
Influence of Cross-Linking and Crystalline Morphology on the Shape-Memory Properties of PET/PEN/PCL Copolyesters Using Trimesic Acid and Glycerol


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PET and PET/PEN/PCL Copolyesters
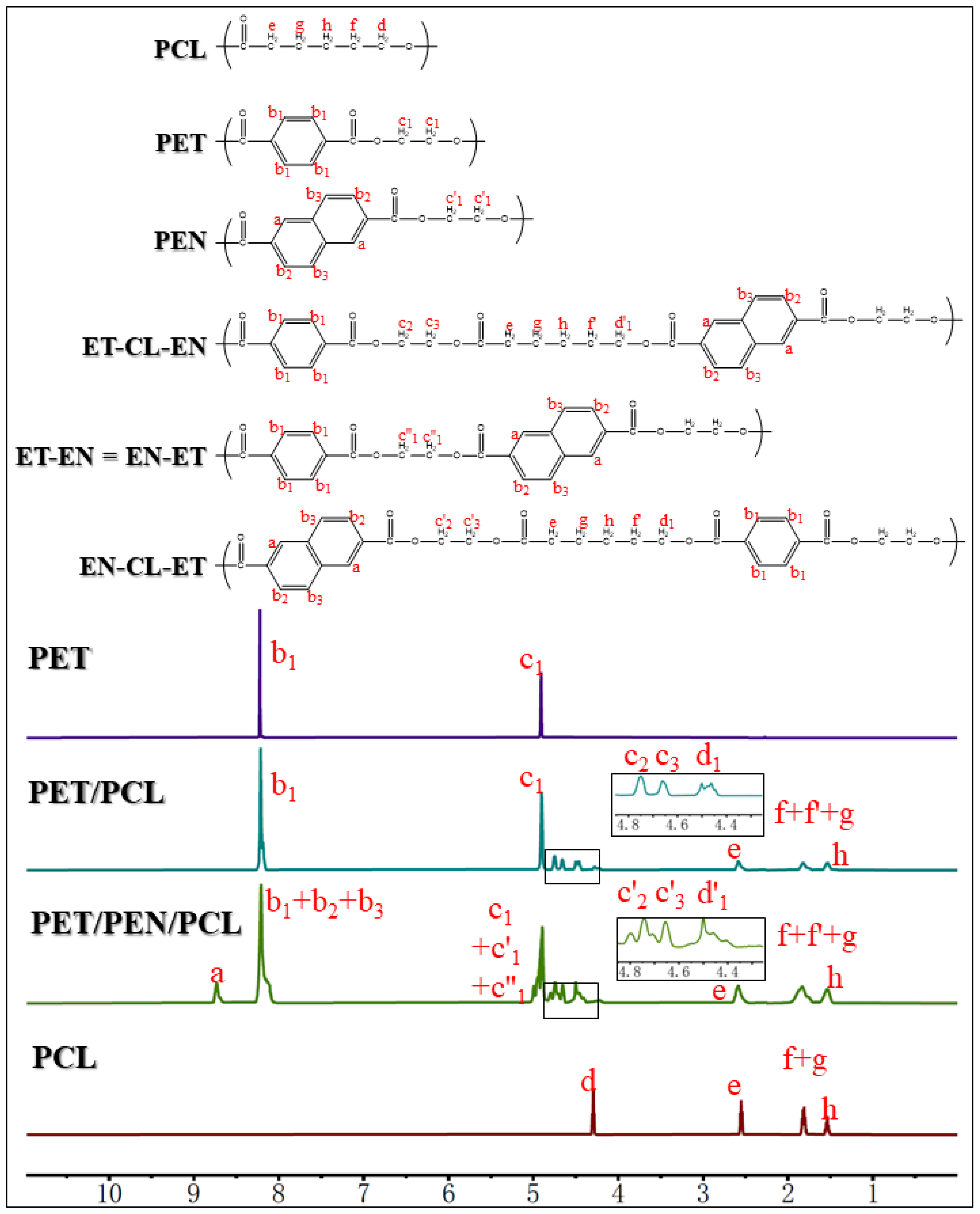
2.3. Nuclear Magnetic Resonance Spectroscopic Analysis
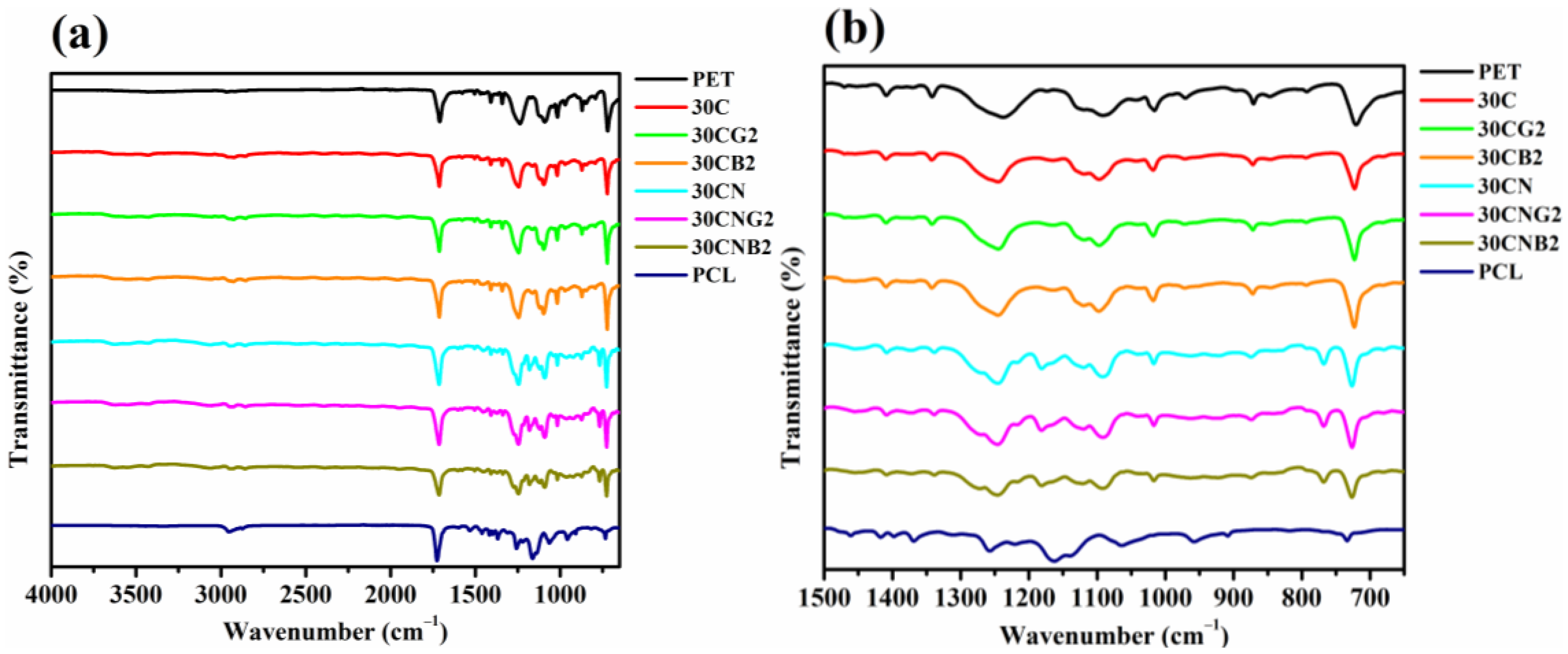
2.4. Fourier Transform Infrared Spectroscopy
2.5. Intrinsic Viscosity
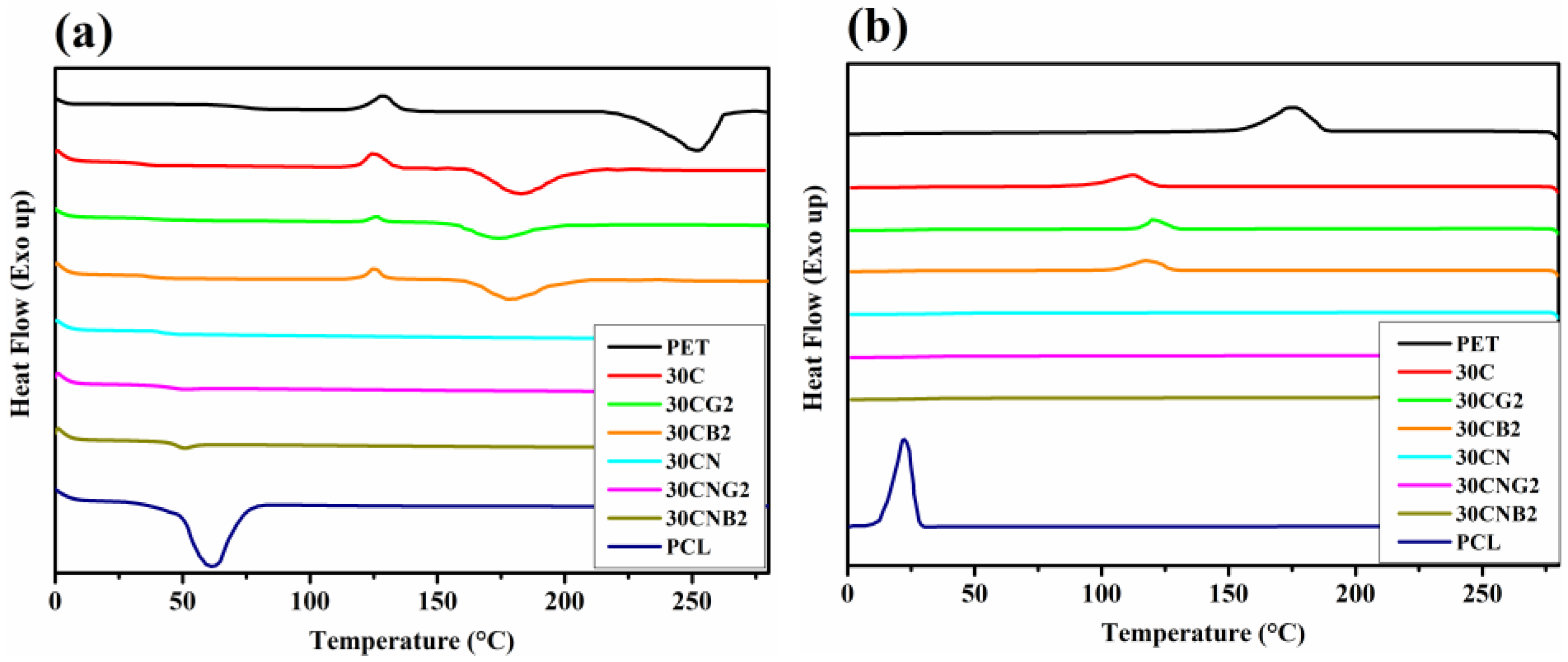
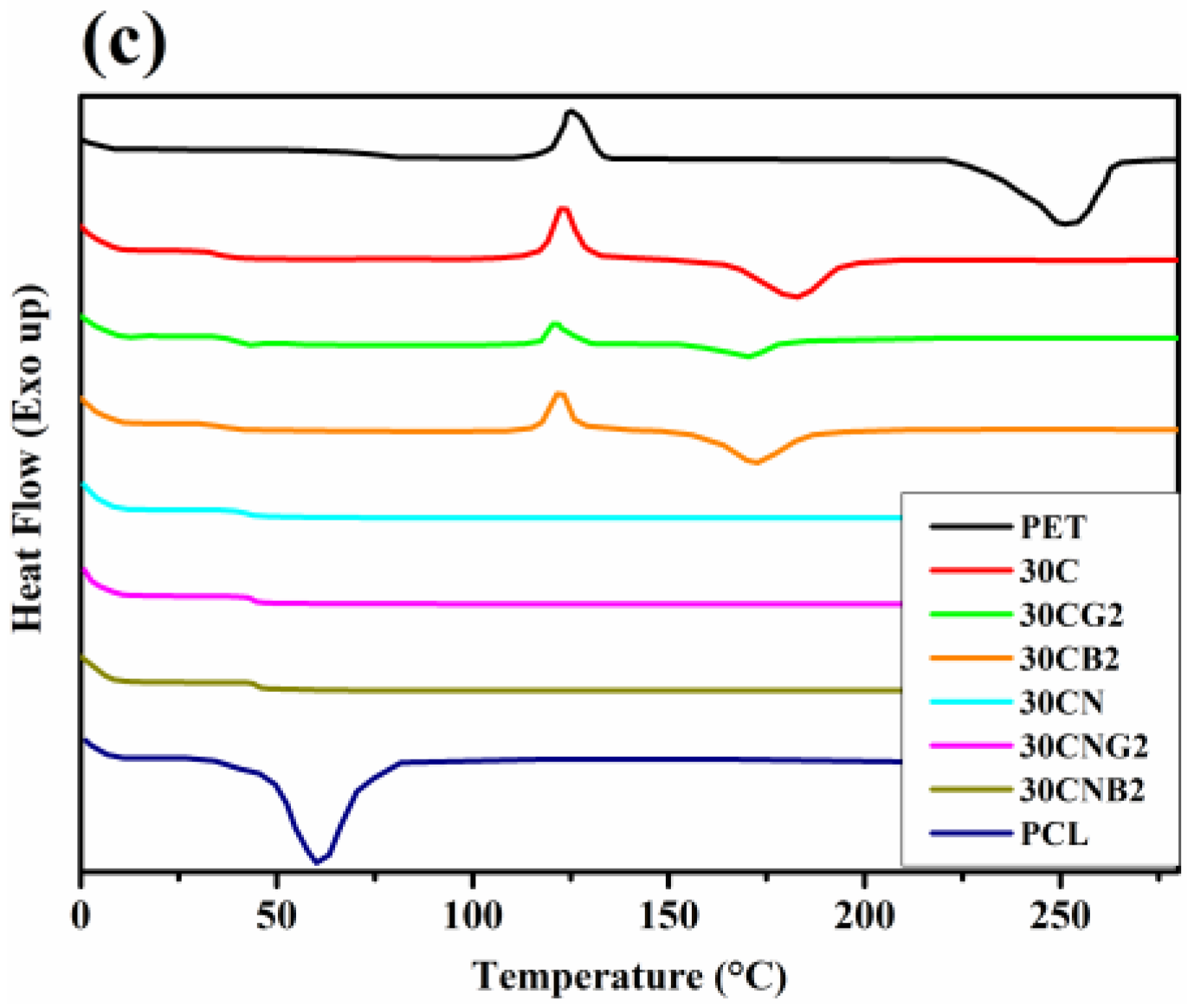
2.6. Differential Scanning Calorimetry
2.7. Dynamic Mechanical Analysis
2.8. Thermogravimetric Analysis
2.9. Equilibrium Swelling Analysis
2.10. Wide-Angle X-ray Diffraction
2.11. Tensile Test
2.12. Enzymatic Degradation
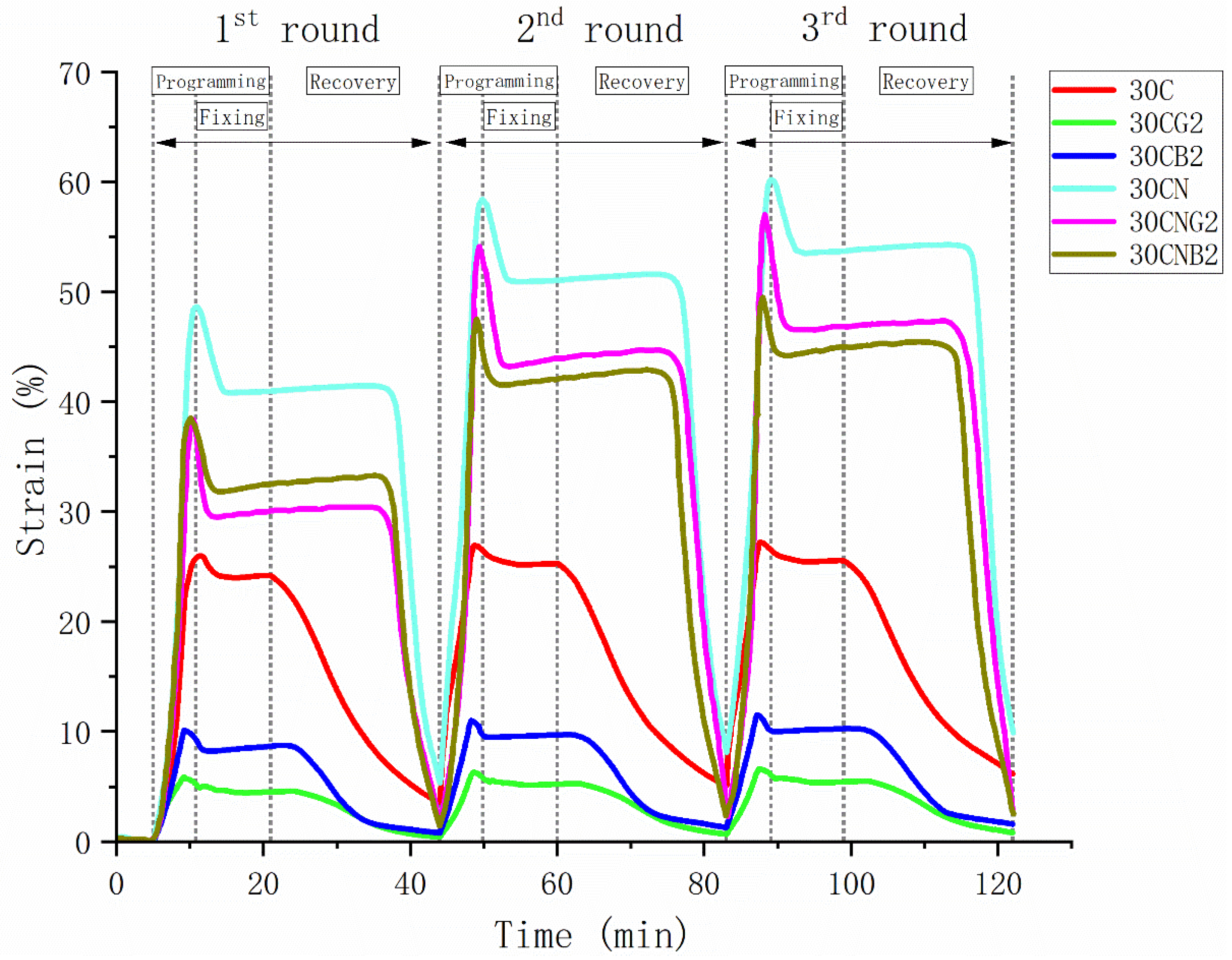
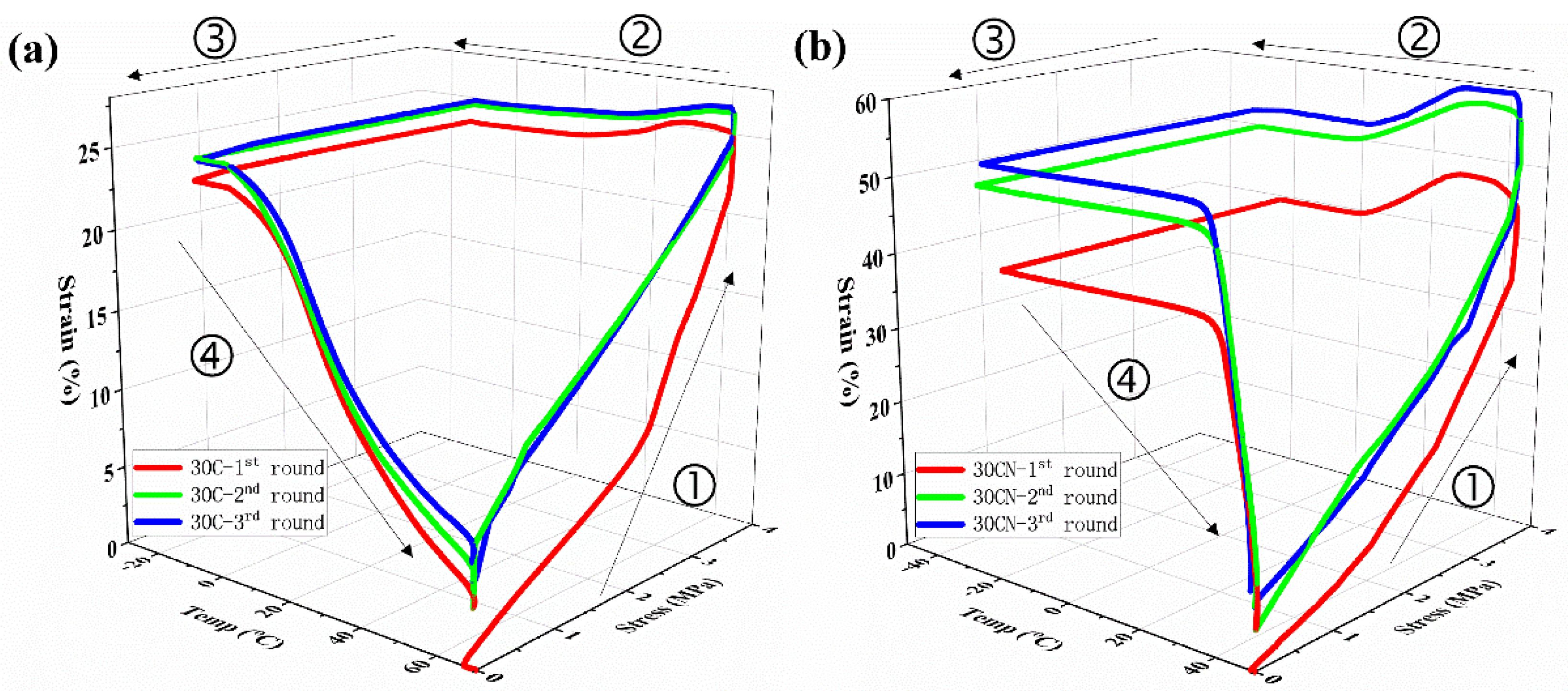
2.13. Shape-Memory Properties
3. Results and Discussion
3.1. Synthesis and Structure Characterization of the Copolyesters
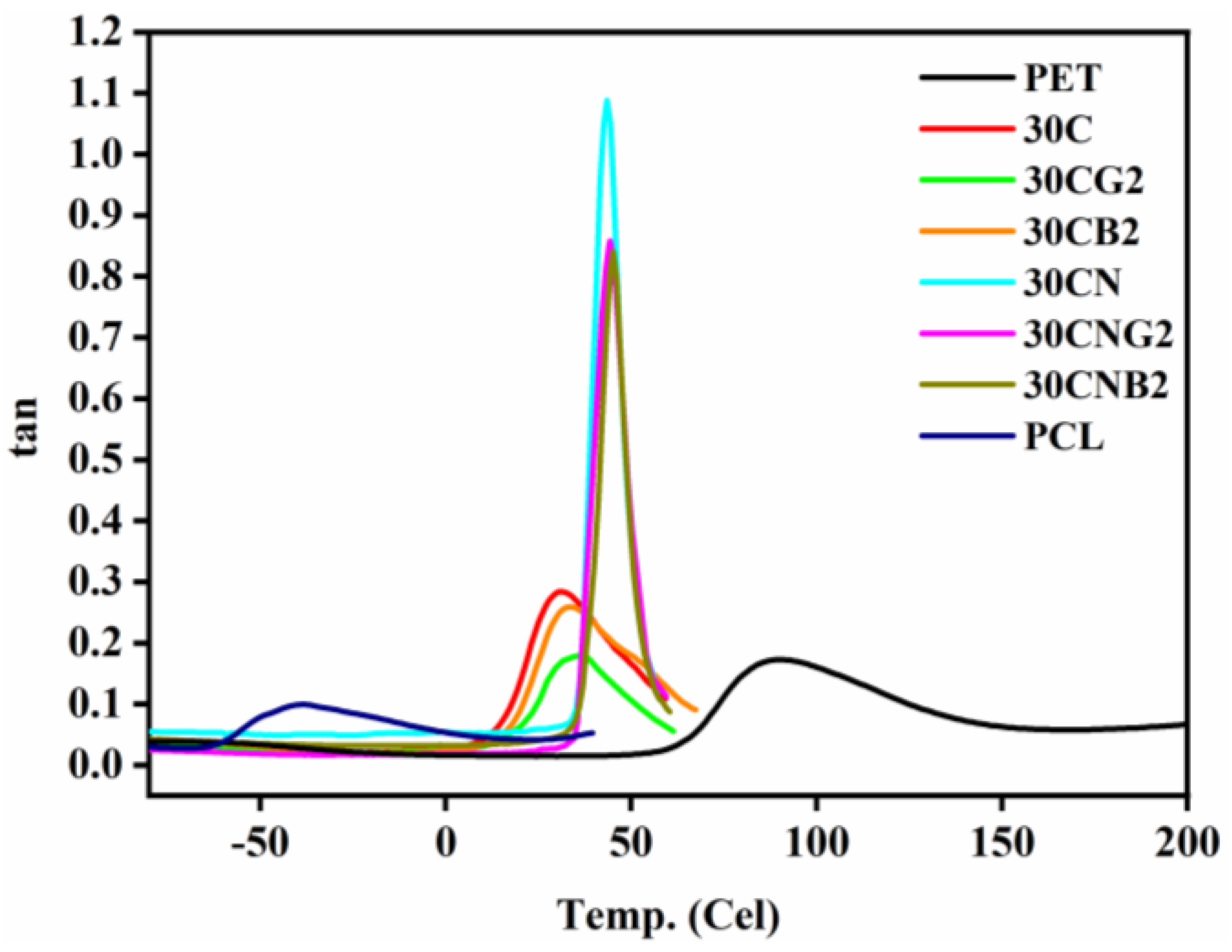
3.2. Thermal Properties of the Copolyesters
3.3. Mechanical Performance of the Copolyesters
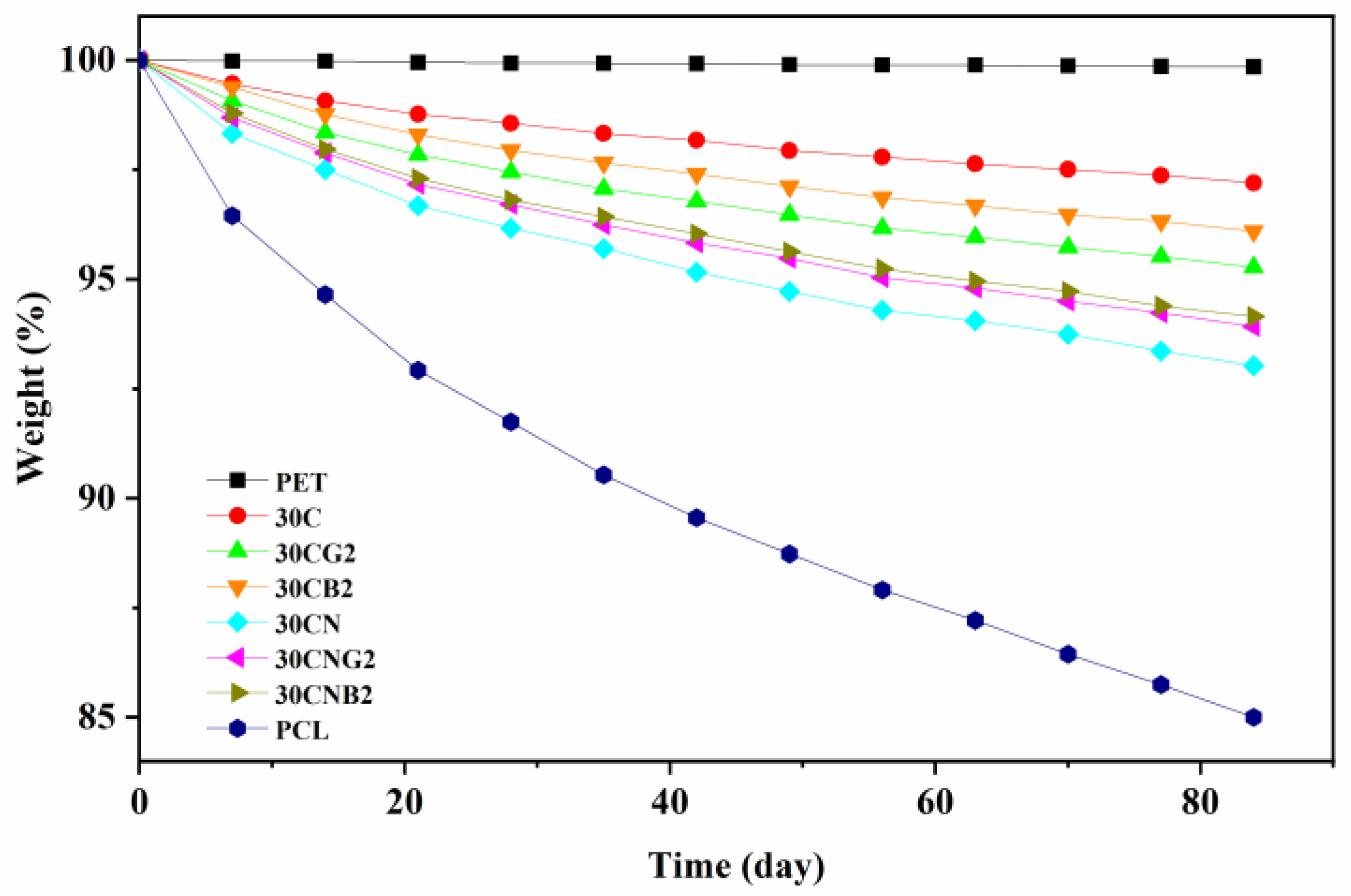
3.4. Controllable Degradation of Cross-Linked Copolyesters
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.S.; Ni, Q.Q.; Fu, S.Y.; Kurashiki, K. Electromagnetic interference shielding effect of nanocomposites with carbon nanotube and shape memory polymer. Compos. Sci. Technol. 2007, 67, 2973–2980. [Google Scholar] [CrossRef]
- Tobushi, H.; Hara, H.; Yamada, E.; Hayashi, S. Thermomechanical properties in a thin film of shape memory polymer of polyurethane series. Smart Mater. Struct. 1996, 5, 483–491. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Y.; Huang, H.; Lu, J. Recent advances in shape-memory polymers: Structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 2012, 37, 1720–1763. [Google Scholar] [CrossRef]
- Behl, M.; Razzaq, M.Y.; Lendlein, A. Multifunctional shape-memory polymers. Adv. Mater. 2010, 22, 3388–3410. [Google Scholar] [CrossRef]
- Du, H.; Liu, L.; Zhang, F.; Leng, J.; Liu, Y. Triple-shape memory effect in a styrene-based shape memory polymer: Characterization, theory and application. Compos. Part B Eng. 2019, 173, 106905. [Google Scholar] [CrossRef]
