
Thermal Conductivities of Uniform and Random Sulfur Crosslinking in Polybutadiene by Molecular Dynamic Simulation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Green–Kubo Method for Calculating Thermal Conductivity
2.2. Degree of Crosslinking
3. Results and Discussion
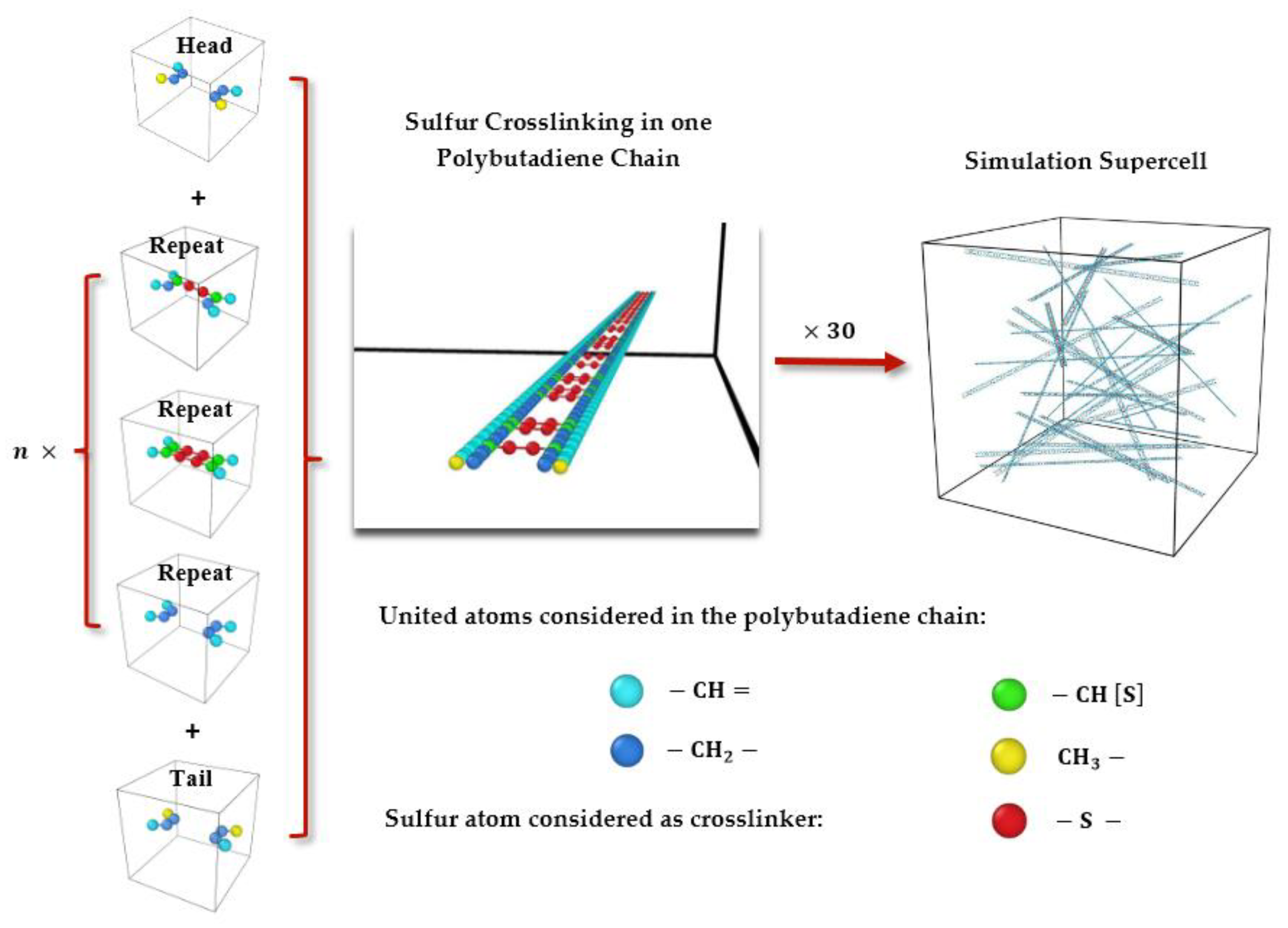
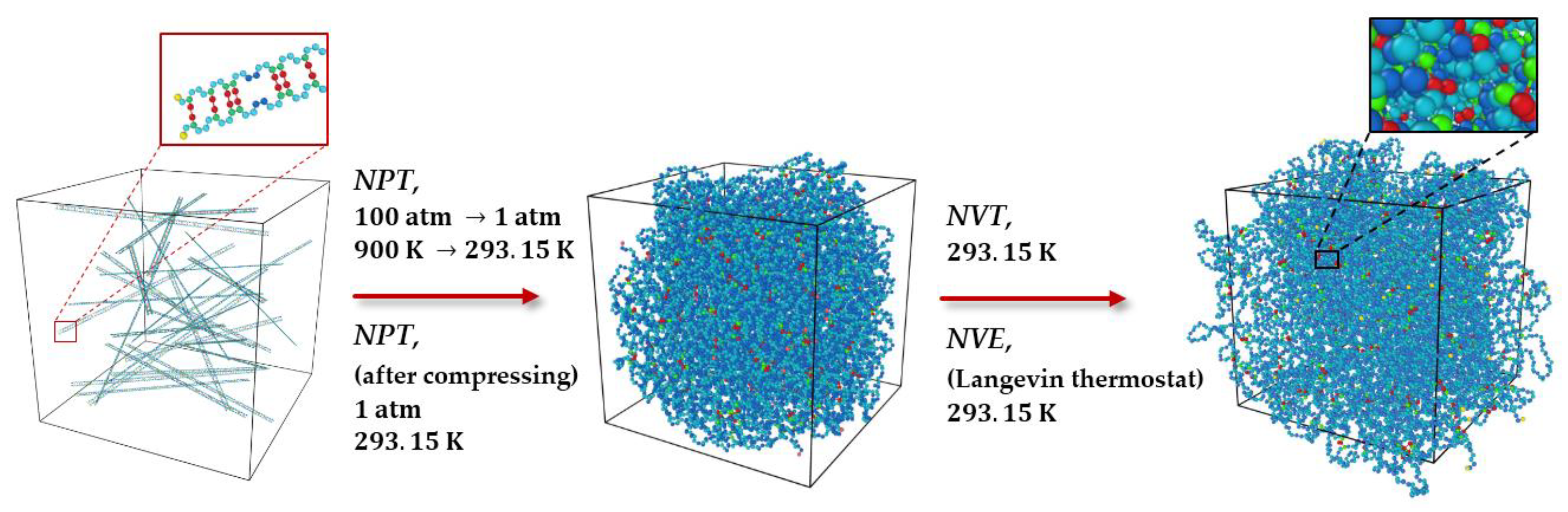
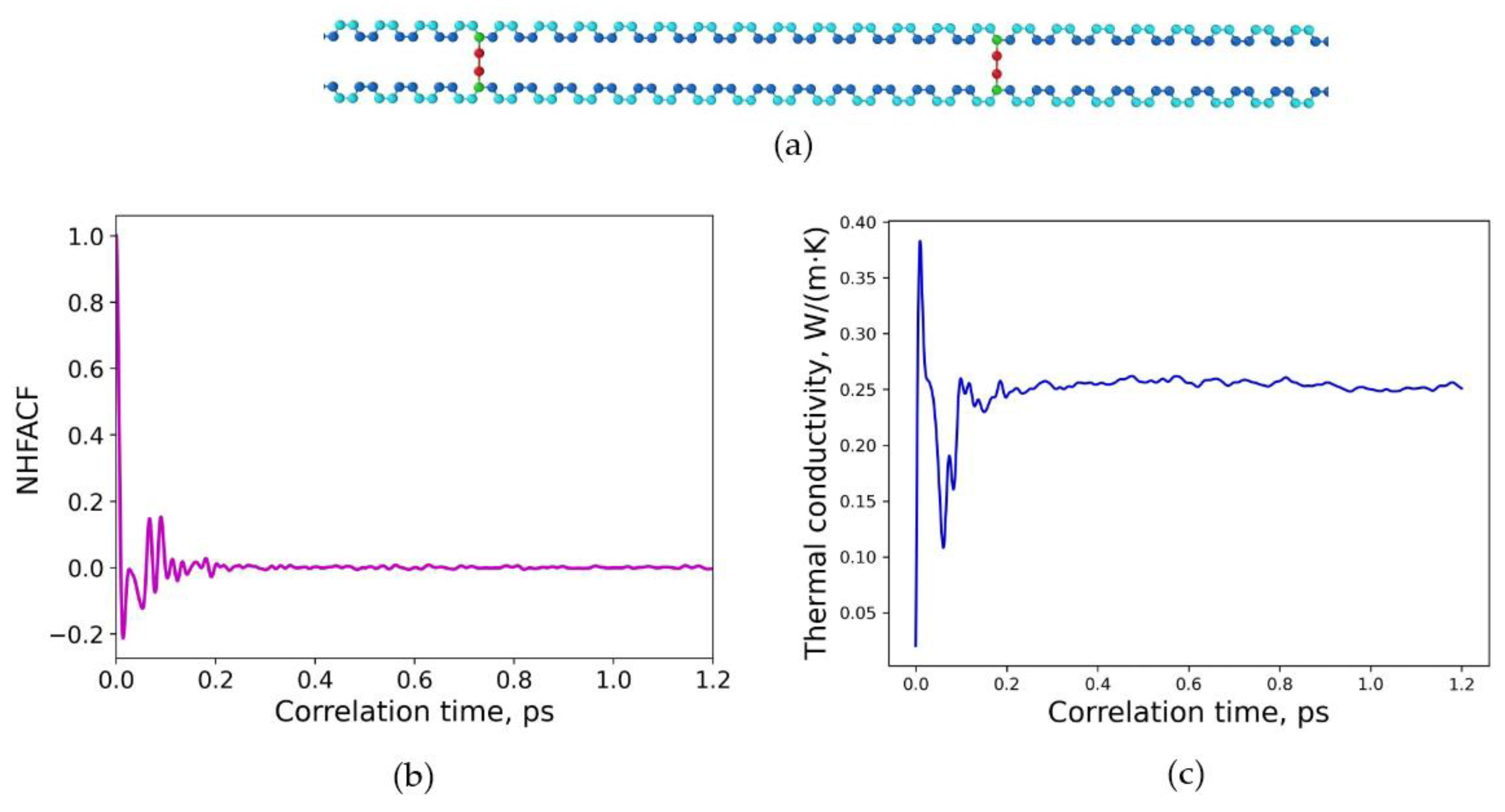
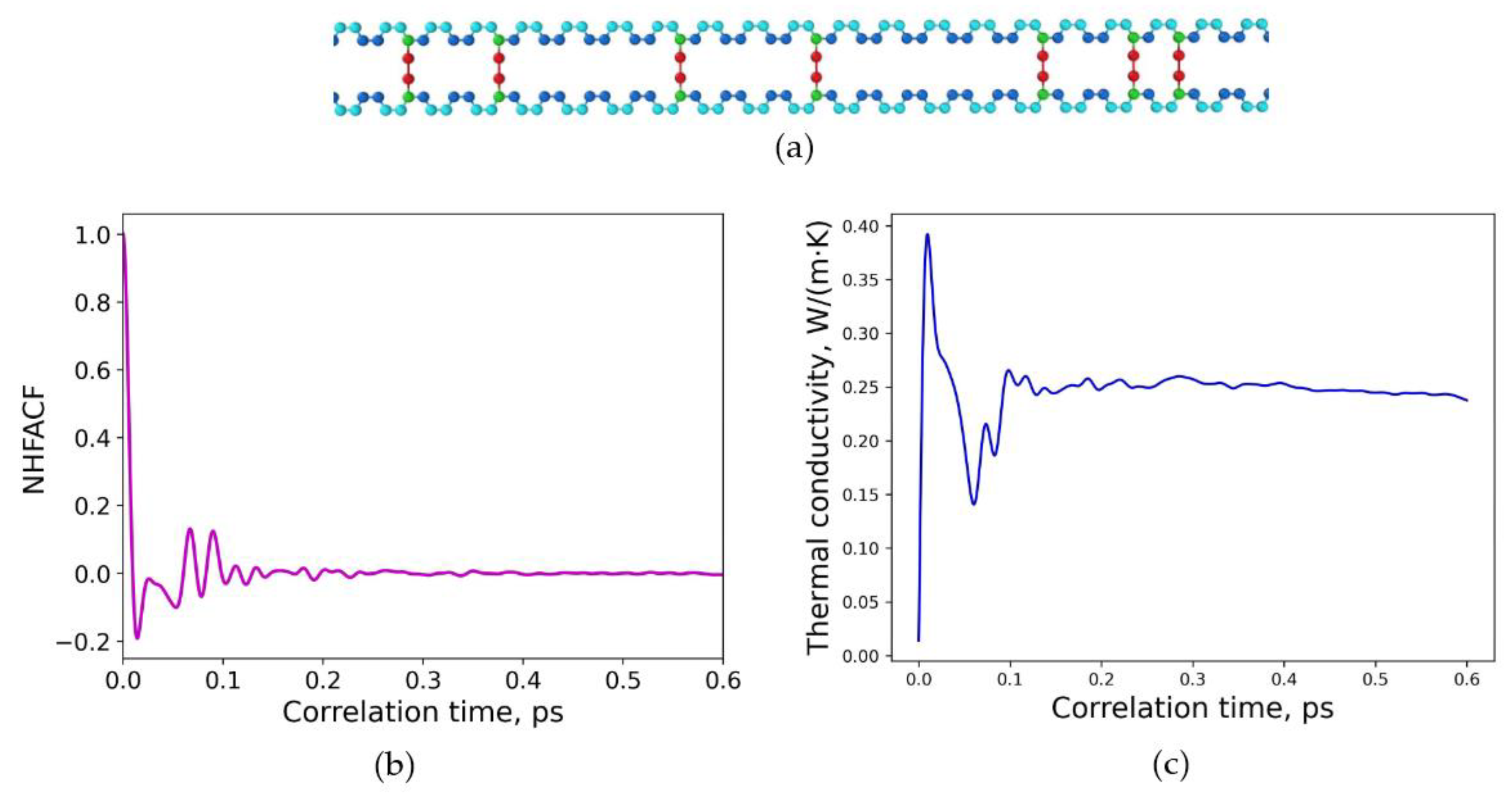
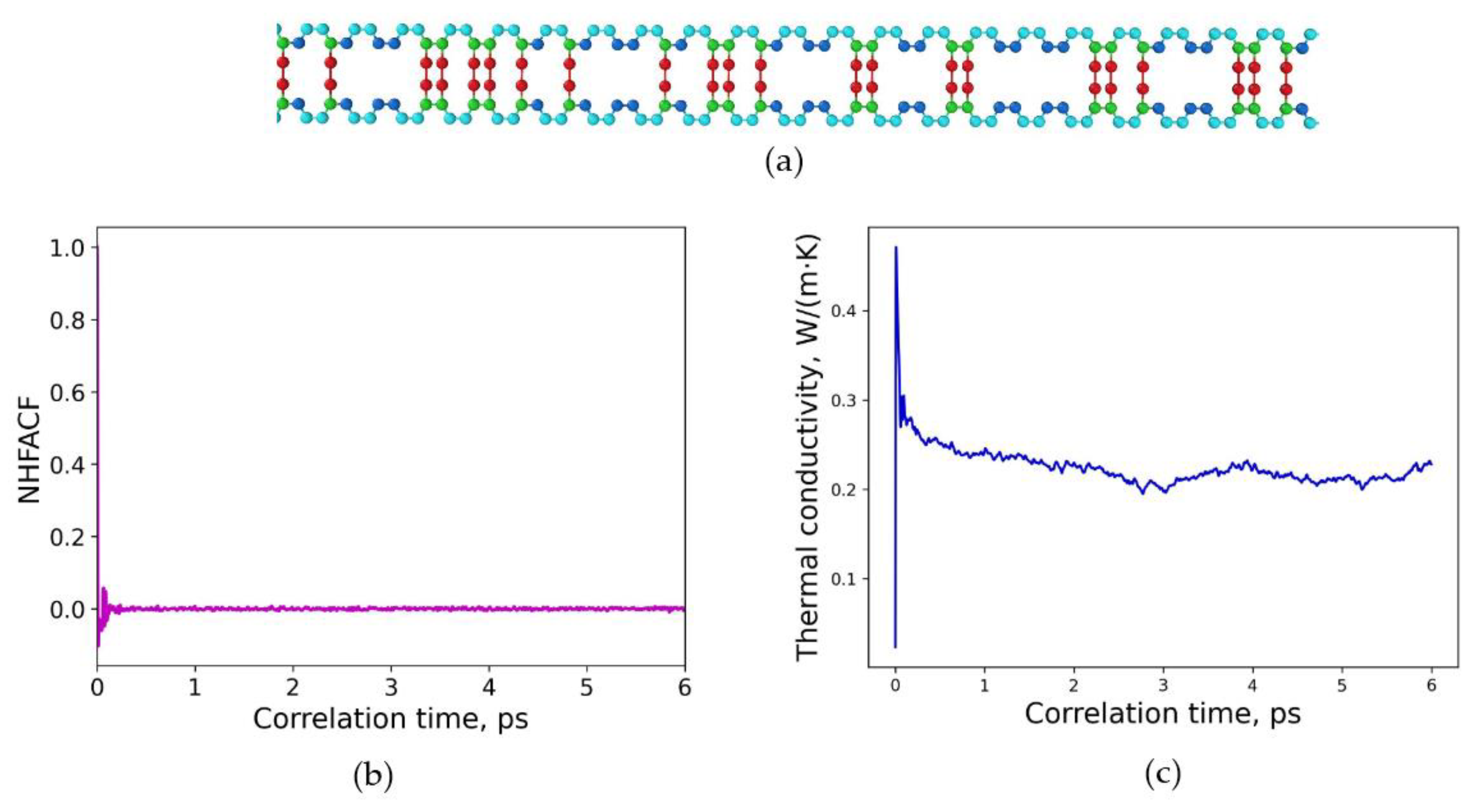
3.1. Polymeric Model Structures for MD Simulations
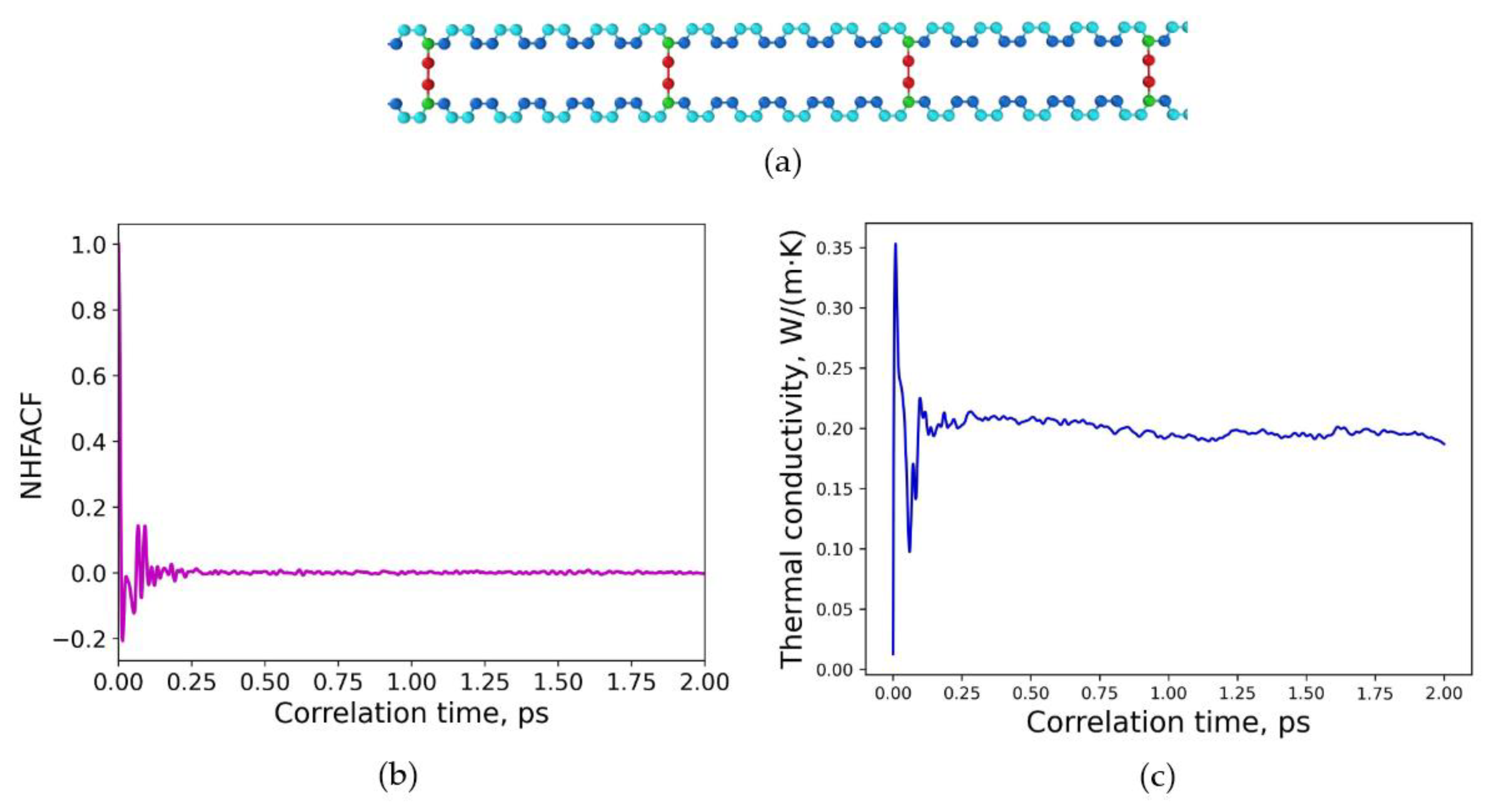
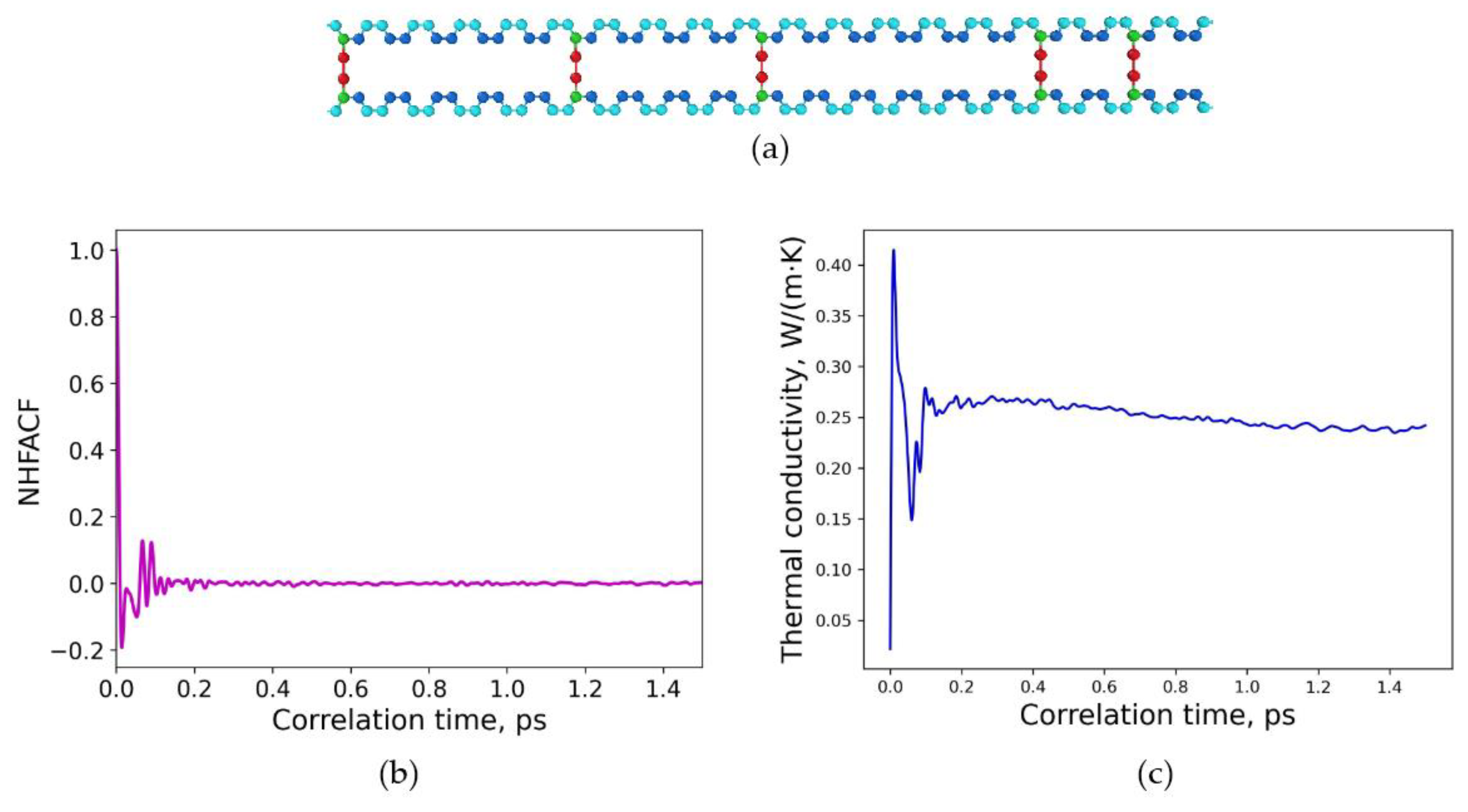
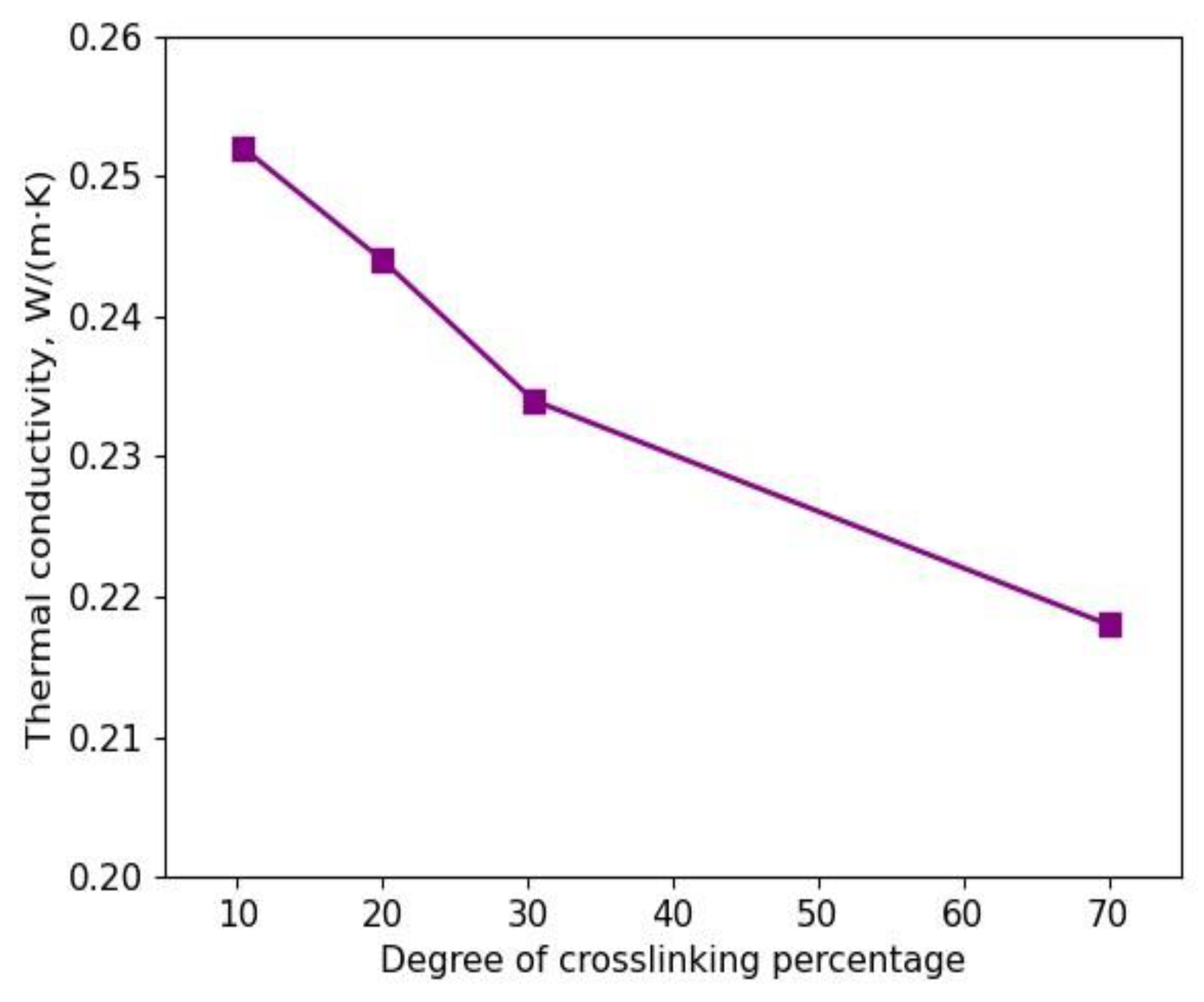
3.2. Determination of Thermal Conductivities for Uniform and Non-Uniform Crosslinked Polymers with Varying Degrees of Sulfur Crosslinking Bridges
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, S.-S.; Choi, S.-J. Influence of Silane Coupling Agent Content on Crosslink Type and Density of Silica-Filled Natural Rubber Vulcanizates. Bull. Korean Chem. Soc. 2006, 27, 1473–1476. [Google Scholar] [CrossRef] [Green Version]
- Doherty, D.C.; Holmes, B.N.; Leung, P.; Ross, R.B. Polymerization Molecular Dynamics Simulations. I. Cross-Linked Atomistic Models for Poly(Methacrylate) Networks. Comput. Theor. Polym. Sci. 1998, 8, 169–178. [Google Scholar] [CrossRef]
