1 | |
 14 | a | +1 | 10 | 5 | 5.72 | 624.71 | 205.35 | −11.5 | −10.1 | −11.5 | −9.9 |
| b | +1 | 11 | 3 | 6.35 | 707.84 | 231.47 | −11.0 | −9.5 | −10.9 | −10.0 | |
| c | 0 | 13 | 2 | 6.23 | 694.80 | 224.49 | −10.8 | −9.9 | −10.5 | −9.8 | |
| d | 0 | 11 | 3 | 7.87 | 700.80 | 231.68 | −10.4 | −10.4 | −11.0 | −10.2 | |
| e | 0 | 12 | 3 | 7.33 | 728.81 | 235.46 | −11.0 | −10.8 | −11.1 | −10.4 | |
 15 | a | +1 | 10 | 5 | 5.72 | 624.71 | 205.35 | −11.3 | −10.0 | −11.3 | −9.8 |
| b | +1 | 11 | 3 | 6.35 | 707.84 | 231.47 | −11.8 | −10.4 | −10.7 | −11.0 | |
| c | 0 | 13 | 2 | 6.23 | 694.80 | 224.49 | −11.8 | −10.6 | −10.5 | −10.7 | |
| d | 0 | 11 | 3 | 7.87 | 700.80 | 231.68 | −11.8 | −10.5 | −10.8 | −9.9 | |
| e | 0 | 12 | 3 | 7.33 | 728.81 | 235.46 | −12.4 | −11.1 | −11.1 | −10.8 | |
 16 | a | +1 | 11 | 5 | 4.49 | 625.69 | 203.19 | −11.3 | −10.1 | −10.1 | −9.8 |
| b | +1 | 12 | 3 | 5.12 | 708.83 | 229.31 | −10.7 | −10.2 | −10.8 | −9.6 | |
| c | 0 | 14 | 2 | 5.00 | 695.78 | 222.33 | −10.5 | −10.1 | −10.4 | −10.0 | |
| d | 0 | 12 | 3 | 6.64 | 701.79 | 229.52 | −10.2 | −10.9 | −10.7 | −10.0 | |
| e | 0 | 13 | 3 | 6.10 | 729.80 | 233.30 | −10.5 | −10.9 | −11.2 | −10.5 | |
 17 | a | +1 | 11 | 5 | 4.49 | 625.69 | 203.19 | −11.1 | −10.2 | −11.8 | −10.6 |
| b | +1 | 12 | 3 | 5.12 | 708.83 | 229.31 | −11.4 | −9.8 | −10.5 | −9.8 | |
| c | 0 | 14 | 2 | 5.00 | 695.78 | 222.33 | −11.5 | −10.7 | −10.6 | −10.4 | |
| d | 0 | 12 | 3 | 6.64 | 701.79 | 229.52 | −11.6 | −10.7 | −10.8 | −10.3 | |
| e | 0 | 13 | 3 | 6.10 | 729.80 | 233.30 | −12.3 | −10.6 | −11.1 | −10.5 | |
 18 | a | +1 | 11 | 5 | 4.49 | 625.69 | 203.19 | −11.3 | −10.1 | −10.1 | −9.8 |
| b | +1 | 12 | 3 | 5.12 | 708.83 | 229.31 | −10.6 | −8.4 | −10.4 | −10.4 | |
| c | 0 | 14 | 2 | 5.00 | 695.78 | 222.33 | −10.4 | −10.0 | −10.4 | −9.8 | |
| d | 0 | 12 | 3 | 6.64 | 701.79 | 229.52 | −10.3 | −9.8 | −10.6 | −9.7 | |
| e | 0 | 13 | 3 | 6.10 | 729.80 | 233.30 | −10.8 | −10.8 | −10.9 | −10.3 | |
 19 | a | +1 | 11 | 5 | 4.49 | 625.69 | 203.19 | −11.4 | −9.4 | −10.3 | −10.6 |
| b | +1 | 12 | 3 | 5.12 | 708.83 | 229.31 | −11.6 | −10.6 | −10.5 | −10.7 | |
| c | 0 | 14 | 2 | 5.00 | 695.78 | 222.33 | −11.5 | −10.5 | −10.6 | −9.8 | |
| d | 0 | 12 | 3 | 6.64 | 701.79 | 229.52 | −11.6 | −11.2 | −10.8 | −9.6 | |
| e | 0 | 13 | 3 | 6.10 | 729.80 | 233.30 | −12.2 | −10.5 | −11.2 | −10.8 | |
 20 | a | +1 | 16 | 5 | 5.14 | 714.78 | 224.74 | −10.2 | −9.5 | −10.3 | −9.7 |
| b | +1 | 17 | 3 | 5.76 | 797.92 | 250.86 | −10.9 | −8.2 | −10.3 | −9.1 | |
| c | 0 | 19 | 2 | 5.64 | 784.87 | 243.88 | −10.9 | −9.6 | −10.1 | −8.9 | |
| d | 0 | 17 | 3 | 7.29 | 790.89 | 251.07 | −10.7 | −10.4 | −9.4 | −9.0 | |
| e | 0 | 18 | 3 | 6.74 | 818.89 | 254.85 | −11.1 | −10.3 | −10.8 | −9.7 | |
 21 | a | +1 | 16 | 5 | 5.14 | 714.78 | 224.74 | −10.8 | −9.8 | −10.6 | −10.1 |
| b | +1 | 17 | 3 | 5.76 | 797.92 | 250.86 | −10.9 | −9.1 | −11.1 | −8.9 | |
| c | 0 | 19 | 2 | 5.64 | 784.87 | 243.88 | −10.8 | −9.0 | −11.0 | −9.1 | |
| d | 0 | 17 | 3 | 7.29 | 790.89 | 251.07 | −11.5 | −9.6 | −10.8 | −8.8 | |
| e | 0 | 18 | 3 | 6.74 | 818.89 | 254.85 | −11.5 | −10.5 | −11.3 | −10.7 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, V.; Rocha, A.; Castro, T.G.; Carvalho, M.A. Synthesis of Novel 2,9-Disubstituted-6-morpholino Purine Derivatives Assisted by Virtual Screening and Modelling of Class I PI3K Isoforms. Polymers 2023, 15, 1703. https://doi.org/10.3390/polym15071703
Lobo V, Rocha A, Castro TG, Carvalho MA. Synthesis of Novel 2,9-Disubstituted-6-morpholino Purine Derivatives Assisted by Virtual Screening and Modelling of Class I PI3K Isoforms. Polymers. 2023; 15(7):1703. https://doi.org/10.3390/polym15071703
Chicago/Turabian StyleLobo, Vítor, Ashly Rocha, Tarsila G. Castro, and Maria Alice Carvalho. 2023. "Synthesis of Novel 2,9-Disubstituted-6-morpholino Purine Derivatives Assisted by Virtual Screening and Modelling of Class I PI3K Isoforms" Polymers 15, no. 7: 1703. https://doi.org/10.3390/polym15071703
APA StyleLobo, V., Rocha, A., Castro, T. G., & Carvalho, M. A. (2023). Synthesis of Novel 2,9-Disubstituted-6-morpholino Purine Derivatives Assisted by Virtual Screening and Modelling of Class I PI3K Isoforms. Polymers, 15(7), 1703. https://doi.org/10.3390/polym15071703