- Hu, W.; Lum, G.Z.; Mastrangeli, M.; Sitti, M. Small-scale soft-bodied robot with multimodal locomotion. Nature 2018, 554, 81–85. [Google Scholar] [CrossRef]
- Qi, X.; Yao, X.; Deng, S.; Zhou, T.; Fu, Q. Water-induced shape memory effect of graphene oxide reinforced polyvinyl alcohol nanocomposites. J. Mater. Chem. A 2014, 2, 2240–2249. [Google Scholar] [CrossRef]
- Yang, F.T.; Chen, Y.M.; Rwei, S.P. Synthesis and Characterization of the Temperature Controllable Shape Memory of Polycaprolactone/Poly (ethylene terephthalate) Copolyester. Fibers Polym. 2022, 23, 2526–2538. [Google Scholar] [CrossRef]
- Behl, M.; Lendlein, A. Shape-memory polymers. Mater. Today 2007, 10, 20–28. [Google Scholar] [CrossRef]
- Peterson, G.I.; Dobrynin, A.V.; Becker, M.L. Biodegradable Shape Memory Polymers in Medicine. Adv. Healthc. Mater. 2017, 6, 1–16. [Google Scholar] [CrossRef]
- Liu, C.; Qin, H.; Mather, P.T. Review of progress in shape-memory polymers. J. Mater. Chem. 2007, 17, 1543–1558. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, B.H.; Li, Z. Biodegradable polyester shape memory polymers: Recent advances in design, material properties and applications. Mater. Sci. Eng. C 2018, 92, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, Z.; Wu, S.; Wei, J.; Xiao, C. Uniaxial orientation and crystallization behavior of amorphous poly (ethylene terephthalate) fibers. J. Polym. Res. 2006, 13, 9–15. [Google Scholar] [CrossRef]
- Jacquel, N.; Saint-Loup, R.; Pascault, J.P.; Rousseau, A.; Fenouillot, F. Bio-based alternatives in the synthesis of aliphatic-aromatic polyesters dedicated to biodegradable film applications. Polymer 2015, 59, 234–242. [Google Scholar] [CrossRef]
- Mouzakis, D.E.; Papke, N.; Wu, J.S.; Karger-Kocsis, J. Fracture toughness assessment of poly (ethylene terephthalate) blends with glycidyl methacrylate modified polyolefin elastomer using essential work of fracture method. J. Appl. Polym. Sci. 2001, 79, 842–852. [Google Scholar] [CrossRef]