- Komarov, P.V.; Yu-Tsung, C.; Shih-Ming, C.; Khalatur, P.G.; Reineker, P. Highly Cross-Linked Epoxy Resins: An Atomistic Molecular Dynamics Simulation Combined with a Mapping/Reverse Mapping Procedure. Macromolecules 2007, 40, 8104–8113. [Google Scholar] [CrossRef]
- Varshney, V.; Patnaik, S.S.; Roy, A.K.; Farmer, B.L. A Molecular Dynamics Study of Epoxy-Based Networks: Cross-Linking Procedure and Prediction of Molecular and Material Properties. Macromolecules 2008, 41, 6837–6842. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Ryde, U. Comparison of Methods for Deriving Atomic Charges from the Electrostatic Potential and Moments. J. Comput. Chem. 1998, 19, 377–395. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Bernardes, C.E.S.; Joseph, A. Evaluation of the OPLS-AA Force Field for the Study of Structural and Energetic Aspects of Molecular Organic Crystals. J. Phys. Chem. A 2015, 119, 3023–3034. [Google Scholar] [CrossRef]
- Yong, C.W. Descriptions and Implementations of DL_F Notation: A Natural Chemical Expression System of Atom Types for Molecular Simulations. J. Chem. Inf. Model. 2016, 56, 1405–1409. [Google Scholar] [CrossRef]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to Molecular Simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Novel Configurational-Bias Monte Carlo Method for Branched Molecules. Transferable Potentials for Phase Equilibria. 2. United-Atom Description of Branched Alkanes. J. Phys. Chem. B 1999, 103, 4508–4517. [Google Scholar] [CrossRef]
- Wick, C.D.; Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 4. United-Atom Description of Linear and Branched Alkenes and Alkylbenzenes. J. Phys. Chem. B 2000, 104, 8008–8016. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Potoff, J.J.; Siepmann, J.I. Monte Carlo Calculations for Alcohols and Their Mixtures with Alkanes. Transferable Potentials for Phase Equilibria. 5. United-Atom Description of Primary, Secondary, and Tertiary Alcohols. J. Phys. Chem. B 2001, 105, 3093–3104. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Potoff, J.J.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes. J. Phys. Chem. B 2004, 108, 17596–17605. [Google Scholar] [CrossRef]
- Yang, L.; Tan, C.-H.; Hsieh, M.-J.; Wang, J.; Duan, Y.; Cieplak, P.; Caldwell, J.; Kollman, P.A.; Luo, R. New-Generation Amber United-Atom Force Field. J. Phys. Chem. B 2006, 110, 13166–13176. [Google Scholar] [CrossRef]
- Tieleman, D.P.; Maccallum, J.L.; Ash, W.L.; Kandt, C.; Xu, Z.; Monticelli, L. Membrane Protein Simulations with a United-Atom Lipid and All-Atom Protein Model: Lipid-Protein Interactions, Side Chain Transfer Free Energies and Model Proteins. J. Phys. Condens. Matter. 2006, 18, S1221–S1234. [Google Scholar] [CrossRef]
- Kukol, A. Lipid Models for United-Atom Molecular Dynamics Simulations of Proteins. J. Chem. Theory Comput. 2009, 5, 615–626. [Google Scholar] [CrossRef] [Green Version]
- González, M.A. Force Fields and Molecular Dynamics Simulations. JDN 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Kawamura, T.; Kangawa, Y.; Kakimoto, K. Investigation of Thermal Conductivity of Nitride Mixed Crystals and Superlattices by Molecular Dynamics. Phys. Status Solidi c 2006, 3, 1695–1699. [Google Scholar] [CrossRef]
- Chantrenne, P.; Raynaud, M.; Baillis, D. Study of Phonon Heat Transfer in Metallic Solids from Molecular Dynamics Simulations. Microscale Thermophys. Eng. 2003, 7, 117–136. [Google Scholar] [CrossRef]
- Terao, T.; Lussetti, E.; Müller-Plathe, F. Nonequilibrium Molecular Dynamics Methods for Computing the Thermal Conductivity: Application to Amorphous Polymers. Phys. Rev. E 2007, 75, 057701. [Google Scholar] [CrossRef] [PubMed]