- Benvenuta Tapia, J.J.; Tenorio-López, J.A.; Martínez-Estrada, A.; Guerrero-Sánchez, C. Application of RAFT-synthesized reactive tri-block copolymers for the recycling of post-consumer R-PET by melt processing. Mater. Chem. Phys. 2019, 229, 474–481. [Google Scholar] [CrossRef]
- Yin, Q.; Yun, X.; Chen, Z.; Cao, W.; Zhang, Q.; Fu, Q. Largely improved tensile properties of poly (ethylene terephthalate) by using glycerin. Chin. J. Polym. Sci. 2007, 25, 319–324. [Google Scholar] [CrossRef]
- Lim, K.Y.; Kim, B.C.; Yoon, K.J. Structural and physical properties of biodegradable copolyesters from poly (ethylene terephthalate) and polycaprolactone blends. J. Appl. Polym. Sci. 2003, 88, 131–138. [Google Scholar] [CrossRef]
- Jikei, M.; Takeyama, Y.; Yamadoi, Y.; Shinbo, N.; Matsumoto, K.; Motokawa, M.; Ishibashi, K.; Yamamoto, F. Synthesis and properties of Poly (L-lactide)-Poly (ε-caprolactone) multiblock copolymers by the self-polycondensation of diblock macromonomers. Polym. J. 2015, 47, 657–665. [Google Scholar] [CrossRef]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef]
- Stanley, N.; Bucataru, G.; Miao, Y.; Favrelle, A.; Bria, M.; Stoffelbach, F.; Woisel, P.; Zinck, P. Brønsted acid-catalyzed polymerization of ε-caprolactone in water: A mild and straightforward route to poly (ε-caprolactone)-graft-water-soluble polysaccharides. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 2139–2145. [Google Scholar] [CrossRef]
- Christodoulou, E.; Klonos, P.A.; Tsachouridis, K.; Zamboulis, A.; Kyritsis, A.; Bikiaris, D.N. Synthesis, crystallization, and molecular mobility in poly (ϵ-caprolactone) copolyesters of different architectures for biomedical applications studied by calorimetry and dielectric spectroscopy. Soft Matter 2020, 16, 8187–8201. [Google Scholar] [CrossRef] [PubMed]
- Gumede, T.P.; Luyt, A.S.; Müller, A.J. Review on PCL, PBS, AND PCL/PBS blends containing carbon nanotubes. Express Polym. Lett. 2018, 12, 505–529. [Google Scholar] [CrossRef]
- Avérous, L.; Pollet, E. Silicate Nano-Biocomposites; Amouroux, J., Ed.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2006; ISBN 9781608052851. [Google Scholar]