- Varshney, V.; Patnaik, S.S.; Roy, A.K.; Farmer, B.L. Heat Transport in Epoxy Networks: A Molecular Dynamics Study. Polymer 2009, 50, 3378–3385. [Google Scholar] [CrossRef]
- Kubo, R.; Toda, M.; Hashitsume, N.; Saito, N. Statistical Physics: Nonequilibrium Statistical Mechanics. In Statistical Physics II: Nonequilibrium Statistical Mechanics; Springer: Berlin, Germany, 1985. [Google Scholar]
- Evans, D.J.; Morriss, G.P. Statistical Mechanics of Nonequilibrium Liquids, 2nd ed.; ANU E Press: Canberra, Australia, 2007; ISBN 978-1-921313-22-6. [Google Scholar]
- McQuarrie, D.A. Mathematical Methods for Scientists and Engineers; University Science Books: Melville, NY, USA, 2003; ISBN 978-1-891389-24-5. [Google Scholar]
- Ladd, A.J.C.; Moran, B.; Hoover, W.G. Lattice Thermal Conductivity: A Comparison of Molecular Dynamics and Anharmonic Lattice Dynamics. Phys. Rev. B 1986, 34, 5058–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sundararaghavan, V.; Browning, A.R. Study of Temperature Dependence of Thermal Conductivity in Cross-Linked Epoxies Using Molecular Dynamics Simulations with Long Range Interactions. Modelling Simul. Mater. Sci. Eng. 2014, 22, 025013. [Google Scholar] [CrossRef] [Green Version]
- Kikugawa, G.; Desai, T.G.; Keblinski, P.; Ohara, T. Effect of Crosslink Formation on Heat Conduction in Amorphous Polymers. J. Appl. Phys. 2013, 114, 034302. [Google Scholar] [CrossRef] [Green Version]
- Jewett, A.I.; Stelter, D.; Lambert, J.; Saladi, S.M.; Roscioni, O.M.; Ricci, M.; Autin, L.; Maritan, M.; Bashusqeh, S.M.; Keyes, T.; et al. Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics. J. Mol. Biol. 2021, 433, 166841. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS-a Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Vasilev, A.; Lorenz, T.; Breitkopf, C. Thermal Conductivity of Polyisoprene and Polybutadiene from Molecular Dynamics Simulations and Transient Measurements. Polymers 2020, 12, 1081. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.; Stoll, E. Molecular-Dynamics Study of a Three-Dimensional One-Component Model for Distortive Phase Transitions. Phys. Rev. B 1978, 17, 1302–1322. [Google Scholar] [CrossRef]
- Vasilev, A.; Lorenz, T.; Kamble, V.G.; Wießner, S.; Breitkopf, C. Thermal Conductivity of Polybutadiene Rubber from Molecular Dynamics Simulations and Measurements by the Heat Flow Meter Method. Materials 2021, 14, 7737. [Google Scholar] [CrossRef]