- Williams, J.M.; Adewunmi, A.; Schek, R.M.; Flanagan, C.L.; Krebsbach, P.H.; Feinberg, S.E.; Hollister, S.J.; Das, S. Bone tissue engineering using polycaprolactone scaffolds fabricated via selective laser sintering. Biomaterials 2005, 26, 4817–4827. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer-Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Mallek, H.; Jegat, C.; Mignard, N.; Taha, M.; Abid, M.; Abid, S. One-step synthesis of PCL-urethane networks using a crosslinking/de-crosslinking agent. J. Macromol. Sci. Part A Pure Appl. Chem. 2013, 50, 728–737. [Google Scholar] [CrossRef]
- Dash, T.K.; Konkimalla, V.B. Poly-є-caprolactone based formulations for drug delivery and tissue engineering: A review. J. Control. Release 2012, 158, 15–33. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, J.; Yeung, K.; Choi, K.; Liu, Y.; Liem, H. Effect of cationic group content on shape memory effect in segmented polyurethane cationomer. J. Appl. Polym. Sci. 2007, 103, 545–556. [Google Scholar] [CrossRef]
- Rabani, G.; Luftmann, H.; Kraft, A. Synthesis and characterization of two shape-memory polymers containing short aramid hard segments and poly (ε{lunate}-caprolactone) soft segments. Polymer 2006, 47, 4251–4260. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, J.L.; Yeung, K.W.; Liu, Y.Q.; Liem, H.M. Influence of ionic groups on the crystallization and melting behavior of segmented polyurethane ionomers. J. Appl. Polym. Sci. 2006, 100, 4603–4613. [Google Scholar] [CrossRef]
- Zhu, G.; Liang, G.; Xu, Q.; Yu, Q. Shape-memory effects of radiation crosslinked poly (ε-caprolactone). J. Appl. Polym. Sci. 2003, 90, 1589–1595. [Google Scholar] [CrossRef]
- Zhu, G.M.; Xu, Q.Y.; Liang, G.Z.; Zhou, H.F. Shape-memory behaviors of sensitizing radiation-crosslinked polycaprolactone with polyfunctional poly (ester acrylate). J. Appl. Polym. Sci. 2005, 95, 634–639. [Google Scholar] [CrossRef]
- Nagata, M.; Kitazima, I. Photocurable biodegradable poly (ε-caprolactone)/poly (ethylene glycol) multiblock copolymers showing shape-memory properties. Colloid Polym. Sci. 2006, 284, 380–386. [Google Scholar] [CrossRef]
- Nagata, M.; Sato, Y. Synthesis and properties of photocurable biodegradable multiblock copolymers based on poly (ε-caprolactone) and poly (L-lactide) segments. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 2426–2439. [Google Scholar] [CrossRef]
- Petrova, T.; Manolova, N.; Rashkov, I.; Li, S.; Vert, M. Synthesis and Characterization of Poly(oxyethylene)-Poly(caprolactone) Multiblock Copolymers. Polym. Int. 1998, 45, 419–426. [Google Scholar] [CrossRef]
- Cohn, D.; Stern, T.; González, M.F.; Epstein, J. Biodegradable poly (ethylene oxide)/poly (ϵ-caprolactone) multiblock copolymers. J. Biomed. Mater. Res. 2002, 59, 273–281. [Google Scholar] [CrossRef]
- Lendlein, A.; Schmidt, A.M.; Schroeter, M.; Langer, R. Shape-memory polymer networks from oligo (є-caprolactone) dimethacrylates. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1369–1381. [Google Scholar] [CrossRef]
- Ping, P.; Wang, W.; Chen, X.; Jing, X. Poly (ε-caprolactone) polyurethane and its shape-memory property. Biomacromolecules 2005, 6, 587–592. [Google Scholar] [CrossRef]
- Ratna, D.; Karger-Kocsis, J. Recent advances in shape memory polymers and composites: A review. J. Mater. Sci. 2008, 43, 254–269. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, C.; Gu, X. Thermosetting polyurethanes with water-swollen and shape memory properties. J. Appl. Polym. Sci. 2002, 84, 1504–1512. [Google Scholar] [CrossRef]
- Luo, Q.; Chen, J.; Gnanasekar, P.; Ma, X.; Qin, D.; Na, H.; Zhu, J.; Yan, N. Highly Cross-Linked and Stable Shape-Memory Polyurethanes Containing a Planar Ring Chain Extender. ACS Appl. Polym. Mater. 2020, 2, 5259–5268. [Google Scholar] [CrossRef]
- Pavel, D.; Shanks, R. Molecular dynamics simulation of diffusion of O2 and CO2 in amorphous poly (ethylene terephthalate) and related aromatic polyesters. Polymer 2003, 44, 6713–6724. [Google Scholar] [CrossRef]
- Lee, T.; Yu, H.; Forrester, M.; Wang, T.; Shen, L.; Liu, H.; Li, J.; Li, W.; Kraus, G.; Cochran, E. Next-Generation High-Performance Bio-Based Naphthalate Polymers Derived from Malic Acid for Sustainable Food Packaging. ACS Sus. Chem. Eng. 2022, 10, 2624–2633. [Google Scholar] [CrossRef]
- Billmeyer, F.W. Methods for estimating intrinsic viscosity. J. Polym. Sci. 1949, 4, 83–86. [Google Scholar] [CrossRef]