- Yamamoto, O.; Kambe, H. Thermal Conductivity of Cross-Linked Polymers. A Comparison between Measured and Calculated Thermal Conductivities. Polym. J. 1971, 2, 623–628. [Google Scholar] [CrossRef]
- Xiong, X.; Yang, M.; Liu, C.; Li, X.; Tang, D. Thermal Conductivity of Cross-Linked Polyethylene from Molecular Dynamics Simulation. J. Appl. Phys. 2017, 122, 035104. [Google Scholar] [CrossRef]
- Rashidi, V.; Coyle, E.J.; Sebeck, K.; Kieffer, J.; Pipe, K.P. Thermal Conductance in Cross-Linked Polymers: Effects of Non-Bonding Interactions. J. Phys. Chem. B 2017, 121, 4600–4609. [Google Scholar] [CrossRef]
- Ni, B.; Watanabe, T.; Phillpot, S.R. Thermal Transport in Polyethylene and at Polyethylene-Diamond Interfaces Investigated Using Molecular Dynamics Simulation. J. Phys. Condens. Matter. 2009, 21, 084219. [Google Scholar] [CrossRef]
- Yu, S.; Park, C.; Hong, S.M.; Koo, C.M. Thermal Conduction Behaviors of Chemically Cross-Linked High-Density Polyethylenes. Thermochim. Acta 2014, 583, 67–71. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Force Field Parameters for Polybutadiene Crosslinked with Sulfur | |||
|---|---|---|---|
| 0.144 | 3.905 | ||
| 0.142 | 3.852 | ||
| 0.107 | 3.697 | ||
| 0.209 | 3.723 | ||
| 0.116 | 3.852 | ||
| 0.088 | 3.697 | ||
| 0.172 | 3.723 | ||
| 0.275 | 3.647 | ||