- Flory, P.J.; Rehner, J. Statistical Mechanics of Cross-Linked Polymer Networks II. Swelling. J. Chem. Phys. 1943, 11, 521–526. [Google Scholar] [CrossRef]
- Barton, A.F.M. CRC Handbook of Solubility Parameters and Other Cohesion Parameters, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1991; ISBN 978-0-8493-0176-6. [Google Scholar]
- Heidarzadeh, N.; Rafizadeh, M.; Taromi, F.A.; del Valle, L.J.; Franco, L.; Puiggalí, J. Biodegradability and biocompatibility of copoly (butylene sebacate-co-terephthalate)s. Polym. Degrad. Stab. 2017, 135, 18–30. [Google Scholar] [CrossRef]
- Pryjmaková, J.; Vokatá, B.; Slepička, P.; Siegel, J. Laser-Processed PEN with Au Nanowires Array: A Biocompatibility Assessment. Int. J. Mol. Sci. 2022, 23, 10953. [Google Scholar] [CrossRef]
- Lee, T.; Forrester, M.; Wang, T.; Shen, L.; Liu, H.; Dileep, D.; Kuehl, B.; Li, W.; Kraus, G.; Cochran, E. Dihydroxyterephthalate—A Trojan Horse PET Counit for Facile Chemical Recycling. Adv. Mater. 2023, 35, 2210154. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Input | Output * | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hard Segment | Soft Segment # | Cross-linker + | Hard Segment | Soft Segment | ||||||
| Acid or Ester | Alcohol | Acid or Ester | Alcohol | |||||||
| TPA | NDC | EG | CL | GC | BTC | TPA | NDC | EG | CL | |
| PET | 1 | 0 | 1.2 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| 30C | 1 | 0 | 1.2 | 0.51 | 0 | 0 | 1 | 0 | 0.65 | 0.30 |
| 30CG2 | 1 | 0 | 1.2 | 0.51 | 0.02 | 0 | 1 | 0 | 0.64 | 0.30 |
| 30CB2 | 1 | 0 | 1.2 | 0.51 | 0 | 0.02 | 1 | 0 | 0.66 | 0.36 |
| 30CN | 0.67 | 0.33 | 1.2 | 0.55 | 0 | 0 | 0.67 | 0.32 | 0.68 | 0.34 |
| 30CNG2 | 0.67 | 0.33 | 1.2 | 0.55 | 0.02 | 0 | 0.67 | 0.32 | 0.67 | 0.36 |
| 30CNB2 | 0.67 | 0.33 | 1.2 | 0.55 | 0 | 0.02 | 0.67 | 0.32 | 0.67 | 0.36 |
| Wavenumber (cm−1) | Type of Vibration |
|---|---|
| 3422 | Overtone C=O |
| 3063 | Benzene CH vibration |
| 2967, 2963 | Amorphous methylene stretch, CH2 |
| 2899 | Crystalline stretch, CH2 |
| 1720 | Carbonyl stretch, C=O |
| 1336, 1120, 871 | Aromatic ring vibration |
| 1239 | Asymmetric stretch, C-O-C |
| 1179, 764 | Naphthalene ring vibration |
| Sample | a | b, b1, b2, b3 | c, c′1, c″1 | c2, c′2, c3, c′3, d1, d′1 | d | e | f, f′,g | h | XEG′ | XCL | n | m + l | R |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8.8 | 8.1~8.3 | 4.9~5.1 | 4.4~4.8 | 4.25 | 2.5 | 1.8 | 1.5 | ||||||
| PET | - | 1.00 | 1.00 | - | - | - | - | - | 0.00 | - | - | - | - |
| 30C | - | 1.00 | 0.65 | 0.49 | 0.07 | 0.17 | 0.35 | 0.15 | 0.35 | 0.23 | 0.86 | 2.86 | 1.52 |
| 30CG2 | - | 1.00 | 0.64 | 0.50 | 0.08 | 0.17 | 0.35 | 0.15 | 0.36 | 0.23 | 0.83 | 2.78 | 1.56 |
| 30CB2 | - | 1.00 | 0.66 | 0.54 | 0.08 | 0.18 | 0.38 | 0.18 | 0.34 | 0.26 | 1.06 | 2.94 | 1.28 |
| 30CN | 0.16 | 1.00 | 0.68 | 0.48 | 0.03 | 0.18 | 0.38 | 0.17 | 0.31 | 0.26 | 1.11 | 3.22 | 1.22 |
| 30CNG2 | 0.16 | 1.00 | 0.67 | 0.52 | 0.03 | 0.18 | 0.38 | 0.18 | 0.32 | 0.27 | 1.14 | 3.12 | 1.21 |
| 30CNB2 | 0.16 | 1.00 | 0.67 | 0.50 | 0.03 | 0.19 | 0.4 | 0.18 | 0.32 | 0.27 | 1.14 | 3.12 | 1.21 |
| PCL | - | - | - | - | 1.00 | 1.00 | 2.00 | 1.00 | - | - | - | - | - |
| Sample | DSC 1st Heating | DSC 1st Cooling | DSC 2nd Heating | DMA | TGA | X * | CD # | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tg | Tcc | ΔHcc | Tm | ΔHm | Tc | ΔHc | Tg | Tcc | ΔHcc | Tm | ΔHm | Tg | tanδ | Td-5% | Char | |||
| (°C) | (°C) | (J/g) | (°C) | (J/g) | (°C) | (J/g) | (°C) | (°C) | (J/g) | (°C) | (J/g) | (°C) | - | (°C) | (%) | |||
| PET | 72.9 | 126.5 | 3.8 | 251.4 | 38.5 | 173.4 | −22.7 | 74.9 | 123.6 | 13.8 | 250.6 | 37.5 | 90.1 | 0.17 | 397.9 | 20.0 | 18.9 | 0 |
| 30C | 34.2 | 124.2 | 3.4 | 183.1 | 22.8 | 111.9 | −4.8 | 34.4 | 122.3 | 11.1 | 182.3 | 16.4 | 31.4 | 0.28 | 376.1 | 12.7 | 4.0 | 0 |
| 30CG2 | 38.9 | 125.5 | 1.2 | 172.1 | 14.1 | 120.2 | −1.5 | 39.8 | 120.3 | 5.1 | 170.0 | 7.9 | 35.2 | 0.18 | 381.0 | 13.1 | 1.2 | 156 |
| 30CB2 | 36.2 | 124.4 | 2.6 | 177.0 | 17.5 | 116.9 | −2.9 | 36.5 | 121.5 | 9.1 | 172.8 | 12.3 | 33.6 | 0.21 | 379.1 | 13.4 | 2.4 | 97 |
| 30CN | 42.5 | - | - | - | - | - | - | 41.6 | - | - | - | - | 43.5 | 1.09 | 383.4 | 17.1 | - | 0 |
| 30CNG2 | 44.4 | - | - | - | - | - | - | 43.6 | - | - | - | - | 44.3 | 0.86 | 386.2 | 17.4 | - | 77 |