| 0.170 | 3.673 | ||
| 0.128 | 3.525 | ||
| 0.175 | 3.905 | ||
| 0.118 | 3.905 | ||
| 0.115 | 3.800 | ||
| 0.066 | 3.500 | ||
| 0.250 | 3.550 | ||
| 260.0 | 1.526 | ||
| 260.0 | 1.526 | ||
| 260.0 | 1.526 | ||
| 166.0 | 2.038 | ||
| 222.0 | 1.810 | ||
| 530.0 | 1.340 | ||
| 317.0 | 1.500 | ||
| 317.0 | 1.500 | ||
| 68.0 | 103.7 | ||
| 70.0 | 118.0 | ||
| 70.0 | 118.0 | ||
| 63.0 | 112.4 | ||
| --- | --- | ||
| --- | --- | ||
| --- | --- | ||
| --- | --- | ||
| –2.5 | 1.25 | 3.1 | |
| 0.0 | –7.414 | 1.705 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alamfard, T.; Lorenz, T.; Breitkopf, C. Thermal Conductivities of Uniform and Random Sulfur Crosslinking in Polybutadiene by Molecular Dynamic Simulation. Polymers 2023, 15, 2058. https://doi.org/10.3390/polym15092058
Alamfard T, Lorenz T, Breitkopf C. Thermal Conductivities of Uniform and Random Sulfur Crosslinking in Polybutadiene by Molecular Dynamic Simulation. Polymers. 2023; 15(9):2058. https://doi.org/10.3390/polym15092058
Chicago/Turabian StyleAlamfard, Tannaz, Tommy Lorenz, and Cornelia Breitkopf. 2023. "Thermal Conductivities of Uniform and Random Sulfur Crosslinking in Polybutadiene by Molecular Dynamic Simulation" Polymers 15, no. 9: 2058. https://doi.org/10.3390/polym15092058