| 30CNB2 | 46.8 | - | - | - | - | - | - | 45.0 | - | - | - | - | 45.1 | 0.84 | 388.2 | 17.7 | - | 89 |
| PCL | - | - | - | 61.0 | 51.7 | 20.6 | −44.8 | - | - | - | 60.7 | 51.1 | 38.4 | 0.09 | 330.5 | 2.6 | 32.1 | 0 |
| Sample | Tensile Strength | Elongation at Break | Yielding Strength | Young’s Modulus | |
|---|---|---|---|---|---|
| MPa | % | MPa | MPa | dL/g | |
| PET | 64.9 | 6.8 | - | 1064.8 | 0.59 |
| 30C | 27.8 | 200.1 | 8.5 | 556.0 | 0.80 |
| 30CG2 | 35.8 | 139.8 | - | 151.2 | 0.94 |
| 30CB2 | 32.8 | 169.8 | 24.0 | 257.9 | 0.85 |
| 30CN | 53.2 | 9.4 | - | 675.4 | 0.70 |
| 30CNG2 | 54.4 | 8.3 | - | 763.5 | 0.76 |
| 30CNB2 | 57.1 | 8.3 | - | 773.1 | 0.78 |
| PCL | 21.8 | 1201.0 | 22.8 | 89.3 | 1.07 |
| Sample | Rf(1) | Rf(2) | Rf(3) | Rr(1) | Rr(2) | Rr(3) |
|---|---|---|---|---|---|---|
| 30C | 93.0 | 93.9 | 93.9 | 85.2 | 93.0 | 94.9 |
| 30CG2 | 79.1 | 82.9 | 82.2 | 90.8 | 94.6 | 97.1 |
| 30CB2 | 85.1 | 88.2 | 89.3 | 90.9 | 94.4 | 96.7 |
| 30CN | 84.2 | 87.4 | 89.3 | 87.0 | 93.9 | 96.1 |
| 30NG2 | 78.1 | 81.5 | 82.1 | 94.8 | 97.9 | 99.0 |
| 30NB2 | 84.3 | 88.2 | 90.8 | 96.0 | 97.9 | 99.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.-T.; Chen, Y.-M.; Rwei, S.-P. Influence of Cross-Linking and Crystalline Morphology on the Shape-Memory Properties of PET/PEN/PCL Copolyesters Using Trimesic Acid and Glycerol. Polymers 2023, 15, 2082. https://doi.org/10.3390/polym15092082
Yang F-T, Chen Y-M, Rwei S-P. Influence of Cross-Linking and Crystalline Morphology on the Shape-Memory Properties of PET/PEN/PCL Copolyesters Using Trimesic Acid and Glycerol. Polymers. 2023; 15(9):2082. https://doi.org/10.3390/polym15092082
Chicago/Turabian StyleYang, Fu-Ting, Yu-Ming Chen, and Syang-Peng Rwei. 2023. "Influence of Cross-Linking and Crystalline Morphology on the Shape-Memory Properties of PET/PEN/PCL Copolyesters Using Trimesic Acid and Glycerol" Polymers 15, no. 9: 2082. https://doi.org/10.3390/polym15092082
APA StyleYang, F.-T., Chen, Y.-M., & Rwei, S.-P. (2023). Influence of Cross-Linking and Crystalline Morphology on the Shape-Memory Properties of PET/PEN/PCL Copolyesters Using Trimesic Acid and Glycerol. Polymers, 15(9), 2082. https://doi.org/10.3390/polym15092082